
Abstract
Watermelon is important fruit vegetable crop affected by both biotic and abiotic stresses. Among the biotic factors, leaf curl and yellowing disease of watermelon is becoming a major threat for the watermelon production in India. Incidence of watermelon leaf curl and yellowing disease in ten districts covering different regions of Karnataka State, India varied from 6.8 to 60.7%. Diseased watermelon plants exhibited symptoms typical to begomovirus infection such as leaf crumpling, leaf curling, yellowing, reduction in leaf size and stunted growth in the surveyed fields. Thirty-four diseased watermelon samples collected from farmers’ fields during the survey gave positive amplification to begomovirus specific primers in PCR and yielded expected 1.2 kb amplicon, which represent its partial genome. No amplification resulted in PCR with primers specific to alpha and betasatellites in all the samples. Complete genome amplification by RCA and sequencing of six isolates was done. Among these, four six isolates resulted in amplification of both DNA-A and DNA-B components and two isolates gave amplification only to DNA-A component. Genome sequence analysis using Sequence Demarcation Tool (SDT) showed the maximum nucleotide identity of 91.5–93.3% and 80.2–84.5% of six watermelon isolates with Tomato leaf curl Palampur virus (ToLCPalV) for DNA-A and DNA-B components, respectively. Phylogenetic analysis provided further evidence in this regard. The recombination breakpoint analysis indicates, watermelon isolates under study are recombinants emerged from the recombination of ToLCPalV and Tomato leaf curl New Delhi virus (ToLCNDV). This is the first report of leaf curl and yellowing disease of watermelon associated with ToLCPalV from India.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Watermelon [Citrullas lanatus (Thumb) Mastum and Nakai, Synonyms: C. vulgaris] is important cucurbitaceous crop belonging to family, Cucurbitaceae and grown across the world. The crop was originated from Southern Africa and cultivated since ancient times (Wehner 2008). It is also known as tarbuj, tarmuj, kalinda, kallangadi and kalindi at different parts of India. In India, watermelon is cultivated in an area of 103 thousand hectares with the production of 25.04 lakh tons (Anonymous 2019). The major watermelon growing states of India are Punjab, Haryana, Karnataka, Assam, Madhya Pradesh, Andhra Pradesh, Maharashtra, Telangana, West Bengal, Odisha, Himachal Pradesh, Uttar Pradesh and Tamil Nadu (Anonymous 2019). However, watermelon is cultivated throughout the year only in Southern states viz., Tamil Nadu, Telangana and Andhra Pradesh. The fruit contains 92% water and 7.5% of carbohydrates, of these 6.2% sugars and 0.4% dietary fiber. Consumption of 100 g watermelon provides 30 kcal of energy and is also a good source of vitamin C and B, minerals, citrulline, carotenoids and flavonoids (Maoto et al. 2019). The crop is prone to many biotic and abiotic stresses resulting in great economic yield losses to its growers (Wehner 2008). The low productivity in this crop is mainly due to various diseases caused by fungi, bacteria, viruses, phytoplasma and nematodes (Wehner 2008). Among these, the viral diseases are major constraint for watermelon cultivation worldwide. A large number of RNA viruses (Jain et al. 2007; Wang et al. 2010) and DNA viruses (Mansoor et al. 2000; Heydarnejad et al. 2013; Shahid 2017; Zhao et al. 2017; Venkataravanappa et al. 2020) were reported on watermelon across the world.
Geminiviruses (family: Geminiviridae) are small single-stranded (ss) DNA viruses causing diseases in economically important crop plants across the world (Navas-Castillo et al. 2011). The circular single-stranded (ss) DNA genome of these viruses is encapsidated into twinned icosahedral particles. The family, Geminiviridae consists of nine genera based on their genome structure, host range and insect vector (Zerbini et al. 2017). Among these, the genus, Begomovirus is considered as the largest group with over 424 species reported so far and are known to be transmitted by different cryptic species of whitefly (Bemisia tabaci) (Gilbertson et al. 2015). Begomoviruses are subdivided into two categories as monopartite and bipartite viruses based on the composition of genome, with earlier having single genome component (homologue of DNA-A) and later having two similar-sized components (DNA-A and DNA-B). Based on the prevalence and distribution of begomoviruses, a monopartite and bipartite begomoviruses are also referred as Old World (OW) and New World (NW), respectively (Zerbini et al. 2017).
Furthermore, monopartite begomoviruses are commonly associated with sub genome segments known as alphasatellites and betasatellites (Zhou 2013), which is not common feature in bipartite viruses. These satellites play major role related to symptom modulation, pathogenesis and also in silencing suppressors (Zhou 2013). However, recently there are reports showing the association of bipartite begomoviruses with DNA satellites (Venkataravanappa et al. 2019).
Every year, new hosts for the begomoviruses are being reported with imposing economic losses (Sangeeta and Tiwari 2017). However, information of about begomovirus infecting watermelon from India is scanty with only one recent report of Tomato leaf curl New Delhi virus (ToLCNDV) associated with watermelon (Venkataravanappa et al. 2020). With this backdrop, the study was conducted to determine the status of begomovirus with watermelon leaf curl and yellowing disease from Karnataka State, India.
Material and methods
Survey for disease incidence and collection of leaf samples
Roving survey was carried during 2017 and 2018 in 10 districts (Belagavi, Vijayapura, Raichure, Kolar, Chikkaballapur, Bengaluru rural, Mandya, Mysuru, Chamarajanagar and Tumkur) of Karnataka State, India, which are considered as major watermelon growing areas to assess the incidence of leaf curl and yellowing disease on watermelon. In each location percent incidence of watermelon leaf curl and yellowing disease was assessed. Ten micro plots of size 10 mt × 10 mt were randomly selected in each surveyed field for the assessment. The disease incidence of leaf curl and yellowing disease of watermelon was assessed by counting number of infected plants visually over healthy plants. From each field, one symptomatic leaf sample from the watermelon plants showing symptoms known to be induced by begomoviruses were collected along with the one asymptomatic sample and were brought to laboratory, Department of Plant Pathology at College of Horticulture, Bengaluru, UHS, Bagalkot, India and used for molecular analysis. Each sample was designated as different isolate with respect to place of collection.
Whitefly transmission
The vector transmission and maintenance of whitefly was done as per the protocol described by Patil et al. (2017). The watermelon isolate-BLR associated with leaf curl and yellowing disease of watermelon was used for the virus transmission study. Healthy seedlings of watermelon (F1 hybrid; NS-295) were raised in insect proof glasshouse. The adult whiteflies were collected in tube using aspirator from the nonviruliferous whitefly colony maintained for the transmission studies. The leaf curl and yellowing disease infected sample was inserted into acquisition tube containing whiteflies and kept undisturbed for 24 h to facilitate the virus acquisition by its vector. After acquisition, viruliferous whiteflies were released on to healthy watermelon seedlings covered with inoculation tube at the rate of 30 per plant and provided inoculation access for 24 h. Then 0.05% Imidacloprid was sprayed on plants to kill whiteflies and were kept in insect proof glasshouse for symptoms expression.
DNA isolation, detection and characterization of begomovirus
To confirm the presence of begomovirus in the watermelon samples showing leaf curl and yellowing disease symptoms, total genomic DNA was isolated from 34 symptomatic and two-asymptomatic leaf samples of watermelon by CTAB method (Lodhi et al. 1994). The PCR was carried out using primers specific to begomovirus (Supplementary Table S1) with the expected amplicon of 1.2 kb (Venkataravanappa et al. 2012). The resulted amplicon representing partial genome of begomoviruses was cloned into cloning vector pTZ57R/T (Fermentas, Germany), transformed into DH5α strain of Escherichia coli as per the standard protocol (Sambrook and Russell 2001), colony PCR was used to confirm positive clones and three clones in each transformation were sequenced at Medauxin Pvt Ltd, Bengaluru, India. After sequence analysis, six watermelon samples were selected for whole genome amplification and these isolates were designated as Belagavi (BGM), Chikkaballapur (CBP), Bengaluru rural (BLR), Chamarajanagar (CMNR), Vijayapura (VJP) and Raichur (RCR), representing different geographical regions. The rolling circle amplification (RCA) method using TempliPhi illustra amplification kit (GE Healthcare, Piscataway, NJ) as per manufactures instructions was employed for amplification of complete genome of six watermelon isolates. The amplified RCA product (2 µl) was subjected for restriction digestion with BamH1 and Kpn1 enzymes to obtain monomeric DNA-A and DNA-B genome components, respectively. The digested products were cloned into pBluescript KS (+) cloning vector (Sambrook and Russell 2001) and transformed into DH5α strain of E. coli. The plasmid DNA was isolated from the three clones from each transformation by using alkaline lysis method and the presence of insert was confirmed by restriction digestion with respective restriction enzymes. Sequencing of clones was done at Medauxin Pvt Ltd, Bengaluru, India by primer walking. PCR was carried out using alphasatellite (Bull et al. 2003) and betasatellite (Briddon et al. 2002) specific primers (Supplementary Table S1) to know the presence of satellite molecules in all the 34 samples.
Sequence and recombination break point analysis
The vector sequences were removed from genome components (both DNA-A and DNA-B) of six watermelon begomovirus isolates using VecScreen software after sequencing. The sequences obtained were assembled using Bio edit (version 7.2) (http://bioedit.software.informer.com) (Hall 1999). Sequence similarities were checked at NCBI database using BLASTn (http://www.ncbi.nlm.nih). The sequences showed highest blast score with present isolates were retrieved from the NCBI database (Brown et al. 2015). The open reading frames (ORFs) in the sequences were identified by ORF finder available at NCBI (http://www.ncbi.nlm.nih.Gov/gorf/gorf.html). The pairwise nucleotide identity of sequences obtained in the current study with retrieved sequences from the GenBank (Supplementary Table S2 and S3) was calculated using Sequence Demarcation Tool (SDT version 1.2) (http://web.cbio.uct.ac.za/~brejnev/) (Muhire et al. 2014) and percent pairwise identity was generated. Phylogenetic trees were generated using MEGA software (version X.0) (https://www.megasoftware.net/) by following the Neighbor-Joining method with 1,000 bootstrap replicates (Kumar et al. 2018). The recombination break point analysis of watermelon isolates sequences along with other selected begomoviruses (Brown et al. 2015) was done using Recombination detection program (RDP), Max Chi, 3Seq, Genecov, Si Scan Bootscan, and Chimaera integrated in RDP4.4 (http://web.cbio.uct.ac.za/~darren/rdp.html) (Martin et al. 2015). DNA-A and DNA-B sequences were subjected to recombination analysis to drive automated recombination scan and the manual checking of automated analysis results. Analysis was allowed by employing Bonferroni correction with confidence greater than 95% (p-value 0.05). To ensure reliability, the begomovirus sequences were considered recombinant when the recombination signal was supported by at least three methods. In RDP analysis, the length of the window was set to 10 variable sites and the step size was set to one nucleotide. p-values were estimated by randomizing the alignment 1,000 times.
Results
Disease incidence and symptoms
Watermelon leaf curl and yellowing disease was observed in all surveyed areas (Supplementary Table S4). Watermelon plants suspected to be infected by begomovirus showed symptoms viz., chlorotic mottle, yellowing, reduction of leaf size, crinkled and malformed leaves, downward and upward leaf curling and stunted growth (Fig. 1). The disease incidence ranged from 6.8 to 60.7% in the surveyed areas with the maximum incidence (60.7%) observed in Chamarajanagar District and minimum incidence in Kolar District (Table 1). Further, the survey data indicated that, all the varieties/hybrids (NS-295, NS-702, Swaraj-45, Black Box, Sugar Queen, Sugar Baby Arka Akash, Arka Muttu, Black Gold, Aishwarya, Kiran, Sweet Dawn, Sugar King and Apoorva) grown in these areas were found susceptible (Table 1).

Watermelon plants showing leaf crumpling, leaf curling, yellowing, reduction in leaf size and stunted growth symptoms under natural conditions
Vector transmission
Watermelon seedlings (F1 hybrid; NS-295) inoculated with BLR-isolate using whiteflies exhibited symptoms such as leaf crinkling, reduction in leaf size, and stunted growth 12–15 days post inoculation (DPI) (Fig. 2). This provided the evidence for vector transmission nature of the virus isolate.

Watermelon seedlings exhibited leaf crinkling, reduction in leaf size and stunted growth under whitefly inoculation condition
Detection of begomovirus
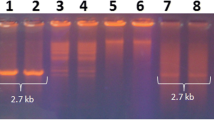
All the 34 infected watermelon samples resulted in the expected 1.2 kb size PCR amplicon specific to begomoviruses. No amplification was observed in asymptomatic plants and also in the PCR specific to alphasatellites and betasatellites. Sequence analysis of amplified PCR products representing the partial genome of begomovirus from all the samples indicated that, they shared nucleotide identity of more than 93% among them and with bipartite begomovirus species, i.e., Tomato leaf curl Palampur virus (ToLCPalV) reported earlier. Because of very high identity, six isolates (BGM, CBP, BLR CMNR, VJP, RCR) representing diverse geographical regions were selected for sequencing of complete genome components. RCA for genome components of selected isolates yielded approximately 2.7 kb DNA-A component in all six isolates and four isolates (BGM, CBP, BLR CMNR) yielded DNA-B, which is also of approximately 2.7 kb size. Whereas, two samples (VJP and RCR) not yielded DNA-B with repeated amplification by both PCR (Rajos et al. 1993; Venkataravanappa et al. 2012) and RCA.
Genome organization of DNA-A watermelon infecting begomovirus
Sequences of DNA-A of six begomovirus isolates (BGM, CBP, BLR CMNR, VJP, RCR) associated with watermelon were 2756 nt in length and deposited in the NCBI GenBank (Accession numbers MT433965-70). SDT analysis of DNA-A sequences of six virus isolates showed 93.5–97.5% nt identity among themselves indicating that they are the isolates of a single species as per the begomoviruses species demarcation criteria (Brown et al. 2015). The genome organization of these isolates is typical to Old World bipartite begomoviruses encoding six open reading frames (ORFs); V2 and CP in viral sense strand and Rep (C1), TrAP (C2), REn (C3), AC4 in antisense orientation in DNA-A. The intergenic region (IR) of 273 nt in length was present in all six watermelon isolates and it shared maximum sequence identities of 88.3 to 98.5% with IR of ToLCPalV infecting crop plants in Indian subcontinent and Iran. Furthermore, iteron sequence, GGTGTCT (Arguello-Astorga et al. 1994) is present in the IR region of all six isolates and has significant identity with iterons of ToLCPalV isolates reported earlier.
The nt sequence of DNA-A of six isolates was compared with selected begomovirus isolates/species comprising both monopartite and bipartite viruses retrieved from the NCBI database. The analysis revealed that six isolates are showing maximum nt identity of 86.9 to 93.3% with isolates of ToLCPalV infecting different crops (Table 2) (Fig. 3b). As per the current ICTV species/strain/variant demarcation criteria (Brown et al. 2015), all the six isolates in the present study are considered as strains of ToLCPalV and proposed descriptor as Tomato leaf curl Palampur virus-[India:Karnataka:Watermelon:2017] and designated as ToLCPalV-[IN:Kar:Wat:17]. The analysis of individual ORFs revealed that four ORFs (C1, C2 C3 and C4) are having highest aa identity with ToLCPalV isolates infecting different crops. However, ORF-AV1 and AV2 had maximum aa identity with ToLCNDV isolates infecting various crop plants (Supplementary Table S5).



Phylogenetic tree showing relationships of the DNA-A component of six watermelon isolates with selected begomoviruses. The phylogenetic trees were constructed employing the MEGA X tool, using the neighbor-joining method with 1000 bootstrap replicates (a). The two dimensional color-coded matrix of pairwise calculated scores of the watermelon isolates (b) using SDT. Pictorial depiction genomic map of DNA-A of watermelon isolates under study and their putative recombination events identified by RDP analysis (c)
Phylogenetic analysis
Phylogenetic analysis showed that six watermelon isolates (BGM, CBP, BLR, CMNR, VJP and RCR) clustered with ToLCPalV isolates infecting different crops in India and Iran (Fig. 3a). Within this cluster, six isolates formed a sub cluster providing the further evidence for strainal difference among them.
Genome organization of DNA-B watermelon infecting begomovirus
The complete nt sequences DNA-B component of four isolates (BGM, BLR, CBP, CMNR) were between 2719 and 2723 nt in length encoding two ORFs, BV1 (nuclear shuttle protein:NSP) and BC1 (movement protein:MP) in sense and antisense strands, respectively. The sequences of these isolates were deposited in NCBI GenBank (Accession numbers MT439868-71). The sequence analysis of DNA-B component of four isolates showed 87.3–99.6% nt identity among themselves. The DNA-B sequence analysis of four isolates with selected begomoviruses sequences was done. The begomoviruses infecting watermelon showed maximum nt identity of 80.2–84.5% with ToLCPalV infecting different crop plants (Table 3) (Fig. 4b). These results are well supported in phylogenetic analysis showing begomoviruses infecting watermelon forming separate cluster with high bootstrap value along within the major cluster having isolates of ToLCPalV infecting different crops (Fig. 4a).


Phylogenetic tree showing relationships of the DNA-B component of four watermelon isolates with selected begomoviruses. The phylogenetic trees were constructed by MEGA X tool, using the neighbor-joining method with 1000 bootstrap replicates (a). The two dimensional color-coded matrix of pairwise calculated scores of the watermelon isolates (b) using SDT. Pictorial depiction genomic map of DNA-B of watermelon isolates under study and their putative recombination events identified by RDP analysis (c)
Further, the aa sequence ORF BV1 and BC1 of four isolates (BLR, VJP, CBP, CMNR) showed maximum aa identity of 73.8–86.9% and 86.1–90.3, respectively with isolates of ToLCPalV infecting different crops (Table 3). The length of the IR region of four watermelon isolates is 273 nt in length, which is similar to several ToLCPalV isolates. The “iterons” GGCGTC was identified in all the four isolates (Arguello-Astorga et al. 1994).
Recombination breakpoint analysis
The analysis showed that six isolates of DNA-A have both intra and inter species recombination (Supplementary Table S6). Four recombination events were observed in BLR, three in VJP, two in BGM, CBP and CMNR isolates and only one recombination event was observed in RCR isolate. This indicates the evidence of recombination in six isolates (BGM, CBP, BLR CMNR, VJP, RCR) with most of the fragments of DNA-A component derived from ToLCPalV infecting cucurbits and ToLCNDV infecting potato or tomato as major and minor parents, respectively to evolve as a novel strain of ToLCPalV infecting watermelon (Supplementary Table S6) (Fig. 3c). Similarly, recombination break point analysis in DNA-B sequences of four the isolates revealed the presence of one recombination event with most of the fragments in DNA-B component derived from ToLCNDV and ToLCPalV as major and minor parents infecting chilli and rumex, respectively (Supplementary Table S6) (Fig. 4c). Overall results showed most of the DNA-A and DNA-B components of begovoirus infecting watermelon (BGM, CBP, BLR CMNR, VJP, RCR) might have been derived from ToLCPalV and ToLCNDV. This is supported by six methods (RDP, Chimera, Geneconv, SiScan, 3Seq and MaxChi) in DNA-A component and four methods (RDP, Chimera, 3Seq and MaxChi) in DNA-B component used for recombination break point analysis (Supplementary Table S6). The maximum recombination events in DNA-A of ToLCPalV was detected by SiScan and 3Seq methods used in the recombination analysis followed by RDP, Geneconv and Chimera (Supplementary Table S6). Similarly, in case of DNA-B, RDP, Chimera, 3Seq and MaxChi detected maximum recombinant events (Supplementary Table S6). Analysis clearly showed that there is a correlation between number of recombination events detected in genome and genetic variability of the virus. Maximum number of recombinant events were detected in both genomic components (DNA-A and DNA-B) by two methods (SiScan and 3Seq) in all the ToLCPalV isolates. Further, the DNA-A of all the isolates had more prone to recombination than the DNA-B, with a maximum number of unique, well-supported recombination events detected by at least two methods with a p value < 0.001. The most of the DNA fragment in DNA-A of ToLCPalV isolates might derived from ToLCPalV infecting cucurbits from Iron as major parent except one isolate (BGM), where it might have derived from ToLCNDV infecting ash gourd from India. Similarly, most of the DNA fragment in DNA-B of ToLCPalV isolates is shown to be descended from the ToLCNDV infecting chilli from Pakistan as major parent. Our results showed a much greater tendency of the DNA-A with number of unique events to recombine compared to the DNA-B.
Discussion
Watermelon is important vegetable fruit crop extensively grown throughout Indian subcontinent. In the watermelon, the diseases caused by both RNA and DNA viruses are the major bottleneck in its production throughout the world (Wehner 2008). Among viral diseases, the leaf curl and yellowing disease of watermelon caused by begomoviruses is an emerging problem on watermelon crop ecosystem in India, considering the imposing damage caused by begomoviruses in other crops (Varma and Malathi 2003). Survey indicated the prevalence of watermelon leaf curl and yellowing disease in Karnataka State, India with varying percent of disease incidence. The varied disease incidence within state might be due to difference in whitefly vector population, growing susceptible cultivars and other factors influencing the crop ecosystem such as climate, surrounding crops etc., In all the surveyed fields, diseased plants produced symptoms such as mosaic, leaf curling, yellowing, reduction in leaf size, curling of leaves and stunted growth similar to the previous reports of begomovirus induced manifestation on watermelon (Mansoor et al. 2000; Shahid et al. 2017; Zhao et al. 2017; Venkatarvanappa et al. 2020). Present study also confirmed the presence of begomovirus in the symptomatic leaf samples through PCR detection providing furthermore evidence for the above mentioned manifestation of symptoms by begomovirus infection in watermelon. Whitefly cryptic species are recognized vectors transmitting the begomoviruses in circulative persistent manner under natural conditions (Gray and Banerjee 1999). Whitefly transmission of begomovirus watermelon (BLR) isolate in the present study was established for biological characterization.
Based on the detection of genome components, among the six isolates, four were considered as bipartite (having both DNA-A and DNA-B) and two were considered as monopartite (having only homologue of DNA-aA) begomoviruses. The alpha and betasatellites association were not found in all the 34 symptomatic samples, which includes the six samples considered for complete genome sequencing and designated as watermelon isolates. This is in contrast with the many earlier reports about the association of monopartite begomoviruses with DNA satellites (Zhou 2013). In the recent past, few reports are available about non-association of monopartite begomoviruses with DNA satellites supporting the results in the current study (Akram et al. 2020).
However, six bipartite [WmCSV, ToLCNDV, Squash leaf curl virus (SLCV), Cucurbit leaf crumple virus (CuLCrV), MeCMV and ToLCPalV] and two monopartite (ChiLCV and TbCSV) begomoviruses reported to be associated with watermelon worldwide (Mansoor et al. 2000; Heydarnejad et al. 2013; Shahid et al. 2017; Zhao et al. 2017; Venkataravanappa et al. 2020). So far only one begomovirus i.e., ToLCNDV is reported on watermelon from India, which is bipartite based on complete genome sequencing (Venkataravanappa et al. 2020). The complete genome and phylogenetic analysis of current begomovirus isolates provided evidence for association of strains of ToLCPalV as per the begomovirus species demarcation criteria (Brown et al. 2015). Evidence for the past recombination was shown in the recombination breakpoint analysis of begomoviruses infecting watermelon with the sequences used for analysis. The mechanism of inter and intra species recombination contributes to the evolution of novel begomoviruses and their subsequent adaptation new host in agricultural eco-system (Lefeuvre et al. 2007).
The understanding of such recombination between different isolates/species of begomoviruses in adaptation to watermelon may provide more insights in this regard. Emergence of novel viral diseases is aided by their high recombination rate, providing the advantage of expanding their host range (Seal et al. 2006).
The ToLCPalV was first reported on tomato during 2008 (Kumar et al. 2008), later it is reported on cucumber, pumpkin, Rumex sp, bitter gourd, muskmelon, chilli, Cucumis melo var. flexuosus, Cucumis callosus var. agrestis, brinjal and basella from India (Namrata et al. 2010; Malathi et al. 2017; Kumari et al. 2019), squash, watermelon, cucumber, common bean and two weed species, Heliotropium europaeum and Chenopodium sp. from Iran (Heydarnejad et al. 2013), bitter gourd, cucumber and muskmelon from Pakistan (Shafiq et al. 2019). Besides this, multiple infections of three different viruses; ToLCPalV, ToLCNDV and SLCCNV in pumpkin (Namrata et al. 2010) and two viruses; ToLCPalV and ToLCNDV in tomato (Gaikwad et al. 2011) were also reported from India. To date, ToLCPalV is known to be distributed in the Indian subcontinent and, central and north eastern Iran.
Looking at the pace of ToLCPalV host range expansion, diseases caused by it will become major production constraint in vegetable crops belonging to Solanaceae, Cucurbitacae, Leguminaceae, Basellaceae and Malvaceae families. This is the first report of association of ToLCPalV with watermelon leaf curl and yellowing disease from India based on biological and molecular approaches.
References
Akram A, Khan AH, Mansoor S, Moffett P, Bridon WR, Saeed M (2020) Detection and molecular characterization of Clerodendron yellow mosaic virus infecting Volkameria inermis in Pakistan. J Plant Pathol. https://doi.org/10.1007/s42161-020-00529-y
Anonymous (2019) Crop wise area and production of horticultural crops in India. http://www.nhb.gov.in
Arguello-Astorga GR, Guevara-Gonzalez LR, Herrera-Estrella LR, Rivera-Bustamante RF (1994) Geminivirus replication origins have a group-specific organization of iterative elements: a model for replication. Virology 203:90–100
Briddon RW, Bull SE, Mansoor S, Amin I, Markham PG (2002) Universal primers for the PCR- mediated amplification of DNA beta—a molecule associated with some monopartite begomoviruses. Mol Biotechnol 20:315–318
Brown JK, Zerbini FM, Navas-Castillo J et al (2015) Revision of begomovirus taxonomy based on pairwise sequence comparisons. Arch Virol 160(6):1593–1619
Bull SE, Briddon RW, Markham PG (2003) Universal primers for the PCR-mediated amplification of DNA 1: a satellite-like molecule associated with begomovirus-DNA β complexes. Mol Biotechnol 23:83–86
Gaikwad KA, Sharma A, Cheema DS (2011) Molecular detection and characterization of leaf curl virus infecting tomato in Punjab, India. Acta Horticulturae (ISHS) 914:153–156
Gilbertson RL, Batuman O, Webster CG, Adkins S (2015) Role of the insect super vectors Bemisia tabaci and Frankliniella occidentalis in the emergence and global spread of plant viruses. Ann Rev Virol 2:67–93
Gray SM, Banerjee N (1999) Mechanisms of arthropod transmission of plant and animal viruses. Microbiol Mol Biol Rev 63:128–148
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Heydarnejad J, Hesari M, Massumi H, Varsani A (2013) Incidence and natural hosts of tomato leaf curl Palampur virus in Iran. Australasian Plant Pathol 42(2):195–203
Jain RK, Bag S, Umamaheswaran K, Mandal B (2007) Natural infection by tospoviruses of cucurbitaceous and fabaceous vegetable crops in India. J Phytopathol 155:22–25
Kumar Y, Hallan V, Zaidi AA (2008) Molecular characterization of a distinct bipartite begomovirus species infecting tomato in India. Virus Genes 37:425–431
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549
Kumari S, Krishnan N, Dubey V, Pandey KK, Singh J (2019) Characterization of recombinant tomato leaf curl Palampur virus causing leaf curl disease of Basella alba L. in India. J Plant Pathol. https://doi.org/10.1007/s42161-019-00464-7
Lefeuvre P, Lett JM, Reynaud B, Martin DP (2007) Avoidance of protein fold disruption in natural virus recombinants. PLoS Pathogen 3:e181
Lodhi MA, Ye GN, Weeden NF, Reisch B (1994) A simple and efficient method for DNA extraction from grapevine cultivars and Vitis species. Plant Mol Biol Rep 12:6–13
Malathi VG, Renukadevi P, Chakraborty S, Biswas KK, Roy A, Sivalingam PN, Venkataravanappa V, Mandal B (2017) Begomoviruses and their satellites occurring in India: distribution, diversity and pathogenesis. In: Mandal B, Rao GP, Baranwal VK, Jain RK (eds) A century of plant virology in. Springer, Singapore, pp 75–178
Mansoor S, Khan SH, Hussain M, Mushtaq N, Zafar Y, Malik KA (2000) Evidence that watermelon leaf curl disease in Pakistan is associated with Tomato leaf curl virus-India, a bipartite begomovirus. Plant Dis 84:102
Maoto MM, Beswa D, Jideani AIO (2019) Watermelon as a potential fruit snack. Int J Food Prop 22(1):355–370
Martin DP, Murrell B, Golden M, Khoosal A, Muhire B (2015) RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol 1:vev003. https://doi.org/10.1093/ve/vev003
Muhire BM, Varsani A, Martin DP (2014) SDT: a virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 9(9):e108277. https://doi.org/10.1371/journal.pone.0108277
Namrata J, Saritha RK, Datta D, Singh M, Dubey RS, Rai AB, Rai M (2010) Molecular characterization of Tomato leaf curl Palampur virus and Pepper leaf curl betasatellite naturally infecting pumpkin (Cucurbita moschata) in India. Indian J Virol 21(2):128–132
Navas-Castillo J, Fiallo-Olive E, Sanchez-Campos S (2011) Emerging virus diseases transmitted by whiteflies. Annu Rev Phytopathol 49:219–248
Patil CV, Ramdas SV, Premchand U, Shankarappa KS (2017) Survey, symptomatology, transmission, host range and characterization of begomovirus associated with yellow mosaic disease of ridge gourd from southern India. Virus Dis 28(2):146–155
Rojas MR, Gilbertson RL, Russell DR, Maxwell DP (1993) Use of degenerate primers in the polymerase chain reaction to detect whitefly-transmitted geminiviruses. Plant Dis 77:340–347
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring
Sangeeta S, Tiwari A (2017) Begomoviruses: occurrence and management in Asia and Africa. Springer Singapore. https://doi.org/10.1007/978-981-10-5984-1
Seal SE, Van Den Bosch F, Jeger MJ (2006) Factors influencing begomovirus evolution and their increasing global significance: implications for sustainable control. Crit Rev Plant Sci 25:23–46
Shafiq M, Ahmad M, Nisar A, Manzoor MT, Arslan Abid A, Mushtaq S, Adeel Riaz A et al (2019) Molecular characterization and phylogenetic analysis of tomato leaf curl Palampur virus, a bipartite begomovirus, associated with Cucumis sativus L. in Pakistan. 3 Biotech 9:204. https://doi.org/10.1007/s13205-019-1727-3
Shahid MS, Al-SadiBriddon AMRW (2017) First report of Chilli leaf curl virus and Tomato leaf curl betasatellite infecting watermelon (Citrullus lanatus) in Oman. Plant Dis 101:1063
Varma A, Malathi VG (2003) Emerging geminivirus problems: a serious threat to crop production. Ann Appl Biol 142(2):145–164
Venkataravanappa V, Lakshminarayanareddy CN, Jalali S, Reddy MK (2012) Molecular characterization of distinct bipartite begomovirus infecting bhendi (Abelmoschus esculentus L.) in India. Virus Genes 44:522–535
Venkataravanappa V, Lakshminarayanareddy CN, Shankarappa KS, Jayappa J, Pandey S, Reddy MK (2019) Characterization of Tomato leaf curl New Delhi virus and DNA-satellites association with mosaic disease of cucumber. Int J Biotechnol Bioeng 5(6):93–109
Venkataravanappa V, Ashwathappa KV, Lakshminarayanareddy CN, Shankarappa KS, Reddy MK et al (2020) Characterization of Tomato leaf curl New Delhi virus associated with leaf curl and yellowing disease of Watermelon and development of LAMP assay for its detection. 3 Biotech 10:282
Wang WL, Zhang H, Yu XQ, Wu YF, Zhang WB, Zhang CP (2010) Establishment and application of multipIex RTPCR for simultaneous detection of five watermelon viruses ZYMV, WMV, TMV, SqMV and CMV. Acta Phytopathol Sin 40:27–32
Wehner TC (2008) Watermelon. In: Prohens J, Nuez F (eds) Handbook of plant breeding; vegetables I: Asteraceae, Brassicaceae, Chenopodiaceae, and Cucurbitaceae. Springer, New York, pp 381–418
Zerbini FM, Briddon RW, Idris A et al (2017) ICTV virus taxonomy profile: geminiviridae. J Gen Virol 98(2):131–133
Zhao LL, Ding M, Zhang XY, Yin YY, Li TT, Zhang ZK (2017) First report of Tobacco curly shoot virus (TbCSV) and its associated satellites from watermelon in china. J Plant Pathol 99(3):761–764
Zhou X (2013) Advances in understanding begomovirus satellites. Ann Rev Phytopathol 51:357–381
Acknowledgements
We are thankful to Dr. J. Jayappa, Dr. T. B. Manjunatha Reddy and Dr. J.S. Aravinda Kumar for their guidance.
Funding
This study is funded by University of Horticultural Sciences, Bagalkot, India.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declared that they have no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hanamasagar, Y., Naganur, P., Shankarappa, K.S. et al. Characterization of Tomato leaf curl Palampur virus associated with leaf curl and yellowing disease of watermelon from India. Indian Phytopathology 74, 1075–1088 (2021). https://doi.org/10.1007/s42360-021-00394-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42360-021-00394-4