
Abstract
Cupriavidus sp. UYMMa02A is a beta-rhizobia strain of the Cupriavidus genus, isolated from nodules of Mimosa magentea in Uruguay. This strain can form effective nodules with several Mimosa species, including its original host. Genome analyses indicate that Cupriavidus sp. UYMMa02A has a highly conserved 35 kb symbiotic island containing nod, nif, and fix operons, suggesting conserved mechanisms for the symbiotic interaction with plant hosts. However, while Cupriavidus sp. UYMMa02A produces functional nodules and promotes Mimosa pudica growth under nitrogen-limiting conditions, nod genes are not induced by luteolin or exposure to Mimosa spp. root exudate. To explore alternative mechanisms implicated in the Cupriavidus-Mimosa interaction, we assessed the proteomic profiles of Cupriavidus sp. UYMMa02A grown in the presence of pure flavonoids and co-culture with M. pudica plants. This approach allowed us to identify 24 differentially expressed proteins potentially involved in bacterial-plant interaction. In light of the obtained results, a possible model for nod-alternative symbiotic interaction is proposed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rhizobia are soil bacteria that engage in symbiotic interactions with legume plants. During this interaction, new specialized organs called nodules are formed in the roots (and sometimes in stems) of host plants. Inside the nodules, bacteria convert atmospheric nitrogen into ammonia in a process known as Symbiotic Nitrogen Fixation (SNF) (Lindström and Mousavi 2020).
The ability to form nitrogen-fixing nodules in symbiosis with legumes is restricted to alpha and beta subgroups of proteobacteria (Andrews and Andrews 2017). The best-characterized rhizobia species; Sinorhizobium meliloti, Rhizobium leguminosarum, or Bradyrhizobium japonicum, belong to the Rhizobiaceae family of alpha-proteobacteria. In beta-proteobacteria, the Burkholderiaceae family is the best characterized and includes just three genera Paraburkholderia, Trinickia, and Cupriavidus (Chen et al. 2001, 2003; Dall’Agnol et al. 2017; Estrada-de los Santos et al. 2018). Both Paraburkholderia and Cupriavidus rhizobia strains were described at the beginning of the twenty-first century. Since then, beta-rhizobia have been described as legume symbionts in America (Bontemps et al. 2010; Andam et al. 2007; dos Reis et al. 2010; Taulé et al. 2012), Africa (Garau et al. 2009; Howieson et al. 2013; Lemaire et al. 2015), Asia (Liu et al. 2012; Gehlot et al. 2013) and Oceania (Parker et al. 2007), primarily associated with legumes of Mimosoid clade, but also with some members of the Papilionoideae (Garau et al. 2009; Lemaire et al. 2015). Despite their worldwide distribution, the molecular mechanisms involved in the interaction between beta-rhizobia and host plants have been analyzed in a few model strains (Amadou et al. 2008; de Campos et al. 2017; Lardi et al. 2017; Klonowska et al. 2018; Bellés-Sancho et al. 2022; Rodríguez‐Esperón et al. 2022). Genome analyses of beta-rhizobia that nodulate Mimosoid clade have shown the presence of a highly conserved and compact genomic region, known as the symbiotic island, that encodes for nod, nif, and fix genes (De Meyer et al. 2016; Zheng et al. 2017). The nod genes code for proteins involved in the synthesis (nodBCHASUQ) and exportation (nodIJ) of nodulation (Nod) factors, as well as their regulation (nodD). In turn, the nif genes encode proteins related to the nitrogenase complex (nifH, nifD, and nifK), regulation (nifA), and maturation processes (nifEDXQ). The fix genes encode membrane proteins required for electron transfer to generate the energy required for the SNF process (fixABCX; fixNOPQ).
In beta-rhizobia, nod genes are induced in the presence of pure flavonoids such as luteolin and apigenin (Marchetti et al. 2011; Rodríguez‐Esperón et al. 2022) or root exudates of the host plant Mimosa pudica (Klonowska et al. 2018). In line with the conservation of this important recognition mechanism, other genes and molecules involved in bacteria-plant interaction have been identified in beta-rhizobia. These include the type-III secretion system (Saad et al. 2012) and synthesis of branched-chain amino acids in Cupriavidus taiwanensis LMG19424 (Chen et al. 2012); type VI secretion system (de Campos et al. 2017; Lardi et al. 2017) and synthesis of exopolysaccharide (EPS) cepacian in Paraburkholderia phymatum STM815 (Liu et al. 2020). Despite these few examples, there is still a lack of knowledge about the molecular mechanisms implicated in the symbiotic interaction between beta-rhizobia and legume hosts.
In Uruguay, beta-rhizobia have been identified as the main symbionts of Parapiptadenia rigida (Taulé et al. 2012) and Mimosa spp. (Platero et al. 2016; Pereira-Gómez et al. 2020). Phylogenetic analyses indicated that Cupriavidus strains symbiotically associated with these plants do not belong to the well-studied C. taiwanensis species but rather to the C. necator and other Cupriavidus species.
Cupriavidus sp. UYMMa02A was isolated from nodules of Mimosa magentea, a native legume found in the southeast region of Uruguay. Through a Multi-Locus Sequence Analysis (MLSA) approach, it was demonstrated that UYMMa02A does not belong to the previously described C. taiwanensis and C. necator, but it may represent a novel rhizobial species within the genus Cupriavidus (Platero et al. 2016). Genome sequencing of Cupriavidus sp. UYMMa02A (Iriarte et al. 2016) indicated the presence of a conserved and compact symbiotic island that encodes for the nod, nif, and fix genes observed in other beta-rhizobia (Amadou et al. 2008; Moulin et al. 2014; De Meyer et al. 2015a, b, 2016). In addition, this strain induces pink nodules on the roots of its original host and other Mimosa species, including M. pudica (Platero et al. 2016).
The present study aimed to analyze the initial steps of the Cupriavidus-Mimosa symbiotic interaction. Firstly, we determined the expression of nod genes in Cupriavidus sp. UYMMa02A strain when cultivated in the presence of pure flavonoids or Mimosa spp. root exudates. Then we analyzed the proteomic changes induced in the bacteria by the presence of pure flavonoids and the plant host. Finally, we integrated the obtained results into a model that reports the changes occurring during the initial steps of the Cupriavidus sp. UYMMa02A—Mimosa pudica symbiotic interaction.
Material and methods
Bacterial strains, plasmids, and growth conditions
The bacteria and plasmids used in this study are listed in Table 1. Escherichia coli strains were grown aerobically at 37 °C in Luria–Bertani (LB) medium. Cupriavidus strains were grown at 30 °C in LB or M9 minimal medium containing 14 mM sodium citrate as a carbon source (Sambrook et al. 1989). When indicated M9 cultures were supplemented with 5 μM luteolin or apigenin. Modified M9 media containing Mimosa spp. root exudates were prepared to replace the original water volume used in M9, by filter-sterilized root exudates. When required the following antibiotics were used for strain selection; ampicillin 100 μg ml−1 (Ap), nitrofurantoin 50 μg ml−1 (Nf), chloramphenicol 25 μg ml−1 (Cf), and tetracycline 8 μg ml−1 (Tc).
Sequence analysis
Homology searches and sequences retrieval were done via Internet server BLAST (NCBI, NIH, Bethesda, MD, USA: http://www.ncbi.nlm.nih.gov) and RAST (Aziz et al. 2008). Sequence alignments were done by Mega v7.0.26 (Kumar et al. 2016) and SnapGene® software (from Insightful Science; available at snapgene.com) was used for graphic presentation.
Reporter strains construction
To evaluate the expression of nodB gene promoter, plasmid pCBM01 containing the pnodB19424-lacZ transcriptional fusion (Marchetti et al. 2010) or plasmid pCZ388 containing a promoterless lacZ (Cunnac et al. 2004) were introduced in Cupriavidus spp. strains by triparental mating as previously described (Rodríguez‐Esperón et al. 2022).
Assessment of pnodB 19424 -lacZ gene expression
Cultures of Cupriavidus spp. strains carrying the plasmids pCBM01 or pCZ388 were grown overnight at 30 °C using a rotary shaker at 200 rpm, in 5 mL of M9 supplemented with Tc. At the end of this time, a 1/100 (v/v) dilution was made in fresh medium supplemented with 5 µM luteolin (≥ 98% TLC, SIGMA, USA) or in a modified M9 prepared with roots exudates and incubated for 18 h at 30 ºC. Uninduced control cultures were included for each assay. Beta-galactosidase assays were performed according to the standard Miller assay (Miller 1972).
Pre-treatment, surface sterilization, and germination of Mimosa spp. seeds
M. pudica seeds were obtained commercially from Outsidepride Seeds, LLC (Oregon, USA). M. magentea seeds were collected from native plant populations growing in Uruguay (Platero et al. 2016). The seeds were submerged in ethanol (95%) for 2 min and dried in filter paper. After they were treated with 10 M sulfuric acid for 20 min, followed by seven washes with sterile distilled water. Finally, the seeds were treated with 4% sodium hypochlorite for 5 min followed by seven washes with sterile distilled water. Surface-sterilized seeds were germinated on 0.8% (wt/vol) agar-water plates, at 30 °C in dark conditions for 2 days.
Roots exudate collection method
Mimosa spp. pre-germinated seeds were sown in a 250 mL test glass jar (20 seeds per flask) containing 20 mL of sterile water and a stainless-steel grate for seedling support. Plants were incubated for five days under a photoperiod of 16 h light/8 h darkness at 26 °C. After that, the water solution containing the roots exudates was collected in 50 mL plastic conical tubes, centrifuged for 5 min at 6,000 g to remove cellular debris and supernatants were filtered-sterilized using 0.45 µm membrane filters (Millipore, USA). Filter-sterilized root exudates were used, instead of water, for preparing modified M9 media. All experiments including root exudates were performed with freshly (same day) prepared root exudates.
Gene expression based on quantitative reverse transcription-PCR (RT-qPCR)
The RT-qPCR experiments were performed with Cupriavidus sp. UYMMa02A growing in M9 with M. pudica root exudates or M9 media. For the bacterial RNA extraction, Cupriavidus sp. UYMMa02A was cultivated in 150 mL of modified M9 with M. pudica root exudates or M9 until reaching the mid-exponential phase (OD600 between 0.6–0.8). Cultures were then incubated for 20 min with 100 µg/mL chloramphenicol and then rapidly cooled by placing the tubes in a water–ice bath. Cells were recovered by centrifuging at 5000g for 5 min at 4 °C. Obtained pellets were suspended in 1.5 mL of lysis buffer (20 mM Tris–HCl Buffer pH 7.6; 50 mM MgCl2; 150 mM NH4Cl) and transferred to 2 mL lysis tubes (MP Biomedicals Lysing Matrix Tubes # Matrix B). Cell lysis was carried out on Fast Prep equipment (MP Biomedicals) using a cycle of 6.0 m/s for 60 s. Finally, lysates were centrifuged at 10,000g for 20 min at 4 °C, and supernatants containing the cell extract were transferred to a new tube. One hundred microliters of the obtained extracts were used for total RNA extraction using a PureLink RNA Mini Kit (Thermo Fisher Scientific #12183018A). Obtained RNA was treated with 5 units of DNase I (Thermo Fisher Scientific EN0521) for 5 min at 37 °C. One microgram of total RNA was converted to cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystem), following the manufacturer’s recommendations. Quantitative PCR (qPCR) analyses were performed essentially as described (Rodríguez-Esperón et al. 2022). The UYMMa02A efg gene (ODV42482.1), which encodes for the elongation factor G and the s14 gene (ODV43065.1), which encodes for the ribosome protein S14, were used as reference genes. The UYMMa02A nodA, nodB, and nodC genes were selected to analyze relative gene expression. Primer Blast software was used to design primer sets for nodA, nodB, nodC, efg, and s14. The following primers were used: efg-for (5′-GCGATCATTTGGGACGAAGC-3′); efg-rev (5′-CGGACTCGACCATCTTCTCG-3′); s14-for (5′-CTGTTCTACGTGTCAG-3′); s14-rev (5′-TGATGTTGATGCGGTGTTC-3′); nodA-for (5′-ACGTCCTCGCTGTGATTCTG-3′); nodA-rev (5′-AGGTCCGTTGCGTTCGATAG-3′); nodB-for (5′-TGGGGCAATTTCAGCTTCCA-3′; nodB-rev (5′-AGCGACTTCGTGTCCTTCAG-3′); nodC-for (5′-CAGAGCTTGCCTCACTTCCA-3′); nodC-rev (5′-TGCATCGTCCTCATAGTCGC-3′). qPCRs were performed on a C1000 Touch Thermal Cycler (Bio-Rad) using the iQTM SYBR Green Supermix (Bio-Rad). qPCRs conditions were as follows: 5 min at 95 °C, 40 cycles of 15 s at 95ºC, 30 s at 60 °C and 30 s at 72 °C. Primer specificity and dimer formation were checked by dissociation curves. A mixture of cDNA from induced and non-induced samples was used for calculating primer efficiency. The relative gene expression level was calculated using the 2−ΔΔCq method, statistical analyses were performed in the InfoStat statistical program (https://www.infostat.com.ar/). Normal distribution of data was confirmed using the Shapiro–Wilk test and one-way ANOVA was used for sample comparisons. Differences were considered statistically significant if the p-value < 0.05.
Assessment of growth promotion capacity
Germinated seeds prepared as described above were transferred into glass tubes containing 15 mL of Jensen’s N-free medium (36) solidified with 0.8% (wt/vol) agar. Seedlings were inoculated with 1 mL of a rhizobial suspension containing 1 × 107 cfu. One milliliter of sterile water was added to negative controls. Plants were grown under a photoperiod of 16 h light/8 h darkness at 26 °C. The presence and coloration of root nodules were evaluated periodically. Three months after inoculation, plants were harvested, and plant height was measured and used as a proxy for the plant growth promotion capacity of inoculated strains. The experiment was repeated three times with at least 10 plants per condition. Differences were considered statistically different if the p-value < 0.01 according to the t-test.
Cupriavidus sp. UYMMa02A-Mimosa pudica co-culture assays
A co-culture device was constructed allowing bacterial-plant signal exchange, but avoiding physical contact between both organisms (Fig. 9). Bacterial containers were assembled using 10 cm dialysis membrane tubing pieces (D9652, Sigma Aldrich). Membranes were washed with distillate water and one of their ends was sealed with a double knot, the other end was attached to a 10 cm silicone tubing and sealed with a plastic seal. For a complete and tight seal, a small glass tube was inserted inside the silicone tubing. Bacterial container sterilization was achieved by gamma-irradiation using a 21 kGy dose in the irradiation facilities of the Uruguayan Technological Laboratory (LATU). Test glass jars of 250 mL containing 50 mL of N-free Howieson (Howieson et al. 1993) liquid media diluted 1/10 (v/v) and polypropylene balls as seedling support, were autoclaved for 20 min at 121 °C. After cooling, the jars were opened in the laminar flow hoods, and bacterial containers were aseptically added, submerging the membrane in the media and the silicone tubing facing the jar lids. A total of 50 M. pudica germinated seeds were sown in each jar and incubated under a photoperiod of 16 h light/8 h darkness. After 5 days, the bacterial containers were filled through the silicone tubing, with 5 mL of 1 × 108 cfu mL−1 of Cupriavidus sp. UYMMa02A, and incubated for another five days under the same conditions of temperature and photoperiod. As control treatments, plant-free systems were used. In these cases, Howieson liquid media was supplemented with 2 mM sodium citrate as a carbon source and 2 mM ammonium chloride as a nitrogen source, to allow bacterial survival. Cell suspensions were carefully transferred from the membranes to clean and sterile plastic tubes and centrifuged for 5 min at 6000g. Bacterial pellets were used for total protein extraction. A minimum of three biological replicates were performed for each sample.
Total protein extraction and solubilization
For protein extraction, bacterial pellets were washed three times with 2 mL of phosphate-buffered saline (PBS) and resuspended in 10% of the original culture volume in PBS containing 1× complete-EDTA-Free protease inhibitor (Roche). Cell lysis was performed by sonication (7 cycles × 30 s) in continuous mode and at a relative power of 4 alternated with 30 s of rest in an ice-water bath, using an ultrasonic homogenizer (Cole-Parmer Instruments Co.). Cell lysates were separated by centrifugation at 12,000g for 15 min at 4 °C and the supernatant containing the total soluble proteins was kept at − 20 °C. At least three biological replicates were performed per sample.
Protein quantification
Protein concentration was estimated by the Bradford method (Bradford 1976) using bovine serum albumin (BSA) for calibration curves. The quality of protein fractions was verified by SDS-PAGE (Laemmli 1970).
Protein precipitation
Previous to two-dimensional (2D) electrophoresis, proteins were precipitated, washed, and concentrated, using the commercial kit “2-D Clean Up Kit” (GE Healthcare, Amersham Biosciences) according to the manufacturer's instructions. Finally, proteins were resuspended in a rehydration buffer (8 M urea, 2 M thiourea, 4% CHAPS, 40 mM DTT, 1.2% (v/v) IPG buffer) pH 8.5.
Protein labelling
Thirty-five µg of total proteins from each condition were labeled using the Refraction-2D™ Labelling Kit (NH DyeAGNOSTICS GmbH, Germany), following the manufacturer's recommendations. The samples were normalized and labeled with the Cy3 dye (λex = 532 nm/λem = 580 nm) or Cy5 (λex = 633 nm/λem = 670 nm), while Cy2 dye (λex = 488 nm/λem = 520 nm) was used to label the internal standard, which consists of a pooled sample comprising equal amounts (10 µg) of all samples to be compared.
2D-DIGE electrophoresis
The labeled samples were mixed in hydration buffer (8 M urea, 2 M thiourea, 2% (p/v) CHAPS, and 1.2% (v/v) IPG-buffer) and loaded on 24-cm Immobiline DryStrips previously hydrated in the same buffer (nonlinear pH range 3–10, GE Healthcare). Isoelectric focusing (IEF) was run using an IPGphor III apparatus (GE Healthcare). The voltage profile used was adapted to run overnight with the following voltage program: constant phase of 500 V/2 h, constant phase of 2000 V/2 h, a linear increase to 4000 V/2 h, a linear increase to 8000 V/2 h, and a constant final phase at 8000 V for 4 h. After that, the IPG strips were allowed to equilibrate for 10 min with mechanical agitation in 1 mL of equilibration buffer (6 M urea, 50 mM Tris–HCl, pH 8.6, 30% glycerol, 2% sodium dodecyl sulfate) containing 1% DDT for 10 min. Then, reactions were quenched by immersing the strips in SDS-PAGE running buffer (25 mM Tris, 192 mM glycine, 0.1% SDS). As a second dimension, proteins were separated by SDS-PAGE gels (12% acrylamide-bisacrylamide) using an Ettan DALT-Six apparatus Electrophoresis System electrophoresis cell maintained at 20 °C with the Multitemp III cooling unit (GE Healthcare). Each strip was placed on the corresponding acrylamide gel and bound to it, using a running buffer containing 0.2% agarose and 0.002% (w/v) bromophenol blue. The runs were carried out at 100 mA and 80 V for the first 5 min and then the voltage was increased to 120 V until the run front reached the end of the gel.
Image analysis
After 2D-DIGE electrophoresis, gels were fixed with ethanol:acetic acid:H2O (5:1:4) solution for 30 min. They were then scanned using a Typhoon 9500 FLA scanner (GE Healthcare), using the parameters recommended by the manufacturer, and analyzed using ImageQuant TL v8.1 software (GE Healthcare). The SameSpots software (Totallab, Newcastle, UK) was used to match and analyze protein spots, allowing the detection, normalization with the internal standard, and quantification of the spots. Differential-in-gel analysis was used to calculate protein abundance alterations between samples on the same gel. The resulting spot maps for each biological replicate were then analyzed through biological variation analysis to provide statistical data on the differential protein expression. Spots that exhibited differences in the level of fluorescence with a p-value ≤ 0.05 and a rate of change ≥ 1.25 were considered as differentially regulated and selected for identification.
Spot picking, protein digestion, and MALDI-TOF/TOF protein identification
To identify the differentially expressed proteins, gels were stained using Coomassie Brilliant Blue G-250 (Bio-Rad, Hercules, CA). Spots presenting significant differences were excised from gels, unstained with a solution of 0.1 M NH4HCO3 in acetonitrile (ACN) 50% (v/v) and in-gel digested overnight using modified sequencing grade trypsin (Promega, Madison, USA). Peptide extraction was performed as previously described (Gil et al. 2019) and samples were desalted using Zip-Tip C18 reverse phase microcolumns (Millipore, Merck, USA) eluted directly on the plate using matrix solution (α-cyano-4-hydroxycinnamic acid in 60% ACN, 0.1% TFA). The mixture was spotted onto an Opti-TOF plate of 384 positions (Ab Sciex). Spectra acquisition was performed on a MALDI-TOF/TOF MS (4800 Analyzer Abi Sciex) operated in positive reflector mode. The collected MS and MS/MS spectra of selected ions were externally calibrated using a standard peptide mix (Applied Biosystems). Protein identification was carried out using Mascot (Matrix Science, London, UK, (http://www.matrixscience.com) in Sequence query mode, using the genome of Cupriavidus sp. UYMMa02A (GCA_001725945.1) or the NCBI NR databases. The search parameters were: Unrestricted taxonomy; allowable trypsin cleavage jumps = 1; partial modifications: oxidation of methionine and alkylation of cysteine by carbamidomethylation; peptide mass tolerance = 0.05 Da and MS/MS tolerance = 0.45 Da. For protein identification at least one MS/MS spectra per protein was required (with a Mascot peptide ion score p < 0.05) and a p-value p < 0.05 in the Mascot protein score. Proteins were classified into COGs functional categories and assigned to KEGG pathways using eggNOG-mapper (Huerta-Cepas et al. 2016) and also classified according to their subcellular location with the CELLO v.2.5 (Cheng et al. 2014). The genomic context of the proteins and the location of their gene sequence at the chromosomal level were analyzed by blast searches against the Cupriavidus sp. UYMMa02A genome annotated in the RAST server (Aziz et al. 2008).
Results
The genome of Cupriavidus sp. UYMMa02A encodes a highly conserved symbiotic island
The draft genome of Cupriavidus sp. UYMMa02A was published in 2016 (Iriarte et al. 2016). Genome comparison among beta-rhizobial strains revealed a high level of synteny and nucleotide identity in operons containing the nod, nif, and fix genes (Fig. 1). However, there were differences in non-symbiotic and transposon-related genes within the symbiotic islands leading to length heterogeneity among them. The symbiotic island of Cupriavidus sp. UYMMa02A was the shortest with a predicted length of 33,908 bp.

Comparison of symbiotic islands of different beta-rhizobia. The size of each symbiotic island is indicated below strain names. Genes belonging to the nod/nif/fix operons are coloured in red/green/blue respectively, and transposons-related genes are coloured in black. Other colours represent genes that do not belong to the aforementioned operons
A closer inspection of the nod operon in Cupriavidus sp. UYMMa02A indicated that eight nod genes, involved in Nod factors biosynthesis and exportation, namely nodBCIJAHSUQ, are arranged in a single operon, while the nodD gene encoding a LysR-type transcriptional regulator, was located in the opposite orientation of nodB gene (Fig. 2). Moreover, a conserved NodD DNA binding motif known as nod-box (Schlaman et al. 1992) was identified in the intergenic region between nodD and nodB genes (Fig. 2). Minor differences in length and sequence were observed among the compared sequences, with the symbiotic islands of Cupriavidus sp. UYMMa02A and C. necator UYPR2.512, showing the highest sequence homology.

Sequence alignment of the nodB-nodD sequence in different Beta-rhizobia strains. Aligned sequences include the intergenic region between nodB and nodD and the first 100 bp of both genes. The black rectangle indicates nod-box localization. Conserved motifs are highlighted in colour letters
The UYMMa02A nod operon is not induced by pure flavonoids
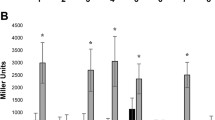
The flavonoids luteolin and apigenin are known to induce the expression of nod genes in rhizobial Cupriavidus sp. strains (Amadou et al. 2008; Marchetti et al. 2010; Rodríguez-Esperón et al. 2022). The observed syntenies lead us to hypothesize that the nod genes of Cupriavidus sp. UYMMa02A would be regulated by the same mechanism. To analyze this, Cupriavidus sp. UYMMa02A was transformed with the pCBM01 plasmid containing a pnodB19424-lacZ transcriptional fusion and beta-galactosidase (B-gal) activity was compared with C. necator UYPR2.512 and C. taiwanensis LMG19424, both harboring the same plasmid. As expected, a strong induction of B-gal activity was observed for C. necator UYPR2.512 and C. taiwanensis LMG19424 in the presence of luteolin. However, Cupriavidus sp. UYMMa02A remained unresponsive to the presence of pure flavonoids (Fig. 3).

Expression of the pnodB19424-lacZ fusion in C. taiwanensis LMG 19424, C. necator UYPR2.512, and Cupriavidus sp. UYMMa02A in response to luteolin
Cupriavidus sp. UYMMa02A nod genes are not induced in the presence of Mimosa spp. root exudates
Considering that nod genes expression is regulated by different compounds in different rhizobial strains (Hungria et al. 1991; Schmidt et al. 1994; Jiménez-Guerrero et al. 2017), we decided to investigate whether M. pudica and M. magentea root exudates could serve as potential inducers for nod genes expression in Cupriavidus sp. UYMMa02A. Root exudates are complex solutions containing different amino acids, organic acids, sugars, and phenolic compounds, which have been shown to induce nod gene expression in C. taiwanensis LMG19424 (Klonowska et al. 2018).
A clear induction of nod gene expression was observed, as indicated by an increase in B-gal activity when C. taiwanensis LMG19424 and C. necator UYPR2.512 were grown in the presence of Mimosa spp. root exudates (Figs. 4 and 5). These results indicate that M. pudica and M. magentea root exudates contain inducers for nod gene expression. However, when Cupriavidus sp. UYMMa02A was exposed to these root exudates, we did not observe any change in B-gal activity, suggesting that nod genes expression was not induced in this strain (Figs. 4 and 5). Similar results were observed when pure flavonoids were used.

Expression of the pnodB19424-lacZ fusion in C. taiwanensis LMG19424, C. necator UYPR2.512, and Cupriavidus sp. UYMMa02A in response to M. pudica root exudates (RE)

Expression of the pnodB19424-lacZ fusion in C. taiwanensis LMG19424, C. necator UYPR2.512, and Cupriavidus sp. UYMMa02A in response to M. magentea root exudates (RE)
Altogether these findings suggest that the pnodB19424-lacZ is not responsive in Cupriavidus sp. UYMMa02A. However, we cannot exclude the possibility that endogenous nod genes in Cupriavidus sp. UYMMa02A nod genes could be induced but were not detected by the assay used. To directly assess this, we analyzed the mRNA levels of nodA, nodB, and nodC in Cupriavidus sp. UYMMa02A in the presence or absence of M. pudica root exudates (Fig. 6). No changes in the relative nod gene expression were observed when Cupriavidus sp. UYMMa02A was grown in the presence of M. pudica root exudates compared to growth in M9 minimal media. These results confirm the absence of nod genes induction in Cupriavidus sp. UYMMa02A.

UYMMa02A nodA, nodB, and nodC expression in response to M. pudica root exudates. The bars represent the relative expression (Fold Change) of the nodA, nodB, and nodC genes to the housekeeping genes elongation factor G (efg) and ribosomal protein S14 (S14) when growing in M9 media supplemented with M. pudica root exudates versus in M9 media without exudates
Cupriavidus sp. UYMMa02A induces functional nodules and promotes M. pudica plant growth in nitrogen-limiting conditions
We have previously shown that Cupriavidus sp. UYMMa02A can form nodules in the roots of several Mimosa sp. including M. pudica plants (Platero et al. 2016). However, there was no direct evidence of this symbiotic interaction's plant growth promotion ability. When M. pudica plants growing in nitrogen-free media were inoculated with Cupriavidus sp. UYMMa02A, we observed the formation of reddish nodules in the roots and a significant increase in plant height (Fig. 7), strongly supporting the proficiency of this symbiotic pair.

M. pudica growth promotion ability of Cupriavidus sp. UYMMa02A. A. Examples of M. pudica development phenotype when inoculated with Cupriavidus sp. UYMMA02A or not inoculated. Insert, detail of the nodules formed by Cupriavidus sp. UYMMa02A in M. pudica roots. B. M. pudica plant height was recorded 30 days post-inoculation with Cupriavidus sp. UYMMa02A or with water as non-inoculated control. Asterisk indicates significant differences (p-value < 0.01) between inoculated plants and non-inoculated plants
Proteomic changes induced by luteolin and apigenin in Cupriavidus sp. UYMMa02A
The lack of induction of nod genes suggests that alternative mechanisms could be involved in the establishment of the Cupriavidus sp. UYMMa02A-M. pudica symbiotic interaction. To determine the significance of this finding, we decided to employ a proteomic approach known as Differential In Gel Expression (DIGE) (Meleady 2018; Mozejko-Ciesielska and Mostek 2019). In the first approach, bacteria were grown in the presence of luteolin or apigenin, and total protein profiles were compared with bacteria growing in the absence of the flavonoids. Two-dimensional electrophoresis allowed us to separate 373 different spots, representing around 4.5% of the CDS encoded by Cupriavidus sp. UYMMa02A genome (Iriarte et al. 2016). DIGE analysis showed 17 spots with altered expression levels in the presence of luteolin while 16 spots were differentially expressed in the presence of apigenin. When we performed a combined analysis of differentially expressed spots in the presence of the flavonoids versus the control condition, a total of 22 differentially expressed spots were detected, 8 overrepresented in the luteolin condition, 8 in the apigenin condition, and 6 in the control condition. After Coomassie blue staining and gel excision, a total of 9 proteins were identified by MALDI-TOF analyses (Table 2). According to COG analyses, 3 of the identified proteins belong to Category E (amino acid transport and metabolism) and 3 to category C (energy conversion and production), one protein to Category I (lipids transport and metabolism), and one belong to category P (inorganic ion transport). Except for the tricarboxylate transporter protein TctC with a periplasmic localization, all the identified proteins were predicted to be cytoplasmic. A model summarizing the major findings using this approach is shown in Fig. 8.

UYMMa02A proteins and pathways observed in response to flavonoids. Proteins differentially expressed in the presence of apigenin and/or luteolin are highlighted in bold. Green and red arrows indicate if proteins were found either upregulated (up direction) or downregulated (down direction), with respect to the control condition. The main cellular processes affected are indicated in bold and cursive text. See the text for a full description
Differential protein expression during Cupriavidus sp. UYMMa02A-M. pudica co-cultures
The next step in elucidating the mechanisms involved in the plant-bacteria interaction was to determine the proteomic responses of Cupriavidus sp. UYMMa02A to the presence of plant host M. pudica. We implemented a co-culture device intended to allow plant-bacteria signal interchange, preventing physical contact between symbionts. In this device, plants were grown hydroponically with bacteria placed in a closed dialysis membrane submerged in the same hydroponic solution (Fig. 9). Protein profile expression under co-culture conditions was compared with bacterial cells cultivated in the same conditions, but in that case, Mimosa plants were not included. The hydroponic solution was supplemented with minimal amounts of carbon and nitrogen sources to allow bacterial survival. Image analyses of the 2D gels captured a total of 674 spots, representing around 8% of the bacterial encode capacity. Thirty-seven differentially expressed spots were determined (26 and 11 spots were overexpressed and repressed respectively). After staining and excision from the gels, 17 different proteins were identified by MALDI-TOF, belonging to 11 different COG categories. Notably, proteins categorized in the categories O (posttranslational modifications and chaperones), G (carbohydrate transport and metabolism), and M (membrane and cell wall) contains both induced and repressed proteins, while proteins in category T (transduction signals mechanisms) were found overexpressed (Table 3). Subcellular localization analysis identified 9 cytoplasmic proteins, 3 periplasmic proteins, and 1 outer membrane protein, while 5 proteins were predicted to be found both at the bacterial cytoplasm and extracellularly. A model integrating the major findings using this approach is presented in Fig. 10.

Plant-microorganism co-culture system. Bacteria were grown in the presence (A) or absence (B) of M. pudica plants. To prevent direct physical interaction between symbionts, the bacteria were placed inside a membrane tubing (C). D Picture of the real co-culture assembled system (I) jar lid; (II) silicone tube connected to the membrane for bacterial inoculation; (III) M. pudica plants; (IV) Mineral plant culture medium containing polypropylene balls for seedling support. As control treatments, plant-free systems were used (B)

Model for the proteomics response of Cupriavidus sp. UYMMa02A-M. pudica co-culture treatment. The illustration shows the main metabolic pathway, biological functions, chemical reactions, enzymes, and other proteins regulated in Cupriavidus sp. UYMMa02A grown in a co-culture system with the plant host M. pudica. Green and red arrows indicate proteins either upregulated (up direction) or downregulated (down direction) with respect to the control condition. Metabolic pathways affected are shadowed in grey. Color shadows were used to cluster affected proteins according to cellular processes; Metabolic reprogramming (in yellow), Stress response (in blue), and Signal transduction (in orange). Proteins implicated in more than one cellular process have been included at shadow intersections. See the text for a full description
Discussion
The presence of highly conserved symbiotic islands in the genome of rhizobial Cupriavidus spp. suggest the existence of conserved symbiotic mechanisms among these strains. Notwithstanding, gene expression analyses have indicated that pure flavonoids or Mimosa spp. root exudates are ineffective in inducing Cupriavidus sp. UYMMa02A nod genes transcription. This is intriguing since it has been shown that the presence of luteolin or apigenin induces the synthesis and exportation of Nod factors in C. taiwanensis LMG19424 (Amadou et al. 2008). In addition, these flavonoids were shown to induce the expression of the pnodB19424-lacZ transcriptional fusion present in pCBM01, in both C. taiwanensis LMG19424 (Marchetti et al. 2010) and C. necator UYPR2.512 (Rodríguez-Esperón et al. 2022). It is well known that the flavonoid effect on nod genes regulation is dependent on the NodD regulatory protein (Schlaman et al. 1992). Different NodD proteins respond to different flavonoids, this mechanism being one of the bases of the rhizobia-legume specificity (Masson-Boivin et al. 2009). Small differences could be observed at Cupriavidus sp. UYMMa02A NodD protein sequence and in the nodB-nodD intergenic sequence in comparison with C. taiwanensis LMG19424 and C. necator UYPR2.512. Thus, it is possible that Cupriavidus sp. UYMMa02A NodD protein does not become activated by the presence of flavonoids or this protein could not recognize the C. taiwanensis LMG19424 nodB promoter region present in the pnodB19424-lacZ used here. To overcome these limitations, we used M. magentea and M. pudica root exudates for nod genes expression analyses, however no changes in the expression levels of nod genes in Cupriavidus sp. UYMMa02A were observed. These results indicated that in Cupriavidus sp. UYMMa02A nod genes may not be involved in the first steps of the symbiotic interaction and suggest the existence of alternative mechanisms to Nod factors for bacterial-plant interaction. It is now known that some rhizobial strains are naturally able to engage in symbiotic interactions with their plant host in a Nod-factor-independent way. Researchers have discovered strains of Bradyrhizobium sp. lacking the canonical nodABC genes that can effectively nodulate Aeschynomene spp. (Giraud et al. 2007; Miché et al. 2010) and Arachis hypogaea (Guha et al. 2022).
The lack of induction of nod genes in Cupriavidus sp. UYMMa02A prompted us to search for alternative mechanisms involved in bacterial-plant interaction. To identify proteins potentially implicated in the initial steps of this process, we analyzed the patterns of protein expression of Cupriavidus sp. UYMMa02A when exposed to pure flavonoids and during plant co-culture conditions. The response to different nod gene-inducing flavonoids and root exudates derived from the plant host has been studied in various rhizobial species, mostly belonging to the alpha-proteobacteria class, using transcriptomic and proteomic approaches (Jiménez-Guerrero et al. 2017; diCenzo et al. 2019). These studies have demonstrated, that in addition to nod genes, flavonoids, and root exudates can influence the expression of several bacterial genes encoded by different bacterial replicons. When comparing the responses to flavonoids, and root exudates, there is only partial overlap, particularly in nod-related genes, indicating that specific mechanisms are regulated in response to distinct stimuli (Capela et al. 2005). In addition, these responses are often specific to the genus and strains of rhizobia (Fagorzi et al. 2021).
Only two works that used omics approaches have been published for the analysis of rhizobial Cupriavidus strains response to flavonoids (Rodríguez-Esperón et al. 2022) or host root exudates (Klonowska et al. 2018). Both works showed the differential expression of hundreds of genes, located at different bacterial replicons, in response to these stimuli. In agreement with this, upon luteolin and apigenin or co-culture conditions, Cupriavidus sp. UYMMa02A experiences significant changes at the proteomic level. Approximately 6% (22 out of 357 spots) of the Cupriavidus sp. UYMMa02A proteome changed in response to the presence of flavonoids, suggesting that these molecules are indeed signal molecules for this Cupriavidus sp. strain. In agreement with the observed lack of induction of the pnodB19424-lacZ transcriptional fusion, we did not detect the differential expression of proteins belonging to the symbiotic Island. Indeed, most of the affected proteins identified were related to the energy conversion and production (C) and amino acid metabolism (E) COG categories, indicating that flavonoids induce metabolic changes in Cupriavidus sp. UYMMa02A. The overexpression of the tricarboxylate transport protein (TctC) could reflect an increase in citrate transport, the only carbon source in the used media. Under these conditions, citrate would be incorporated into the Cupriavidus metabolism directly through the citrate cycle (TCA or Krebs cycle), serving as an energy source and a carbon skeleton source. However, the concomitant diminution of the E2 component of the 2-oxoglutarate dehydrogenase complex would slow down the conversion of 2-oxo-glutarate (2-OG) to Succinyl-CoA. This change would result in the accumulation of 2-OG, a key biosynthetic intermediate that connects carbon and nitrogen metabolism. The 2-OG molecule is also involved in modulating enzyme activity and detoxifying reactive oxygen species (ROS) (Huergo and Dixon 2015). Considering this, our result may indicate that some of the metabolic changes observed after exposure to luteolin and apigenin in Cupriavidus sp. UYMMa02A could be related to an oxidative stress response. In that sense, Mailloux and collaborators have shown that in response to oxidative stress, Pseudomonas fluorescence increased the activity of an NADP-dependent isocitrate dehydrogenase while decreasing the activity of the 2-oxoglutarate dehydrogenase complex, leading to the accumulation of 2-OG (Mailloux et al. 2007). Supporting this hypothesis, we also observed a rise in the DNA starvation/stationary phase protection protein (DpsA), which belongs to the ferritin superfamily and is involved in DNA binding and protection during starvation and/or oxidative stress conditions (Martinez and Kolter 1997; Gambino and Cappitelli 2016; Orban and Finkel 2022). Since luteolin and apigenin have been reported to scavenge ROS (Wu et al. 2015; Salehi et al. 2019; Caporali et al. 2022), it is unlikely that the flavonoids themselves are causing oxidative stress. Instead, the flavonoids may act as signals, advertising that oxidative could occur during the forthcoming interaction with plant roots, as the local production of ROS is a common feature observed during plant-bacteria interactions (Oldroyd et al. 2011; Gourion et al. 2015; Janczarek et al. 2015a, b).
The increased levels of the bifunctional enzyme Aspartate-Transaminase/Aspartate-4-decarboxylase (AsdA) and the branched chain amino acids aminotransferase (IlvE) in the presence of luteolin and apigenin suggest that flavonoids impact amino acid metabolism in Cupriavidus sp. UYMMa02A. AsdA catalyzes the transamination of oxaloacetate to l-aspartate and the subsequent decarboxylation of aspartate to alanine (Kakimoto et al. 1969; Chen et al. 2000). The exportation of alanine and aspartate, through an amino acid cycle between bacteroids and plants, has been postulated to be important for SNF (Prell and Poole 2006; Prell et al. 2009). Besides this, the increase in the IlvE aminotransferase levels indicates the activation of branched-chain amino acids (BCAA) biosynthesis in response to flavonoids. BCAAs have been shown to have an essential role in the formation of effective symbiosis between the beta-rhizobia Paraburkholderia phyamtum STM815, and C. taiwanensis LMG19424 with M. pudica plants (Chen et al. 2012). Furthermore, our recent findings have demonstrated that C. necator UYPR2.512 induces the expression of genes related to BCAAs transport in response to luteolin (Rodríguez-Esperón et al. 2022). Nonetheless, it should be considered that the statistical support for ilvE protein is borderline, and further experiments will be needed to clarify its role as well as the role of BCAAs in the Cupriavidus sp. UYMMa02A-plant interaction.
A third process influenced by the presence of flavonoids in Cupriavidus sp. UYMMa02A is the possible activation of the glyoxylate cycle (Kornberg 1966). In this cycle, isocitrate is converted to succinate and glyoxylate by the enzyme isocitrate lyase (ICL) followed by the synthesis of malate from glyoxylate and acetyl-CoA by malate synthase (MS). The glyoxylate cycle enables growth using C2 compounds by bypassing the CO2-generating steps of the TCA cycle while generating the necessary products for gluconeogenesis and other biosynthetic purposes (Dunn et al. 2009). Our hypothesis is supported by the following arguments: (a) The activation of this cycle would explain the observed drop in the levels of the E2 component of the 2-oxoglutarate dehydrogenase complex, as this enzyme is by-passed in the cycle (b) The glyoxylate cycle would fulfill the increased demand for 2-oxoglutarate, the substrate of the induced aspartate transaminase/aspartate 4-decarboxylase. Since each turn of the cycle needs two molecules of acetyl-CoA, the third piece of evidence supporting our proposal is the observed increase in the enoyl-CoA hydratase (ECH) levels. This enzyme catalyzes the second step in the beta-oxidation pathways of fatty acid metabolism (Moskowitz and Merrick 1969) and would supply the needed acetyl-CoA molecules to fuel the glyoxylate cycle. While some of the changes induced by the assayed flavonoids in Cupriavidus sp. UYMMa02A can be linked to metabolic processes implicated in the symbiotic interaction between rhizobia and host plants, additional experiments should be performed to determine the significance of these findings in interactions involving Cupriavidus rhizobial strains.
To improve the simulation of the initial steps of the symbiotic interaction between Cupriavidus sp. UYMMa02A and M. pudica, we implement a co-culture device. Under these experimental conditions, we observed a broader bacterial response, as reflected by the number and distribution of proteins with altered expression among COG categories. Many of the proteins with changed expression were predicted to be located at periplasmic or extracellular space, suggesting that Cupriavidus sp. UYMMa02A undergoes changes at the membrane and periplasmic levels during the co-culture conditions. Surface remodeling has been well documented in many rhizobia models during their interaction with plant hosts. Variations in exopolysaccharides (EPS), lipopolysaccharides (LPS), and capsular polysaccharides (KPS) composition are required for the proper recognition of rhizobial cells by plant hosts, and for evading the natural plant immune system (Janczarek et al. 2015a, b). Under our experimental conditions, we observed the augmented expression of FabI, an enoyl-ACP reductase involved in fatty-acid biosynthesis, which could reflect an upregulation of lipogenesis for membrane remodeling. Membranes are the primary barriers for cellular interaction with the environment, influencing the uptake and efflux of small molecules. The observed increase in OmpC levels in Cupriavidus sp. UYMMa02A during the co-culture could indicate the need to enhance the transport of nutrients, signal molecules, or other metabolites across the bacterial outer membrane. The concomitant levels rise in the periplasmic binding component of an ABC transport system (XylF) putatively involved in ribose/xylose/arabinose/galactose acquisition, suggests an enhanced transport capacity for these sugars. Xylose and ribose have been detected in M. pudica root exudate (Klonowska et al. 2018), implying that these sugars may support Cupriavidus sp. UYMMa02A metabolism in the rhizosphere of M. pudica. On the other hand, we also observed an increase in the levels of a protein predicted to encode the periplasmic adaptor subunit of the resistance-nodule-division (RND) family efflux transporter. In Gram-negative bacteria, RND efflux systems work in concert with outer membrane porins, like OmpC, to detoxify deleterious compounds (Nies 2003; Klonowska et al. 2020). The importance of RND systems has been demonstrated in many rhizobial species, where they play a crucial role in survival and competence in the rhizosphere, affecting nodule formation and nitrogen fixation (Klonowska et al. 2012; Santos et al. 2014). Moreover, as observed here, some of these systems were shown to be inducible both by flavonoids and plant host root exudates. Similarly, Klonowska and collaborators found that C. taiwanensis LMG19424 and P. phymatum STM815 RND systems are induced by M. pudica root exudates (Klonowska et al. 2018). Our results suggest that this system would also be important for the growth of Cupriavidus sp. UYMMa02A in the presence of M. pudica root exudates. As mentioned, one role for these efflux systems is the detoxification of harmful compounds. In that sense, the RAST annotation server indicates that the identified protein would be a membrane fusion protein of the CzcB family, which is part of the CzcCBA Heavy metal efflux (HME) RND system (Janssen et al. 2010). CzcCBA systems were first described in C. metallidurans CH34 and shown to confer resistance to Co, Zn, and Cd. Our evidence suggests that Cupriavidus sp. UYMMa02A activates the extrusion of metals from its cytoplasm, perhaps preventing the formation of reactive oxygen species (ROS). The increase in Cupriavidus sp. UYMMa02A peroxiredoxin levels support the hypothesis that ROS are formed during this interaction. Peroxiredoxin is a periplasmic protein implicated in the detoxification of ROS and peroxynitrite, also detected in Rhizobium leguminosarum, C. taiwanensis, and P. phymatum rhizobial strains growing in the presence of root exudates (Ramachandran et al. 2011; Klonowska et al. 2018). Another stress-related induced protein is cyclophilin. Cyclophilins are Peptidyl-prolyl cis/trans isomerases (PPiase, EC: 5.2.1.8) enzymes found in all kingdoms of life. Since these proteins catalyze a rate-limiting step in protein folding, they play a critical role in protein homeostasis. Diverse studies have demonstrated the participation of these proteins in signal transduction, biofilm formation, motility, and adaptation to stress (Skagia et al. 2016; Dimou et al. 2017; Thomloudi et al. 2017). Thomloudi and collaborators showed that the heterologous expression of the two cyclophilins isoforms of Sinorhizobium meliloti 1021 in E. coli enhance bacterial survival under stress condition, supporting the role of these proteins in stress adaptation in bacteria. In addition, a PPiase mutant in Azorhizobium caulinodans ORS571 impairs its symbiosis with Sesbania rostrata, reducing nodule size and completely abolishing its nitrogen fixation ability (Suzuki et al. 2007). Along with the increase in the protein levels of the Efflux RND periplasmic adaptor subunit, peroxiredoxin, and PPiase, we also observed an increase in the universal stress protein A (UspA) levels. Universal stress proteins are small cytoplasmic bacterial proteins whose expression is enhanced when the cell is exposed to diverse stress agents and has been implicated in cell survival during prolonged exposure to stress conditions (Nyström and Neidhardt 1994). Induced expression of genes encoding proteins of this family has been observed during R. leguminosarum growing in the rhizosphere of pea, alfalfa, and sugar beet (Ramachandran et al. 2011), as well as in C. taiwanensis and P. phymatum growing in the presence of M. pudica root exudates (Klonowska et al. 2018). Altogether, the evidence presented here strongly suggests that during the first steps of the symbiosis, Cupriavidus sp. UYMMa02A is under considerable stress pressure, a common feature in rhizobia-legumes symbiosis.
Finally, we were able to detect the induction of proteins with the potential to transduce host-secreted signals, such as the protein kinase PrkA protein and the Nucleotide dikinase (NdK) protein. PrkA is a member of the conserved serine/threonine protein kinase family widely distributed among Bacteria, Archaea, and Eukarya (Stancik et al. 2018). Serine/Threonine kinases (STKs) act in concert with Serine/Threonine phosphatase (STPs) to introduce or remove phosphate modifications in proteins at these residues. Reversible protein phosphorylation is one of the main mechanisms allowing bacteria to respond to environmental stimuli. Post-translational phosphorylation/dephosphorylation influences the activity of modified proteins by inducing conformational changes and regulating protein–protein interactions. Thus, STKs and STPs influence many different signal transduction pathways in bacteria. In R. leguminosarum, disruption of the pssZ gene, encoding an STK, impacts exopolysaccharide synthesis, surface properties, and symbiosis with clover (Lipa et al. 2018). Despite this example, very few studies address the role of STKs in the symbiosis between rhizobia and legumes. The induction of the Cupriavidus sp. UYMMa02A PrkA in the presence of the host plant M. pudica, suggests that this STK could have an important role during the initial steps of the symbiotic interaction and demands additional studies to determine its precise role.
On the other hand, Ndks are housekeeping enzymes whose primary role is maintaining the cellular homeostasis of nucleoside triphosphate (NTP) and their deoxy derivatives (dNTPs) pools by catalyzing the reversible γ-phosphate transfer from NTPs (or dNTPs) to NDPs (or dNDPs) (Berg and Joklik 1953). In bacteria, the transfer of high-energy phosphates occurs by a ping-pong mechanism that involves the formation of a phosphor-histidine intermediate at a conserved histidine residue (Lascu and Gonin 2000). Moreover, it has been shown that His-Phosphorylated Ndks can phosphorylate other proteins at histidine residues modulating their activities, and affecting a myriad of bacterial processes (Lu et al. 1996; Attwood and Wieland 2015). For example, the E. coli Ndk can transfer its phosphate to the sensor proteins EnvZ and CheA, and then the phosphorylated kinases transfer the high-energy phosphate to their cognate response regulators OmpR and CheY, respectively, which are implicated in osmosis and chemical sensing (Lu et al. 1996). In other bacteria Ndk homologs have been implicated in the regulation of biofilms formation, the function of type 3 secretion systems, the modulation of quorum sensing, and the response to oxidative stress, playing a critical role in bacteria and host interactions (Yu et al. 2017). Interestingly, in some bacteria, Ndk can also be found extracellular and membrane-associated. In Pseudomonas aeruginosa, it has been observed that membrane-associated Ndk correlates with the status of bacterial growth (Shankar et al. 1996). Analysis of the Cupriavidus sp. UYMMa02A Ndk sequence suggests that this protein could have both intracellular and periplasmic locations. The results presented here, suggest that this protein would be involved in the interaction between the rhizobial Cupriavidus sp. UYMMa02A and the plant host M. pudica. Further experiments are needed to determine the exact role of this protein in the interaction, as the role of Ndks in beneficial plant-bacteria interaction has not been studied in detail.
Conclusions
The genome of Cupriavidus sp. strain UYMMa02A encodes a conserved symbiotic island that includes a complete nodDBCIJHASUQ gene cluster with a nod-box sequence in the promoter region of nodB. However, no induction of the Cupriavidus sp. UYMMa02A nod genes was evidenced in response to pure flavonoids or Mimosa spp. root exudates. These results suggest that Cupriavidus sp. UYMMA02A may employ Nod-independent mechanisms to stablish symbiosis. Using a quantitative proteomic approach, we detected significant proteomic changes in Cupriavidus sp. UYMMa02A when exposed to flavonoids or root exudates. Twenty-four differentially expressed proteins were identified covering diverse bacterial processes ranging from basic metabolism and transport functions to stress response and signal transduction. In the presence of the plant host, the major bacterial responses were related to amino acid metabolism and oxidative stress, which are common features in many rhizobia-legume interactions. Besides this, the increased levels of the proteins PrkA and Ndk proteins during the co-culture conditions indicated the involvement of these versatile proteins in the symbiotic interaction. While further research is necessary to determine the precise roles of the identified proteins, our study presents a new model for interrogating the symbiotic interaction between beta-rhizobia and plant hosts.
Data availability
Data is available upon request to the authors.
References
Amadou C, Pascal G, Mangenot S (2008) Genome sequence of the β-rhizobium Cupriavidus taiwanensis and comparative genomics of rhizobia. Genome Res 18:1472–1483
Andam CP, Mondo SJ, Parker MA (2007) Monophyly of nodA and nifH genes across Texan and Costa Rican populations of Cupriavidus nodule symbionts. Appl Environ Microbiol 73:4686–4690
Andrews M, Andrews ME (2017) Specificity in legume-rhizobia symbioses. Int J Mol Sci 18:705
Attwood PV, Wieland T (2015) Nucleoside diphosphate kinase as protein histidine kinase. Naunyn Schmiedebergs Arch Pharmacol 388:153–160
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA et al (2008) The RAST server: rapid annotations using subsystems technology. BMC Genom 9:75
Bellés-Sancho P, Liu Y, Heiniger B, von Salis E, Eberl L, Ahrens CH et al (2022) A novel function of the key nitrogen-fixation activator NifA in beta-rhizobia: repression of bacterial auxin synthesis during symbiosis. Front Plant Sci. https://doi.org/10.3389/fpls.2022.991548
Berg P, Joklik WK (1953) Transphosphorylation between nucleoside polyphosphates. Nature 172:1008–1009
Bontemps C, Elliott GN, Simon MF, Dos Reis Júnior FB, Gross E, Lawton RC et al (2010) Burkholderia species are ancient symbionts of legumes. Mol Ecol 19:44–52
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Capela D, Carrere S, Batut J (2005) Transcriptome-based identification of the Sinorhizobium meliloti NodD1 regulon. Appl Environ Microbiol 71:4910–4913
Caporali S, De Stefano A, Calabrese C, Giovannelli A, Pieri M, Savini I et al (2022) Anti-inflammatory and active biological properties of the plant-derived bioactive compounds luteolin and luteolin 7-glucoside. Nutrients 14:1–19
Chen CC, Chou TL, Lee CY (2000) Cloning, expression and characterization of L-aspartate β-decarboxylase gene from Alcaligenes faecalis CCRC 11585. J Ind Microbiol Biotechnol 25:132–140
Chen W, Laevens S, Lee T, Coenye T, Vos PD, Mergeay M, Vandamme P (2001) Ralstonia taiwanensis sp. nov., isolated from root nodules of Mimosa species and sputum of a cystic fibrosis patient. Int J Syst Evol Microbiol 51:1729–1735
Chen W-M, Moulin L, Bontemps C, Vandamme P, Béna G, Boivin-Masson C (2003) Legume symbiotic nitrogen fixation by beta-proteobacteria is widespread in nature. J Bacteriol 185:7266–7272
Chen W-M, Prell J, James EK, Sheu D-S, Sheu S-Y (2012) Biosynthesis of branched-chain amino acids is essential for effective symbioses between betarhizobia and Mimosa pudica. Microbiology 158:1758–1766
Cheng C-S, Su C-W, Chang W-C, Huang K-C (2014) CELLO2GO: a web server for protein subCELlular LOcalization prediction with functional gene ontology annotation. PLoS ONE 9:99368
Cunnac S, Boucher C, Genin S (2004) Characterization of the cis-acting regulatory element controlling HrpB-mediated activation of the type III secretion system and effector genes in Ralstonia solanacearum. J Bacteriol 186:2309–2318
Dall’Agnol RF, Bournaud C, de Faria SM, Bena G, Moulin L, Hungria M (2017) Genetic diversity of symbiotic Paraburkholderia species isolated from nodules of Mimosa pudica (L.) and Phaseolus vulgaris (L.) grown in soils of the Brazilian Atlantic Forest (Mata Atlantica). FEMS Microbiol Ecol. https://doi.org/10.1093/femsec/fix027
de Campos SB, Lardi M, Gandolfi A, Eberl L, Pessi G (2017) Mutations in two Paraburkholderia phymatum type VI secretion systems cause reduced fitness in interbacterial competition. Front Microbiol 8:2473
De Meyer SE, Fabiano E, Tian R, Van Berkum P, Seshadri R, Reddy T et al (2015a) High-quality permanent draft genome sequence of the Parapiptadenia rigida-nodulating Cupriavidus sp. strain UYPR2.512. Stand Genom Sci 10:13
De Meyer SE, Parker M, Van Berkum P, Tian R, Seshadri R, Reddy TBK et al (2015b) High-quality permanent draft genome sequence of the Mimosa asperata-nodulating Cupriavidus sp. strain AMP6. Stand Genomic Sci 10:9–11
De Meyer SE, Briscoe L, Martínez-Hidalgo P, Agapakis CM, de-Los Santos PE, Seshadri R et al (2016) Symbiotic Burkholderia species show diverse arrangements of nif/fix and nod genes and lack typical high-affinity cytochrome cbb3 oxidase genes. Mol Plant Microbe Interact 29:609–619
diCenzo GC, Zamani M, Checcucci A, Fondi M, Griffitts JS, Finan TM, Mengoni A (2019) Multidisciplinary approaches for studying rhizobium–legume symbioses. Can J Microbiol 65:1–33
Dimou M, Venieraki A, Katinakis P (2017) Microbial cyclophilins: specialized functions in virulence and beyond. World J Microbiol Biotechnol 33:1–8
dos Reis FB, Simon MF, Gross E, Boddey RM, Elliott GN, Neto NE et al (2010) Nodulation and nitrogen fixation by Mimosa spp. in the Cerrado and Caatinga biomes of Brazil. New Phytol 186:934–946
Dunn MF, Ramírez-Trujillo JA, Hernández-Lucas I (2009) Major roles of isocitrate lyase and malate synthase in bacterial and fungal pathogenesis. Microbiology 155:3166–3175
Estrada-delossantos P, Palmer M, Chávez-Ramírez B, Beukes C, Steenkamp ET, Briscoe L et al (2018) Whole genome analyses suggests that Burkholderia sensu lato contains two additional novel genera (Mycetohabitans gen. nov., and Trinickia gen. nov.): implications for the evolution of diazotrophy and nodulation in the Burkholderiaceae. Genes (basel). 9:389
Fagorzi C, Bacci G, Huang R, Cangioli L, Checcucci A, Fini M et al (2021) Nonadditive transcriptomic signatures of genotype-by-genotype interactions during the initiation of plant-rhizobium symbiosis. mSystems. https://doi.org/10.1128/mSystems.00974-20
Gambino M, Cappitelli F (2016) Mini-review: biofilm responses to oxidative stress. Biofouling 32:167–178
Garau G, Yates RJ, Deiana P, Howieson JG (2009) Novel strains of nodulating Burkholderia have a role in nitrogen fixation with papilionoid herbaceous legumes adapted to acid, infertile soils. Soil Biol Biochem 41:125–134
Gehlot HS, Tak N, Kaushik M, Mitra S, Chen W-M, Poweleit N et al (2013) An invasive Mimosa in India does not adopt the symbionts of its native relatives. Ann Bot 112:179–196
Gil M, Lima A, Rivera B, Rossello J, Urdániz E, Cascioferro A et al (2019) New substrates and interactors of the mycobacterial serine/threonine protein kinase PknG identified by a tailored interactomic approach. J Proteom 192:321–333
Giraud E, Moulin L, Vallenet D, Barbe V, Cytryn E, Avarre J-C et al (2007) Legumes symbioses: absence of Nod genes in photosynthetic bradyrhizobia. Science 316:1307–1312
Gourion B, Berrabah F, Ratet P, Stacey G (2015) Rhizobium-legume symbioses: the crucial role of plant immunity. Trends Plant Sci 20:186–194
Guha S, Molla F, Sarkar M, Ibañez F, Fabra A, DasGupta M (2022) Nod factor-independent “crack-entry” symbiosis in dalbergoid legume Arachis hypogaea. Environ Microbiol 24:2732–2746
Hanahan D (1983) Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166(4):557–580. https://doi.org/10.1016/s0022-2836(83)80284-8
Howieson JG, Robson AD, Ewing MA (1993) External phosphate and calcium concentrations, and Ph, but not the products of rhizobial nodulation genes, affect the attachment of rhizobium meliloti to roots of annual medics. Soil Biol Biochem 25:567–573
Howieson JG, De Meyer SE, Vivas-Marfisi A, Ratnayake S, Ardley JK, Yates RJ (2013) Novel Burkholderia bacteria isolated from Lebeckia ambigua—a perennial suffrutescent legume of the fynbos. Soil Biol Biochem 60:55–64
Huergo LF, Dixon R (2015) The emergence of 2-oxoglutarate as a master regulator metabolite. Microbiol Mol Biol Rev 79:419–435
Huerta-Cepas J, Szklarczyk D, Forslund K, Cook H, Heller D, Walter MC et al (2016) eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res 44:286–293
Hungria M, Joseph CM, Phillips DA (1991) Rhizobium nod gene inducers exuded naturally from roots of common bean (Phaseolus vulgaris L.). Plant Physiol 97:759–764
Iriarte A, Platero R, Romero V, Fabiano E, Sotelo-Silveira JR (2016) Draft genome sequence of Cupriavidus UYMMa02A, a novel beta-rhizobium species. Genome Announc 4:e01258-e1316
Janczarek M, Rachwał K, Cieśla J, Ginalska G, Bieganowski A (2015a) Production of exopolysaccharide by Rhizobium leguminosarum bv. trifolii and its role in bacterial attachment and surface properties. Plant Soil 388:211–227
Janczarek M, Rachwał K, Marzec A, Grządziel J, Palusińska-Szysz M (2015b) Signal molecules and cell-surface components involved in early stages of the legume–rhizobium interactions. Appl Soil Ecol 85:94–113
Janssen PJ, Van Houdt R, Moors H, Monsieurs P, Morin N, Michaux A et al (2010) The complete genome sequence of cupriavidus metallidurans strain CH34, a master survivalist in harsh and anthropogenic environments. PLoS ONE 5:e10433
Jiménez-Guerrero I, Acosta-Jurado S, del Cerro P, Navarro-Gómez P, López-Baena F, Ollero F et al (2017) Transcriptomic studies of the effect of nod gene-inducing molecules in rhizobia: different weapons, one purpose. Genes (basel). https://doi.org/10.3390/genes9010001
Kakimoto T, Kato J, Shibatani T, Nishimura N, Chibata I (1969) Crystalline l-aspartate β-decarboxylase of Pseudomonas dacunhae. J Biol Chem 244:353–358
Kessler B, de Lorenzo V, Timmis KN (1992) A general system to integrate lacZ fusions into the chromosomes of gram-negative eubacteria: regulation of the Pm promoter of the TOL plasmid studied with all controlling elements in monocopy. Mol Gen Genet MGG 233:293–301
Klonowska A, Chaintreuil C, Tisseyre P, Miché L, Melkonian R, Ducousso M et al (2012) Biodiversity of Mimosa pudica rhizobial symbionts (Cupriavidus taiwanensis, Rhizobium mesoamericanum) in New Caledonia and their adaptation to heavy metal-rich soils. FEMS Microbiol Ecol 81:618–635
Klonowska A, Melkonian R, Miché L, Tisseyre P, Moulin L (2018) Transcriptomic profiling of Burkholderia phymatum STM815, Cupriavidus taiwanensis LMG19424 and Rhizobium mesoamericanum STM3625 in response to Mimosa pudica root exudates illuminates the molecular basis of their nodulation competitiveness and symbiotic ev. BMC Genom 19:105
Klonowska A, Moulin L, Ardley JK, Braun F, Gollagher MM, Zandberg JD et al (2020) Novel heavy metal resistance gene clusters are present in the genome of Cupriavidus neocaledonicus STM 6070, a new species of Mimosa pudica microsymbiont isolated from heavy-metal-rich mining site soil. BMC Genom 21:214
Kornberg HL (1966) The role and control of the glyoxylate cycle in Escherichia coli. Biochem J 99:1–11
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Laemmli U (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lardi M, Liu Y, Purtschert G, de Campos SB, Pessi G (2017) Transcriptome analysis of Paraburkholderia phymatum under nitrogen starvation and during symbiosis with Phaseolus vulgaris. Genes (basel). 8:389
Lascu I, Gonin P (2000) The catalytic mechanism of nucleoside diphosphate kinases. J Bioenerg Biomembr 32:237–246
Lemaire B, Dlodlo O, Chimphango S, Stirton C, Schrire B, Boatwright JS et al (2015) Symbiotic diversity, specificity and distribution of rhizobia in native legumes of the Core Cape Subregion (South Africa). FEMS Microbiol Ecol 91:1–17
Lindström K, Mousavi SA (2020) Effectiveness of nitrogen fixation in rhizobia. Microb Biotechnol 13:1314–1335
Lipa P, Vinardell JM, Kopcińska J, Zdybicka-Barabas A, Janczarek M (2018) Mutation in the pssZ gene negatively impacts exopolysaccharide synthesis, surface properties, and symbiosis of Rhizobium leguminosarum bv. trifolii with clover. Genes (basel). 9:369
Liu X, Wei S, Wang F, James EK, Guo X, Zagar C et al (2012) Burkholderia and Cupriavidus spp. are the preferred symbionts of Mimosa spp. in Southern China. FEMS Microbiol Ecol 80:417–426
Liu Y, Bellich B, Hug S, Eberl L, Cescutti P, Pessi G (2020) the exopolysaccharide cepacian plays a role in the establishment of the Paraburkholderia phymatum—Phaseolus vulgaris symbiosis. Front Microbiol 11:1600
Lu Q, Park H, Egger LA, Inouye M (1996) Nucleoside-diphosphate kinase-mediated signal transduction via histidyl-aspartyl phosphorelay systems in Escherichia coli. J Biol Chem 271:32886–32893
Mailloux RJ, Bériault R, Lemire J, Singh R, Chénier DR, Hamel RD, Appanna VD (2007) The tricarboxylic acid cycle, an ancient metabolic network with a novel twist. PLoS ONE 2:e690
Marchetti M, Capela D, Glew M, Cruveiller S, Chane-Woon-Ming B, Gris C et al (2010) Experimental evolution of a plant pathogen into a legume symbiont. PLoS Biol 8:e1000280
Marchetti M, Catrice O, Batut J, Masson-Boivin C (2011) Cupriavidus taiwanensis bacteroids in Mimosa pudica Indeterminate nodules are not terminally differentiated. Appl Environ Microbiol 77:2161–2164
Martinez A, Kolter R (1997) Protection of DNA during oxidative stress by the nonspecific DNA-binding protein Dps. J Bacteriol 179:5188–5194
Masson-Boivin C, Giraud E, Perret X, Batut J (2009) Establishing nitrogen-fixing symbiosis with legumes: how many rhizobium recipes? Trends Microbiol 17:458–466
Meleady P (2018) Two-dimensional gel electrophoresis and 2D-DIGE. Methods Mol Biol 1664:3–14
Miché L, Moulin L, Chaintreuil C, Contreras-Jimenez JL, Munive-Hernández JA, del Carmen Villegas-Hernandez M et al (2010) Diversity analyses of Aeschynomene symbionts in Tropical Africa and Central America reveal that nod-independent stem nodulation is not restricted to photosynthetic bradyrhizobia. Environ Microbiol 12:2152–2164
Miller JH (1972) Assay of B-galactosidase In: Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Moskowitz GJ, Merrick JM (1969) Metabolism of poly-β-hydroxybutyrate. II. Enzymic synthesis of D-(-)-β-hydroxybutyryl coenzyme A by an enoyl hydrase from Rhodospirillum rubrum. Biochemistry 8:2748–2755
Moulin L, Klonowska A, Caroline B, Booth K, Vriezen JAC, Melkonian R et al (2014) Complete genome sequence of Burkholderia phymatum STM815(T), a broad host range and efficient nitrogen-fixing symbiont of Mimosa species. Stand Genom Sci 9:763–774
Mozejko-Ciesielska J, Mostek A (2019) A 2D-DIGE-based proteomic analysis brings new insights into cellular responses of Pseudomonas putida KT2440 during polyhydroxyalkanoates synthesis. Microb Cell Fact 18:1–13
Nies DH (2003) Efflux-mediated heavy metal resistance in prokaryotes. FEMS Microbiol Rev 27:313–339
Nyström T, Neidhardt FC (1994) Expression and role of the universal stress protein, UspA, of Escherichia coli during growth arrest. Mol Microbiol 11:537–544
Oldroyd GED, Murray JD, Poole PS, Downie JA (2011) The rules of engagement in the legume-rhizobial symbiosis. Annu Rev Genet 45:119–144
Orban K, Finkel SE (2022) Dps is a universally conserved dual-action DNA-binding and ferritin protein. J Bacteriol 204:1–23
Parker MA, Wurtz AK, Paynter Q (2007) Nodule symbiosis of invasive Mimosa pigra in Australia and in ancestral habitats: a comparative analysis. Biol Invasions 9:127–138
Pereira-Gómez M, Ríos C, Zabaleta M, Lagurara P, Galvalisi U, Iccardi P et al (2020) Native legumes of the Farrapos protected area in Uruguay establish selective associations with rhizobia in their natural habitat. Soil Biol Biochem 148:107854
Platero R, James EK, Rios C, Iriarte A, Sandes L, Zabaleta M et al (2016) Novel Cupriavidus strains isolated from root nodules of native Uruguayan Mimosa species. Appl Environ Microbiol 82:3150–3164
Prell J, Poole P (2006) Metabolic changes of rhizobia in legume nodules. Trends Microbiol 14:161–168
Prell J, White JP, Bourdes A, Bunnewell S, Bongaerts RJ, Poole PS (2009) Legumes regulate Rhizobium bacteroid development and persistence by the supply of branched-chain amino acids. Proc Natl Acad Sci USA 106:12477–12482
Ramachandran VK, East AK, Karunakaran R, Downie JA, Poole PS (2011) Adaptation of Rhizobium leguminosarum to pea, alfalfa and sugar beet rhizospheres investigated by comparative transcriptomics. Genome Biol 12:R106
Rodríguez-Esperón MC, Eastman G, Sandes L, Garabato F, Eastman I, Iriarte A et al (2022) Genomics and transcriptomics insights into luteolin effects on the beta-rhizobial strain Cupriavidus necator UYPR2.512. Environ Microbiol 24:240–264
Saad MM, Crèvecoeur M, Masson-Boivin C, Perret X (2012) The type 3 protein secretion system of Cupriavidus taiwanensis strain LMG19424 compromises symbiosis with Leucaena leucocephala. Appl Environ Microbiol 78:7476–7479
Salehi B, Venditti A, Sharifi-Rad M, Kręgiel D, Sharifi-Rad J, Durazzo A et al (2019) The therapeutic potential of Apigenin. Int J Mol Sci 20:1305
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
Santos MR, Marques AT, Becker JD, Moreira LM (2014) The Sinorhizobium meliloti EmrR regulator is required for efficient colonization of Medicago sativa root nodules. Mol Plant Microbe Interact 27:388–399
Schlaman HRM, Okker RJH, Lugtenberg BJJ (1992) Regulation of nodulation gene expression by nodD in rhizobia. J Bacteriol 174:5177–5182
Schmidt PE, Broughton WJ, Werner D (1994) Nod factors of Bradyrhizobium-japonicum and Rhizobium sp. NGR234 induce flavonoid accumulation in soybean root exudate. Mol Plant Microbe Interact 7:384–390
Shankar S, Kamath S, Chakrabarty AM (1996) Two forms of the nucleoside diphosphate kinase of Pseudomonas aeruginosa 8830: altered specificity of nucleoside triphosphate synthesis by the cell membrane-associated form of the truncated enzyme. J Bacteriol 178:1777–1781
Skagia A, Zografou C, Vezyri E, Venieraki A, Katinakis P, Dimou M (2016) Cyclophilin PpiB is involved in motility and biofilm formation via its functional association with certain proteins. Genes Cells 21:833–851
Stancik IA, Šestak MS, Ji B, Axelson-Fisk M, Franjevic D, Jers C et al (2018) Serine/threonine protein kinases from bacteria, archaea and Eukarya share a common evolutionary origin deeply rooted in the tree of life. J Mol Biol 430:27–32
Suzuki S, Aono T, Lee KB, Suzuki T, Liu CT, Miwa H et al (2007) Rhizobial factors required for stem nodule maturation and maintenance in Sesbania rostrata-Azorhizobium caulinodans ORS571 symbiosis. Appl Environ Microbiol 73:6650–6659
Taulé C, Zabaleta M, Mareque C, Platero R, Sanjurjo L, Sicardi M et al (2012) New betaproteobacterial Rhizobium strains able to efficiently nodulate Parapiptadenia rigida (Benth.) Brenan. Appl Environ Microbiol 78:1692–1700
Thomloudi E-E, Skagia A, Venieraki A, Katinakis P, Dimou M (2017) Functional analysis of the two cyclophilin isoforms of Sinorhizobium meliloti. World J Microbiol Biotechnol 33:28
Wu PS, Yen JH, Kou MC, Wu MJ (2015) Luteolin and apigenin attenuate 4-hydroxy-2-nonenal-mediated cell death through modulation of UPR, Nrf2-ARE and MAPK pathways in PC12 cells. PLoS ONE 10:1–23
Yu H, Rao X, Zhang K (2017) Nucleoside diphosphate kinase (Ndk): a pleiotropic effector manipulating bacterial virulence and adaptive responses. Microbiol Res 205:125–134
Zheng J, Wang R, Liu R, Chen J, Wei Q, Wu X et al (2017) The structure and evolution of beta-rhizobial symbiotic genes deduced from their complete genomes. Immunome Res 13:131
Acknowledgement
The authors would like to thank to Prof. Catherine Masson-Boivin for providing plasmids pCZ388 and pCBM01.
Funding
Funding was provided by Agencia Nacional de Investigación e Innovación (grant no. FCE_1_2014_1_104338, FCE_1_2017_1_136082, FCE_1_2019_1_156520), Programa de desarrollo de las ciencias básicas, PEDECIBA (grant no. 2018), and FONTAGRO (grant no. ID 30).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Rodríguez-Esperón, C., Sandes, L., Eastman, I. et al. Nodulation in the absence of nod genes induction: alternative mechanisms involved in the symbiotic interaction between Cupriavidus sp. UYMMa02A and Mimosa pudica. Environmental Sustainability 6, 383–401 (2023). https://doi.org/10.1007/s42398-023-00286-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42398-023-00286-5