
Abstract
Primary membranous nephropathy (PMN) is an autoimmune disease caused by the attack of autoantibodies against podocyte antigens leading to the in situ production of immune complexes. However, the etiology is unknown and the pathogenesis is still far from being completely elucidated. MN is prevalently idiopathic or primary, but in about 20–30% of cases it is secondary to chronic infections, systemic diseases, exposure to drugs, or malignancy. The differentiation between primary and secondary MN may be difficult, particularly when MN precedes signs and symptoms of the original disease, as in some cases of cancer or systemic lupus erythematosus. The natural course of PMN is variable, but in the long term 40–60% of patients with nephrotic syndrome progress to end-stage renal disease (ESRD) or die from thrombotic or cardiovascular events. PMN is a treatable disease. Patients with asymptomatic proteinuria should receive supportive care. Immunosuppressive treatments should be given to patients with nephrotic syndrome or risk of progression. The most frequently adopted treatments rely on cyclical therapy alternating steroids with a cytotoxic agent every other month, i.e., rituximab at different doses, or calcineurin inhibitors plus low-dose steroids. A good rate of response may be obtained but relapses can occur. Randomized controlled trials, with adequate size, long-term follow-up, and fair definition of endpoints are needed to identify treatment with the best therapeutic index.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The term membranous nephropathy (MN) was proposed years ago to define a glomerular disease clinically characterized by proteinuria, usually in nephrotic range, and histologically by an apparent thickening of the glomerular basement membrane (GBM) [1]. Further studies with electron microscopy and immunofluorescence showed that this apparent thickening of the GBM was actually due to subepithelial deposits of immune complexes in which the antibodies were mainly represented by immunoglobulin G4 (IgG4), at least in the so-called primary form. After the discovery of many antigens (see below) the etiology remains unknown only in few cases and the disease is called primary, but in 20–30% of cases MN is called secondary, being associated with other diseases, exposure to drugs, or infections. Progress in elucidating the pathophysiology of MN has been made in the last years but some doubts still remain. The course of primary MN (PMN) is variable and it may be difficult to predict the long-term natural outcome. Different treatments may favor remission of proteinuria and stabilization of kidney function, but which therapy might offer the best efficacy and safety is still uncertain. In this manuscript we will discuss the main, unsolved topics in idiopathic MN.
Is it possible to differentiate primary from secondary MN?
Secondary MN may be associated with chronic infections (particularly hepatitis B or C), autoimmune diseases (particularly lupus nephropathy), exposure to drugs or toxic agents (such as nonsteroidal antiinflammatory drugs or mercury compounds) or other conditions (including kidney or bone marrow transplantation, etc.) [2] (Table 1). The clinical history may lead to a correct diagnosis, but in several cases it can be difficult to recognize the original disease or the responsible drug. In these instances, kidney biopsy may help. At light microscopy, it is possible to observe endocapillary proliferation and cellular infiltration in some types of secondary MN, including those associated with lupus or cancer, while these findings are usually absent in PMN. At immunofluorescence, IgG4 are predominant in PMN, while IgG 1–3 deposits are frequent in secondary MN [3, 4]. IgA, IgM and C1q deposits are rare in PMN and frequent in secondary forms, particularly in lupus nephritis. Staining with phospholipase A2 receptor (PLA2R) is frequently positive in PMN [5, 6] but it can also be seen in sarcoidosis, hepatitis B and C or cancer [7,8,9,10]. On electron microscopy, subepithelial electron-dense deposits are typical of PMN, while subendothelial mesangial, and subepithelial deposits can be seen in some forms of secondary MN. Tubuloreticular inclusions can be found in patients with lupus [11].
A main problem is represented by the fact that in some patients with cancer, sarcoidosis, or lupus the signs and symptoms of MN may precede those of the original disease by weeks or even years. Cancer may occur in 10% of patients with untreated MN either at the time of kidney biopsy or one year later [12]. A misdiagnosis of cancer is a true nightmare. Thus, invasive and noninvasive procedures to detect a possible underlying cancer may be suggested, especially in patients older than 50 years, heavy smokers or in those with negative anti-PLA2R antibodies [13, 14]. However, this approach is not shared by all investigators who feel that the cost of investigating cancer and other possible causes of secondary MN would become excessive.
Etiopathogenesis of primary MN
The etiology of PMN is unknown by definition. The disease might be triggered by viral infection or exposure to toxic agents, including hydrocarbons, but the search for the agent(s) responsible for the disease remains difficult.
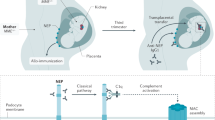
A great deal of progress has been made in understanding the patophysiology of idiopathic MN. Experimental models showed that in Heymann nephritis the disease is caused by an in situ deposit of immune complexes resulting from the reaction of a circulating antibody against an antigen planted in the subepithelial position. In 2002, Debiec et al. [15] showed that MN in the newborn of a neutral endoproteinase (NEP)–deficient mother was mediated by maternal anti-NEP antibodies which formed immune complexes with NEP on the podocyte membranes of the infant. This was the first demonstration that antibodies against a podocyte antigen can induce MN in humans by mechanisms similar to those proposed for Heymann nephritis [16].
A few years later, Beck et al. [17] showed that 70% of adults with PMN exhibited circulating IgG4 antibodies against the M-type PLA2R expressed on the podocyte surface. Other reports confirmed the presence of anti-PLA2R antibodies in 65–80% of patients with MN. However, PLA2R is not the only antigen in MN. In 2–10% of patients with MN, circulating antibodies are directed against thrombospondin 1 domain containing 7 A expressed on the podocyte surface [18,19,20]. These antibodies are pathogenic [21, 22] and can be found not only in PMN, but also in MN associated with cancer [6, 10, 23]. Novel antigens of primary and secondary MN have recently been detected by mass spectrometry or immunochemistry: exostosin 1 and 2 in type V lupus nephritis, NELL-1, semaphorin 3B, protocadherin-7, serinprotease AHTRA1, contactin1 [24,25,26].
Research on the epitope of PLA2R demonstrated that the main epitope is located in an outer cysteine-rich (CysR) domain [27]. Two inner epitopes have also been found, respectively in the C-type lectin domains 1 and 7:CTLD1, and CTLD7 [28, 29]. While the disease progresses, the target epitope may change from outer CysR to the inner CTDL1 to CTDL7. This epitope spreading can be associated with a poor prognosis [30]. Patients showing antibodies against the inner epitopes tend to be elderly and resistant to therapy [31].
In the last years, genetic studies have proliferated [32,33,34,35,36]. A recent genome-wide association study (GWAS) for primary MN in 3782 cases and 9038 controls of East Asian and European ancestries reported that the disease was associated with three classical HLA alleles: DRB1*1501 in East Asians (OR = 3.81), DQA1*0501 in Europeans (OR = 2.88), and DRB1*0301 in both ethnicities (OR = 3.50). GWAS loci explained 32% of MN risk in East Asians and 25% in Europeans, and re-classified 20–37% of the cases that were antibody-negative by the serum anti-PLA2R ELISA diagnostic test. Two previously unreported loci, NFKB1 and IRF4 were also discovered [37].
One may postulate that viral infection or other exogenous factor(s) might induce a conformational change of the epitopes in some proteins of the podocytes (more frequenty PLA2R) in genetically predisposed individuals. The abnormal epitope may be recognized by toll-like receptors as a damage-associated molecular pattern (DAMP), with activation of proinflammatory molecules and cells of the innate immunity. In the inflammatory microenvironment dendritic cells recognize the DAMP as an antigen, become mature and migrate to lymph nodes where they present the antigen to the T cells, thereby activating the adaptive immunity. Activated T cells proliferate and differentiate into effector Th1 and Th17 or T reg. The collaboration between T and B cells activates B cells to produce plasmablasts and antibodies (IgG4) directed against the podocyte antigen, with in situ formation of immmune complexes and activation of complement. The late complement components C5b-C9 induce the production of reactive oxygen species, cytokines, extracellular matrix, and proteolysis of synaptopodin and nephrin These changes result in disruption of actin cytoskeleton, podocyte effacement and proteinuria. C5b-C9 may also induce podocyte hypertrophy and proliferation leading to glomerular sclerosis [38]. One may ask why complement is activated, since IgG4 cannot activate complement [39]. However, it has been demonstrated that glycosylated IgG4 can activate the lectin pathway and induce podocyte injury in a podocyte culture model [40].
In summary, the current research demonstrated that PMN is an autoimmune disease in which the antigen is located in the podocytes and is reached by circulating antibodies with in situ formation of immune complexes that activate complement. However doubts remain. What is the etiology of idiopathic MN? What causes the loss of tolerance to PLA2R and other endogenous molecules? Can polymorphism of PLA2R (or other molecules) create a conformational change of epitopes, representing a target for autoantibodies? How many genetic factors regulate antigens in MN?
Prediction of the natural course of MN
Many studies have tried to identify parameters that could predict the long-term outcome of the disease. Only a few histologic and clinical risk factors for progression are used in clinical practice. Old studies could not find any correlation between the stage of glomerular lesions in the initial biopsy and the long-term results [41, 42]. The presence of segmental glomerular sclerosis can be associated with high risk of renal failure [43]. Instead, the presence of severe interstitial fibrosis and tubular atrophy is associated with a poor outcome [44]. High intensity C3 deposition can also predict renal function deterioration [45].
Increased serum creatinine levels at presentation presage the later development of end stage renal disease (ESRD), unless renal insufficiency is functional and reversible, as in the cases of hypovolemia, excessive dosage of diuretics, or drug-associated acute interstitial nephritis. Based on retrospective analysis of three international centers, Cattran et al. reported that patients with normal serum creatinine and proteinuria remaining < 4 g/day over 6 months have a low risk of progression; those with normal or near normal serum creatinine and proteinuria ranging between 4 and 8 g/day over 6 months have a 55% probability of being in ESRD at 10 years; patients with abnormal or deteriorating serum creatinine and proteinuria > 8 g per day over 6 months have a 66–80% probability of ESRD within 10 years [46]. After the discovery of anti-PLA2R antibodies, DeVries et al. suggested considering these antibodies as predictors of progression [47]. Other investigators confirmed that high baseline or increasing anti-PLA2R antibody levels predict poor outcome [48,49,50]. Accordingly, the most recent KDIGO recommendations decided to incorporate PLA2R antibody > 50 RU/ml among the criteria that indicate a high risk of progression: i.e.—estimated glomerular filtration rate (eGFR) < 60 ml/min /1.73 m2 and/or proteinuria < 8 g/day for at least 6 months—or normal eGFR, proteinuria > 3.5 g/day without a > 50% decrease after 6 months of renin-angiotensin system inhibitors and at least serum albumin < 2.5 g/l and/or PLA2R antibodies > 50 RU/ml [51]. Patients who obtained complete remission, either spontaneously or after treatment, have excellent chances of stable kidney function in the long-term. A fair outcome was also observed with partial remission, although not as good as for complete response [52, 53].
The current opinion is that about 1/3 of patients with PMN may enter spontaneous remission, 1/3 show persistent proteinuria and 1/3 progress to ESRD [54]. However, if one considers the outcome of PMN at 10 years after clinical onset it appears that at least half of them died or required regular renal replacement therapy [55]. Thus the rule of thirds may be replaced by the rule of halves and MN cannot be considered a benign disease not requiring immunosuppressive therapy but rather a main cause of ESRD, at least in patients with persistent nephrotic syndrome or very high levels of PLA2R antibodies.
What treatment for PMN?
The most recent KDIGO guidelines recommend that all patients with PMN and proteinuria should receive optimal supportive care [51]. Immunosuppressive therapy should be limited to patients at risk for disease progression or with serious complications of nephrotic syndrome. The initial treatment may consist of rituximab or cyclophosphamide and corticosteroids, or tacrolimus-based therapy for ≥ 6 months, depending on the estimate of risk. Longitudinal monitoring of anti-PLA2R antibody levels after starting therapy may be useful for evaluating treatment response in patients with MN, and can be used to guide adjustments to therapy.
In 1984, a randomized controlled trial demonstrated that a 6-month treatment based on alternating corticosteroids with chlorambucil every other month significantly increased the probability of nephrotic syndrome remission and protected kidney function [56] These results were confirmed by two other trials with the same therapeutic regimen [57, 58]. An overall estimate of these studies reported that 9% of participants complained of serious adverse events, the most frequent being infections and gastric intolerance [59]. To evaluate whether cyclophosphamide could be better tolerated than chlorambucil, a multicenter randomized trial compared the efficacy and safety of 6-month regimens alternating corticosteroids with either of the two drugs in PMN. Participants who received cyclophosphamide (2 mg/kg/day) obtained a numerically higher number of remissions than those assigned to chlorambucil (0.2 mg/kg/day) and lower number of side effects [60]. A few years later, a multicenter Indian trial compared 6-month cyclical therapy with cyclophospamide alternated with steroids vs symptomatic treatment. The mean follow-up was 11 years. Altogether, 72% of treated patents entered complete or partial remission vs 35% of controls. At 10 years, 89% of treated patients and 65% of controls were alive without dialysis, the differences being highly significant. Hypertension, hypercholesterolemia and edema were significantly less frequent in the treated arm. Serious infections occurred in 15% of treated participants vs 24% of controls. No cases of malignancy were reported [61]. Recently, three randomized, controlled trials compared cyclical therapy with other treatments. An Italian multicenter trial involving 74 patients with PMN reported a better trend to remission at one year with steroid-cyclosphosphamide therapy compared to intravenous rituximab 1 g at days 1 and 15. At two years the probability of total and complete remission was the same in the two groups (82% and 43% in cyclical therapy vs 83% and 42% in rituximab). Serious adverse events occurred in 19% of patients in the rituximab arm and in 14% in the cyclical-regimen [62]. An Indian collaborative trial reported the superior efficacy of cyclical therapy vs tacrolimus combined with corticosteroids. At the end of a 6-year follow-up period, 62% and 28% of participants maintained relapse-free remission in the cyclical therapy and tacrolimus-glucocorticoid groups, respectively. There was no significant difference in the proportion of patients who had a 40% decline in eGFR, death, or ESRD. None of the patients treated with the cyclical regimen reported malignancy [63]. A Spanish multicenter trial enrolled patients with PMN to receive six-month cyclical treatment with corticosteroids and cyclophosphamide or tacrolimus (full-dose for six months and tapering for another three months) and rituximab (one gram at month six). The cumulative rate of remission was 84% for participants assigned to the steroid-cyclophosphamide vs 58% for those assigned to the tacrolimus-rituximab group. Complete remission at 24 months occurred in 26 participants (60%) in the cyclical therapy and in 11 participants (26%) in the tacrolimus-rituximab group. Relapse occurred in one patient in the corticosteroid-cyclophosphamide group, and in three patients in the tacrolimus-rituximab group. Serious adverse events were similar in both groups. However, only one dose of rituximab was given in the tacrolimus group [64]. Taken together, the results of the available controlled trials with combined corticosteroid-cyclophosphamide therapy would appear to show that after an arithmetic average of 54 months, 45% of treated participants obtained complete remission and another 38% achieved partial remission (Table 2).
Some measures should be taken to prevent serious complications. Methylprednisolone pulse administration is usually safe but exceptionally, seizures or anaphylactic reactions may occur when steroids are infused too rapidly into a central vein. Cardiac arrhythmias are rare events that may be favored by hypokalemia. Transient hyperglycemia is frequent. During cyclophosphamide therapy, white blood cells should be measured every 7–10 days, and the dosage should be adjusted in case of leukopenia. This can minimize the risk of infections. To prevent malignancy, the total doses should be less than 360 mg/kg or 2 mg/kg/day for 6 months [65]. Thus, it is unsafe to repeat the standard cyclical regimen (3 months of cyclophosphamide 2 mg/kg/day for each 6-months cycle, resulting in a cumulative dose of 180 mg/kg for ech cycle) more than twice. Therefore, the cumulative dose of cyclophosphamide for each cycle is 180 mg/kg. The risk of gonadal toxicity is mainly related to the use of high dosages. It has been suggested not to exceed a cumulative dose of 250 mg/kg in children and adolescents [66]. If higher doses are needed, semen cryopreservation is recommended in males, while the use of leuprolide acetate can reduce the risk of ovarian damage in females [67].
Rituximab is a monoclonal antibody with a high affinity for the CD20 antigen. It was first used by Remuzzi et al. in eight cases of PMN [68]. The drug was administered intravenously at a dose of 375 mg/m2 every week for four doses. After 20 weeks mean proteinuria significantly decreased from 8.6 to 3.6 g/day. Since then, a number of observational studies have been carried out. A review of those studies showed that rituximab was given at low or high doses and the follow-ups were different [50, 69,70,71,72,73,74,75,76,77]. The cumulative results showed that of 353 enrolled patients, 102 (28.8%) entered complete remission, and another 147 (41.6%) had partial remission (Table 3). A first multicenter controlled trial randomized 75 patients with PMN to renin-angiotensin system inhibitors (RASi) plus rituximab—375 mg/m2 intravenously on days 1 and 8- or RASi alone. At 6 months, 13 (35%) patients given RASi and rituximab and 8 (21%) controls achieved complete or partial remission. In a post-hoc analysis after a mean period of 23 months the remission rate rose to 24/37 patients (65%) in the rituximab group vs 13/38 (34%) in controls. Complete remission rates were respectively, 19% vs 2.6%. The use of rituximab had no impact on safety [78]. Another multicenter trial assigned 130 patients with PMN to receive intravenous rituximab 1 g × 2 repeated after 6 months or cyclosporine 3.5 mg/kg/day for 12 months. At one year the cumulative incidence of complete plus partial remission did not differ between the two groups (60% vs 52%). At 24 months the number of remissions remained stable in the rituximab arm (60%, of which 35% were complete remissions), while it fell to 20% in the cyclosporine group [79]. As mentioned above, two other randomized controlled trials reported, respectively, the superiority of steroid-cyclosphosphamide therapy compared to tacrolimus and a single dose of rituximab [64] or the equivalence between a cyclical therapy with steroids and cyclophosphamide and rituximab [62]. In these trials the side effects of rituximab were mainly related to reactions to the first dose. Of note, the mean follow-up was similar in all these studies, i.e., 23–24 months. Strangely, rituximab-treated participants in these trials showed important differences in the rate of cumulative and complete remissions, ranging between 60 and 83%, and between 19 and 42%, respectively.
Some questions remain unanswered with rituximab. Does the efficacy depend on the different doses of rituximab? Should the doses be based on their effects on circulating B cells? Should rituximab be used as monotherapy or together with other immunosuppressive agents to reinforce the efficacy?
Calcineurin inhibitors (CNIs)
Both cyclosporine and tacrolimus have been largely used in PMN. A number of observational studies demonstrated the efficacy of CNIs in reducing proteinuria and in inducing remission in patients with normal renal function. However, in many patients proteinuria relapsed after discontinuation of CNIs, and the MENTOR trial showed that cyclosporine obtained fewer remissions than rituximab [79]. Similar results have been reported with tacrolimus. Good rates of response have been followed by relapses of proteinuria after discontinuation of the drug. A prospective randomized trial assessed the role of monotherapy with tacrolimus in PMN. Twenty-five patients were randomized to receive tacrolimus (0.05 mg/kg/day) over 12 months with a 6-months taper, whereas 23 patients were assigned to the control group. The probability of remission at 18 months was 94% in the treatment group vs 35% in the control group. The decrease in proteinuria was significantly greater in the treatment group. Nephrotic syndrome reappeared in almost half of the patients who were in remission by the 18th month after tacrolimus discontinuation [80]. A long-term Indian trial and the STARMEN trial concluded that tacrolimus plus steroids or low-dose rituximab achieved fewer responses than cyclical therapy with cyclophosphamide [63, 64]. A pilot study reported that combined treatment with cyclosporine and rituximab can reduce the risk of relapse. In 13 patients with PMN and elevated proteinuria, induction therapy with rituximab plus cyclosporine was given for 6 months, followed by a second cycle of rituximab and tapering of cyclosporine over an 18 months maintenance phase. Complete or partial remission was attained in 92% of participants and 54% were in complete remission at 12 months [81]. The ability of CNIs to modify the risk of ESRD in the long-term remains unproven. There are no data on the use of voclosporin in PMN. Preliminary studies with this new CNI in lupus nephritis reported that voclosporin could reduce proteinuria and maintain stable kidney function [82].
In summary, CNIs may maintain a role in the management of PMN in patients who do not respond or result intolerant to rituximab or cyclical therapy. Given the high relapse rate, long-term therapy is necessary. To reduce the risk of nephrotoxicity, serum creatinine and blood pressure should be frequently monitored, the lowest possible dosage of CNIs should be used for maintenance, and repeat renal biopsy should be performed in those undergoing long-term therapy.
Further treatments
The adrenocorticotropic hormone (ACTH) is composed of 39 amino acids. The first 13 amino acids may form a melanocortin peptide that, after binding to its receptor, can exert anti-inflammatory and immunomodulating effects. Both synthetic and natural ACTH have been used in PMN. Berg et al. [83] administered intramuscular synthetic ACTH, 1 mg twice a week for one year, to 5 patients with PMN and mean proteinuria fell from 7.87 to 0.09 g/day in 3 of them. In the other two participants proteinuria dropped from 13.2 to 0.51 g per day. In Italy, a small multicenter study on 32 participants with PMN and nephrotic syndrome compared cyclical therapy for 6 months and synthetic ACTH 1 mg twice a week for one year. At the end of a mean follow-up of 22 months 12 of the 16 participants assigned to cyclical therapy and 14 of the 16 assigned to ACTH responded with complete or partial remission [84]. In an observational study, 20 patients received a subcutaneous dose of 40 or 80 IU twice weekly of H.P. Acthar(®) Gel. The drug was well tolerated and a decrease in proteinuria > 50% was observed in 2/3 of the patients. Anti-PLA2R antibodies cleared in some, but not all patients [85]. A retrospective study compared a cohort of patients given synthetic ACTH for 12 months with a historical group of patients treated with cyclophosphamide and steroids for 6–12 months. ACTH was less effective in inducing remission and was associated with many adverse events [86]. A systematic review reported that in 149 pts with MN treated with synthetic or natural ACTH complete or partial remission occurred in 69% at 12 months, and in 95% at 24 months. The drug was well tolerated. However, all papers but one were retrospective and many of them were Abstracts [87]. Further studies are needed before recommending the use of ACTH as a first approach in patients with PMN.
Mycophenolate salts are prodrugs that release active mycophenolic acid. This inhibits the de novo pathway of guanosine nucleotide synthesis of T and B cells. A retrospective study compared the effects of mycophenolate mofetil (MMF) plus corticosteroids vs a historical group treated with cyclophosphamide plus corticosteroids. Both treatments lasted one year. After 23 months of follow-up, complete (12% vs 22%) and partial (53% vs 62%) remissions did not significantly differ between the two groups but relapses (57% vs 15%) were significantly more frequent in patients receiving MMF [88]. A randomized controlled trial assigned 36 participants with PMN to receive symptomatic therapy or MMF at a dose of 2 g per day over 12 months. At the end of the study no difference between the two arms was found in mean proteinuria, or partial and complete remissions. Serious adverse effects were observed in 20% of participants receiving MMF [89]. In a Korean trial 39 participants with PMN were treated with MMF plus corticosteroids or cyclosporine plus corticosteroids. At 48 weeks, 76.1% of the MMF group and 66.7% of the cyclosporine group had achieved remission. There was no difference in eGFR between the two groups [90]. There are insufficient data to sustain the role of MMF as a single agent in the management of PMN.
Belimumab is a monoclonal antibody that inhibits the BLyS protein which is necessary for B cell stimulation. In a prospective, open-label, study, 14 participants with PMN and persistent nephrotic-range proteinuria received up to 2 years of belimumab monotherapy (10 mg/kg, every 4 weeks). Proteinuria decreased from 724 to 130 mg/mmol after 24 months. Participants with abnormal albumin and/or cholesterol at baseline reached normal or near normal levels by the last follow-up. Adverse events were consistent with those expected in this population [91].
Ofatumumab is a human anti-CD20 monoclonal antibody that proved to be effective in patients with PMN complicated by rituximab-induced serum sickness [92]. Ofatumumab associated with double-filtration plasmapheresis to obtain antibody depletion has been proposed as a novel therapeutic option in patients with severe PMN [93].
Obinutuzumab is a type II anti-CD20 monoclonal antibody approved for use in chronic lymphocytic leukemia. In a study, obinutuzumab was administered to ten patients with MN resistant to previous treatments including rituximab. Four patients entered complete remission and five had partial remission. The drug was well tolerated [94]. In another study, three patients with PMN resistant to rituximab were given obinutuzumab. All patients obtained complete immunologic response and two of them had partial remission [95].
Bortezomib, a proteasome inhibitor, has been successfully used in anecdotal cases of PMN [96,97,98].
In a prospective study, 43 patients with PMN received hydroxychloroquine and RASi and 83 patients received RASi alone. At 6 months, both the clinical remission rate and percentage change in proteinuria were significantly higher in the hydroxychloroquine group [99].
What to do in patients with kidney insufficiency?
Most observational and randomized studies have been carried out in patients with normal or subnormal kidney function. Only a few studies reported the results of different treatments in patients with severe renal insufficiency.
In the UK, a multicenter, randomized trial was conducted in patients with PMN and a serum creatinine concentration of less than 3.4 mg/dL and at least a 20% decline in estimated creatinine clearance. The 106 participants were randomly assigned to receive no further treatment, 6 months of cyclical therapy with prednisolone and chlorambucil, or 12 months of cyclosporine at a dose of 5 mg/kg/days. The risk of a further 20% decline in kidney function was significantly lower in the prednisolone and chlorambucil group (58%) than in the cyclosporine (81%) or the supportive care groups (87%). Side effects were more frequent and two cases of malignancy occurred in the steroid-chlorambucil group [100]. A main drawback of this trial was that the doses of chlorambucil and cyclosporine had to be adapted to the reduction of kidney function in order to prevent side effects. In addition, clinical control of participants was poor since no measures were taken in patients who did not tolerate the prescribed treatment [101].
In a retrospective study, 13 patients with PMN and mean eGFR of 16 ml/min received 2 weekly infusions of 375 mg/m2 of rituximab or 2 rituximab infusions of 1 g/day 2 weeks apart. Ten treatment courses led to an increase in eGFR and remission of nephrotic syndrome after a median follow-up of 40.8 months in nine patients, while in the other four patients treatments were unsuccessful, and patients needed regular hemodialysis within 1 year [102].
In an Indian study, 64 patients with PMN and an eGFR < 60 ml/min were treated with rituximab or cyclical therapy with corticosteroids and cyclophosphamide. Participants were followed up for a mean period of 24 months. At the end of the study 30 (47%) patients were in remission and 8 had progressed to ESRD. Rituximab and cyclical therapy were equally effective, but rituximab had fewer adverse events [103].
Conclusions
In the last decades enormous advancements have been made in our knowledge of the physiopathology, long-term outcome, and management of PMN. Many clinicians, pathologists and scientists from different countries have participated in this progress. Yet, the etiopathogenesis of PMN has not been completely elucidated; the genetic influence on disease susceptibility is poorly defined, and the available treatments are far from optimal as a number of patients still progress to ESRD or suffer from the complications of severe nephrotic syndrome. Thus, after almost 80 years of exciting studies the story of PMN is not over. Let us hope that continuing research in this field will fill the gaps that still limit a complete understanding of this disease.
Availability of data and material
Not applicable.
References
Bell ET (1946) Renal diseases. Lea & Febiger, Philadelphia
Moroni G, Ponticelli C (2020) Secondary membranous nephropathy. A narrative review. Front Med (Lausanne) 7:611317
Hemminger J, Nadasdy G, Satoskar A, Brodsky SV, Nadasdy T (2016) IgG subclass staining in routine renal biopsy material. Am J Surg Pathol 40(5):617–626
Ohtani H, Wakui H, Komatsuda A, Okuyama S, Masai R, Maki N, Kigawa A, Sawada K, Imai H (2004) Distribution of glomerular IgG subclass deposits in malignancy-associated membranous nephropathy. Nephrol Dial Transplant 19(3):574–579
Hoxha E, Kneißler U, Stege G, Zahner G, Thiele I, Panzer U, Harendza S, Helmchen UM, Stahl RA (2012) Enhanced expression of the M-type phospholipase A2 receptor in glomeruli correlates with serum receptor antibodies in primary membranous nephropathy. Kidney Int 82(7):797–804
Beck LH Jr (2017) PLA2R and THSD7A: disparate paths to the same disease? J Am Soc Nephrol 28(9):2579–2589
Stehlé T, Audard V, Ronco P, Debiec H (2015) Phospholipase A2 receptor and sarcoidosis-associated membranous nephropathy. Nephrol Dial Transplant 30(6):1047–1050
Xie Q, Li Y, Xue J, Xiong Z, Wang L, Sun Z, Ren Y, Zhu X, Hao CM (2015) Renal phospholipase A2 receptor in hepatitis B virus-associated membranous nephropathy. Am J Nephrol 41(4–5):345–353
Ramachandran R, Yadav AK, Kumar V, Varma A, Taneja S, Nada R, Gupta KL, Jha V (2018) PLA2R related primary membranous nephropathy in a hepatitis C positive patient. Nephrology (Carlton) 23(3):288
Yasuda I, Tokuyama H, Hashiguchi A, Hasegawa K, Uchiyama K, Ryuzaki M, Yasuda M, Mizuno R, Ishidoya S, Wakino S, Itoh H (2021) Malignancy-associated membranous nephropathy with PLA2R double-positive for glomeruli and carcinoma. CEN Case Rep 10(2):281–286
Kudose S, Santoriello D, Bomback AS, Stokes MB, D’Agati VD, Markowitz GS (2019) Sensitivity and specificity of pathologic findings to diagnose lupus nephritis. Clin J Am Soc Nephrol 14(11):1605–1615
Lefaucheur C, Stengel B, Nochy D, Martel P, Hill GS, Jacquot C, Rossert J, GN-PROGRESS Study Group (2006) Membranous nephropathy and cancer: epidemiologic evidence and determinants of high-risk cancer association. Kidney Int 70(8):1510–1507
Ponticelli C, Glassock RJ (2014) Glomerular diseases:membranous nephropathy- a modern view-. Clin J Am Soc Nephrol 9(3):609–616
Plaisier E, Ronco P (2020) Screening for cancer in glomerular diseases. Clin J Am Soc Nephrol 15(6):886–888
Debiec H, Guigonis V, Mougenot B, Decobert F, Haymann JP, Bensman A, Deschênes G, Ronco PM (2002) Antenatal membranous glomerulonephritis due to anti-neutral endopeptidase antibodies. N Engl J Med 346(26):2053–2060
Ronco P, Debiec H (2005) Molecular pathomechanisms of membranous nephropathy: from Heymann nephritis to alloimmunization. J Am Soc Nephrol 16(5):1205–1213
Beck LH Jr, Bonegio RG, Lambeau G, Beck DM, Powell DW, Cummins TD, Klein JB, Salant DJ (2009) M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med 361(1):11–21
Tomas NM, Beck LH Jr, Meyer-Schwesinger C, Seitz-Polski B, Ma H, Zahner G, Dolla G, Hoxha E, Helmchen U, Dabert-Gay AS, Debayle D, Merchant M, Klein J, Salant DJ, Stahl RAK, Lambeau G (2014) Thrombospondin type-1 domain-containing 7A in idiopathic membranous nephropathy. N Engl J Med 371(24):2277–2287
Iwakura T, Ohashi N, Kato A, Baba S, Yasuda H (2015) Prevalence of enhanced granular expression of thrombospondin type-1 domain-containing 7A in the glomeruli of Japanese patients with idiopathic membranous nephropathy. PLoS ONE 10(9):e0138841
Wang J, Cui Z, Lu J, Probst C, Zhang YM, Wang X, Qu Z, Wang F, Meng LQ, Cheng XY, Liu G, Debiec H, Ronco P, Zhao MH (2017) Circulating antibodies against thrombospondin type-I domain-containing 7A in Chinese patients with idiopathic membranous nephropathy. Clin J Am Soc Nephrol 12(10):1642–1651
Tomas NM, Hoxha E, Reinicke AT, Fester L, Helmchen U, Gerth J, Bachmann F, Budde K, Koch-Nolte F, Zahner G, Rune G, Lambeau G, Meyer-Schwesinger C, Stahl RA (2016) Autoantibodies against thrombospondin type 1 domain-containing 7A induce membranous nephropathy. J Clin Invest 126(7):2519–2532
Anders HJ, Ponticelli C (2016) Glomerular disease: membranous nephropathy and the Henle-Koch postulates. Nat Rev Nephrol 12(8):447–448
Baker LW, Jimenez-Lopez J, Geiger XJ, Aslam N (2021) Malignancy-associated membranous nephropathy with positive anti-PLA2R autoantibodies: coincidence or connection. Case Rep Nephrol Dial 11(3):334–339
Sethi S (2021) New “Antigens” in membranous nephropathy. J Am Soc Nephrol 32(2):268–278
Al-Rabadi LF, Caza T, Trivin-Avillach C, Rodan AR, Andeen N, Hayashi N, Williams B, Revelo MP, Clayton F, Abraham J, Lin E, Liou W, Zou CJ, Ramkumar N, Cummins T, Wilkey DW, Kawalit I, Herzog C, Storey A, Edmondson R, Sjoberg R, Yang T, Chien J, Merchant M, Arthur J, Klein J, Larsen C, Beck LH Jr (2021) Serine protease HTRA1 as a novel target antigen in primary membranous nephropathy. J Am Soc Nephrol 32(7):1666–1681
Le Quintrec M, Teisseyre M, Bec N, Delmont E, Szwarc I, Perrochia H, Machet MC, Chauvin A, Mavroudakis N, Taieb G, Lanfranco L, Rigothier C, José B, Concetta C, Geneste C, Pernin V, Larroque C, Devaux J, Beyze A (2021) Contactin-1 is a novel target antigen in membranous nephropathy associated with chronic inflammatory demyelinating polyneuropathy. Kidney Int 100(6):1240–1249
Fresquet M, Jowitt TA, Gummadova J, Collins R, O’Cualain R, McKenzie EA, Lennon R, Brenchley PE (2015) Identification of a major epitope recognized by PLA2R autoantibodies in primary membranous nephropathy. J Am Soc Nephrol 26:302–313
Kao L, Lam V, Waldman M, Glassock RJ, Zhu Q (2015) Identification of the immunodominant epitope region in phospholipase A2 receptor-mediating autoantibody binding in idiopathic membranous nephropathy. J Am Soc Nephrol 26(2):291–301
Seitz-Polski B, Dolla G, Payré C, Girard CA, Polidori J, Zorzi K, Birgy-Barelli E, Jullien P, Courivaud C, Krummel T, Benzaken S, Bernard G, Burtey S, Mariat C, Esnault VL, Lambeau G (2016) Epitope spreading of autoantibody response to PLA2R associates with poor prognosis in membranous nephropathy. J Am Soc Nephrol 27:1517–1533
Seit-Polski B, Debiec H, Rousseau A, Dahan K, Zaghrini C, Payré C, Esnault VLM, Lambeau G, Ronco P (2018) Phospholipase A2 receptor 1 epitope spreading at baseline predicts reduced likelihood of remission of membranous nephropathy. J Am Soc Nephrol 29:401–408
Akiyama S, Imai E, Maruyama S (2019) Immunology of membranous nephropathy. F1000Res 8: F1000 Faculty Rev-734
Stanescu HC, Arcos-Burgos M, Medlar A, Bockenhauer D, Kottgen A, Dragomirescu L, Voinescu C, Patel N, Pearce K, Hubank M, Stephens HA, Laundy V, Padmanabhan S, Zawadzka A, Hofstra JM, Coenen MJ, den Heijer M, Kiemeney LA, Bacq-Daian D, Stengel B, Powis SH, Brenchley P, Feehally J, Rees AJ, Debiec H, Wetzels JF, Ronco P, Mathieson PW, Kleta R (2011) Risk HLA-DQA1 and PLA(2)R1 alleles in idiopathic membranous nephropathy. N Engl J Med 364(7):616–626
Saeed M, Beggs ML, Walker PD, Larsen CP (2014) PLA2R-associated membranous glomerulopathy is modulated by common variants in PLA2R1 and HLA-DQA1 genes. Genes Immun 15(8):556–561
Le WB, Shi JS, Zhang T, Liu L, Qin HZ, Liang S, Zhang YW, Zheng CX, Jiang S, Qin WS, Zhang HT, Liu ZH (2017) HLA-DRB1*15:01 and HLA-DRB3*02:02 in PLA2R-related membranous nephropathy. J Am Soc Nephrol 28(5):1642–1650
Gupta S, Köttgen A, Hoxha E, Brenchley P, Bockenhauer D, Stanescu HC, Kleta R (2018) Genetics of membranous nephropathy. Nephrol Dial Transplant 33(9):1493–1502
Kamyshova ES, Bobkova IN, Gorelova IA, Кakhsurueva PA, Filatova EE (2018) Genetic determinants of the development and course of membranous nephropathy. Ter Arkh 90(6):105–111
Xie J, Liu L, Mladkova N, Li Y, Ren H, Wang W, Cui Z, Lin L, Hu X, Yu X, Xu J, Liu G, Caliskan Y, Sidore C, Balderes O, Rosen RJ, Bodria M, Zanoni F, Zhang JY, Krithivasan P, Mehl K, Marasa M, Khan A, Ozay F, Canetta PA, Bomback AS, Appel GB, Sanna-Cherchi S, Sampson MG, Mariani LH, Perkowska-Ptasinska A, Durlik M, Mucha K, Moszczuk B, Foroncewicz B, Pączek L, Habura I, Ars E, Ballarin J, Mani LY, Vogt B, Ozturk S, Yildiz A, Seyahi N, Arikan H, Koc M, Basturk T, Karahan G, Akgul SU, Sever MS, Zhang D, Santoro D, Bonomini M, Londrino F, Gesualdo L, Reiterova J, Tesar V, Izzi C, Savoldi S, Spotti D, Marcantoni C, Messa P, Galliani M, Roccatello D, Granata S, Zaza G, Lugani F, Ghiggeri G, Pisani I, Allegri L, Sprangers B, Park JH, Cho B, Kim YS, Kim DK, Suzuki H, Amoroso A, Cattran DC, Fervenza FC, Pani A, Hamilton P, Harris S, Gupta S, Cheshire C, Dufek S, Issler N, Pepper RJ, Connolly J, Powis S, Bockenhauer D, Stanescu HC, Ashman N, Loos RJF, Kenny EE, Wuttke M, Eckardt KU, Köttgen A, Hofstra JM, Coenen MJH, Kiemeney LA, Akilesh S, Kretzler M, Beck LH, Stengel B, Debiec H, Ronco P, Wetzels JFM, Zoledziewska M, Cucca F, Ionita-Laza I, Lee H, Hoxha E, Stahl RAK, Brenchley P, Scolari F, Zhao MH, Gharavi AG, Kleta R, Chen N, Kiryluk K (2020) The genetic architecture of membranous nephropathy and its potential to improve non-invasive diagnosis. Nat Commun 11(1):1600
Couser WG, Nangaku M (2006) Cellular and molecular biology of membranous nephropathy. J Nephrol 19(6):699–705
Salant DJ (2019) Unmet challenges in membranous nephropathy. Curr Opin Nephrol Hypertens 28(1):70–76
Haddad G, Lorenzen JM, Ma H, de Haan N, Seeger H, Zaghrini C, Brandt S, Kölling M, Wegmann U, Kiss B, Pál G, Gál P, Wüthrich RP, Wuhrer M, Beck LH, Salant DJ, Lambeau G, Kistler AD (2021) Altered glycosylation of IgG4 promotes lectin complement pathway activation in anti-PLA2R1-associated membranous nephropathy. J Clin Invest 131(5):e140453
Ramzy MH, Cameron JS, Turner DR, Neild GH, Ogg CS, Hicks J (1981) The long-term outcome of idiopathic membranous nephropathy. Clin Nephrol 16(1):13–199
Abe S, Amagasaki Y, Konishi K, Kato E, Iyori S, Sakaguchi H (1986) Idiopathic membranous glomerulonephritis: aspects of geographical differences. J Clin Pathol 39(11):1193–1198
Morita M, Mii A, Shimizu A, Yasuda F, Shoji J, Masuda Y, Ohashi R, Nagahama K, Kaneko T, Tsuruoka S (2015) Glomerular endothelial cell injury and focal segmental glomerulosclerosis lesion in idiopathic membranous nephropathy. PLoS One 10(4):e0116700
Chen Y, Tang L, Feng Z, Cao X, Sun X, Liu M, Liu S, Zhang X, Li P, Wei R, Qiu Q, Cai G, Chen X (2014) Pathological predictors of renal outcomes in nephrotic idiopathic membranous nephropathy with decreased renal function. J Nephrol 27(3):307–316
Oto OA, Demir E, Mirioglu S, Dirim AB, Ozluk Y, Cebeci E, Basturk T, Ucar AR, Soltanova L, Nuriyev K, Kilicaslan I, Yazici H, Caliskan Y (2021) Clinical significance of glomerular C3 deposition in primary membranous nephropathy. J Nephrol 34(2):581–587
Cattran DC, Pei Y, Greenwood CM, Ponticelli C, Passerini P, Honkanen E (1997) Validation of a predictive model of idiopathic membranous nephropathy: its clinical and research implications. Kidney Int 51(3):901–907
De Vriese AS, Glassock RJ, Nath KA, Sethi S, Fervenza FC (2017) A proposal for a serology-based approach to membranous nephropathy. J Am Soc Nephrol 28(2):421–430
Reinhard L, Zahner G, Menzel S, Koch-Nolte F, Stahl RAK, Hoxha E (2020) Clinical relevance of domain-specific phospholipase A2 receptor 1 antibody levels in patients with membranous nephropathy. J Am Soc Nephrol 31(1):197–207
Deng L, Huang Q, Wang J, Luo K, Liu J, Yan W, Jiang F, Xu G (2022) Efficacy and safety of different immunosuppressive therapies in patients with membranous nephropathy and high PLA2R antibody Titer. Front Pharmacol 12:786334
Ruggenenti P, Debiec H, Ruggiero B, Chianca A, Pellé T, Gaspari F, Suardi F, Gagliardini E, Orisio S, Benigni A, Ronco P, Remuzzi G (2015) Anti-phospholipase A2 receptor antibody titer predicts post-rituximab outcome of membranous nephropathy. J Am Soc Nephrol 26(10):2545–2558
Rovin BH, Adler SG, Barratt J, Bridoux F, Burdge KA, Chan TM, Cook HT, Fervenza FC, Gibson KL, Glassock RJ, Jayne DRW, Jha V, Liew A, Liu ZH, Mejía-Vilet JM, Nester CM, Radhakrishnan J, Rave EM, Reich HN, Ronco P, Sanders JF, Sethi S, Suzuki Y, Tang SCW, Tesar V, Vivarelli M, Wetzels JFM, Lytvyn L, Craig JC, Tunnicliffe DJ, Howell M, Tonelli MA, Cheung M, Earley A, Floege J (2021) Executive summary of the KDIGO 2021 guideline for the management of glomerular diseases. Kidney Int 100(4):753–779
Ponticelli C, Passerini P, Altieri P, Locatelli F, Pappalettera M (1992) Remissions and relapses in idiopathic membranous nephropathy. Nephrol Dial Transplant 7(Suppl 1):85–90
Troyanov S, Wall CA, Miller JA, Scholey JW, Cattran DC, Toronto Glomerulonephritis Registry Group (2004) Idiopathic membranous nephropathy: definition and relevance of a partial remission. Kidney Int 66(3):1199–1205
Cattran D (2002) Membranous nephropathy. Quo vadis? Kidney Int 61:349–350
van den Brand JA, Hofstra JM, Wetzels JF (2012) Prognostic value of risk score and urinary markers in idiopathic membranous nephropathy. Clin J Am Soc Nephrol 7(8):1242–1248
Ponticelli C, Zucchelli P, Imbasciati E, Cagnoli L, Pozzi C, Passerini P, Grassi C, Limido D, Pasquali S, Volpini T et al (1984) Controlled trial of methylprednisolone and chlorambucil in idiopathic membranous nephropathy. N Engl J Med 310(15):946–950
Ponticelli C, Zucchelli P, Passerini P, Cagnoli L, Cesana B, Pozzi C, Pasquali S, Imbasciati E, Grassi C, Redaelli B et al (1989) A randomized trial of methylprednisolone and chlorambucil in idiopathic membranous nephropathy. N Engl J Med 320(1):8–13
Ponticelli C, Zucchelli P, Passerini P, Cesana B (1992) Methylprednisolone plus chlorambucil as compared with methylprednisolone alone for the treatment of idiopathic membranous nephropathy. The Italian Idiopathic Membranous Nephropathy Treatment Study Group. N Engl J Med 327(9):599–603
Passerini P, Ponticelli C (2003) Corticosteroids, cyclophosphamide, and chlorambucil therapy of membranous nephropathy. Semin Nephrol 23(4):355–361
Ponticelli C, Altieri P, Scolari F, Passerini P, Roccatello D, Cesana B, Melis P, Valzorio B, Sasdelli M, Pasquali S, Pozzi C, Piccoli G, Lupo A, Segagni S, Antonucci F, Dugo M, Minari M, Scalia A, Pedrini L, Pisano G, Grassi C, Farina M, Bellazzi R (1998) A randomized study comparing methylprednisolone plus chlorambucil versus methylprednisolone plus cyclophosphamide in idiopathic membranous nephropathy. J Am Soc Nephrol 9(3):444–450
Jha V, Ganguli A, Saha TK, Kohli HS, Sud K, Gupta KL, Joshi K, Sakhuja V (2007) A randomized, controlled trial of steroids and cyclophosphamide in adults with nephrotic syndrome caused by idiopathic membranous nephropathy. J Am Soc Nephrol 18(6):1899–1904
Scolari F, Delbarba E, Santoro D, Gesualdo L, Pani A, Dallera N, Mani LY, Santostefano M, Feriozzi S, Quaglia M, Boscutti G, Ferrantelli A, Marcantoni C, Passerini P, Magistroni R, Alberici F, Ghiggeri GM, Ponticelli C, Ravani P, RI-CYCLO Investigators (2021) Rituximab or cyclophosphamide in the treatment of membranous nephropathy: the RI-CYCLO randomized trial. J Am Soc Nephrol 32(4):9729–9782
Ramachandran R, Kumar V, Bharati J, Rovin B, Nada R, Kumar V, Rathi M, Jha V, Gupta KL, Kohli HS (2021) Long-term follow-up of cyclical cyclophosphamide and steroids versus tacrolimus and steroids in primary membranous nephropathy. Kidney Int Rep. 6(10):2653–2660
Fernández-Juárez G, Rojas-Rivera J, Logt AV, Justino J, Sevillano A, Caravaca-Fontán F, Ávila A, Rabasco C, Cabello V, Varela A, Díez M, Martín-Reyes G, Diezhandino MG, Quintana LF, Agraz I, Gómez-Martino JR, Cao M, Rodríguez-Moreno A, Rivas B, Galeano C, Bonet J, Romera A, Shabaka A, Plaisier E, Espinosa M, Egido J, Segarra A, Lambeau G, Ronco P, Wetzels J, Praga M, STARMEN Investigators (2021) The STARMEN trial indicates that alternating treatment with corticosteroids and cyclophosphamide is superior to sequential treatment with tacrolimus and rituximab in primary membranous nephropathy. Kidney Int 99(4):986–998
Faurschou M, Sorensen IJ, Mellemkjaer L, Loft AG, Thomsen BS, Tvede N, Baslund B (2008) Malignancies in Wegener’s granulomatosis: incidence and relation to cyclophosphamide therapy in a cohort of 293 patients. J Rheumatol 35(1):100–105
Latta K, von Schnakenburg C, Ehrich JH (2001) A meta-analysis of cytotoxic treatment for frequently relapsing nephrotic syndrome in children. Pediatr Nephrol 16(3):271–282
Hasky N, Uri-Belapolsky S, Goldberg K, Miller I, Grossman H, Stemmer SM, Ben-Aharon I, Shalgi R (2015) Gonadotrophin-releasing hormone agonists for fertility preservation: unraveling the enigma? Hum Reprod 30(5):1089–1101
Remuzzi G, Chiurchiu C, Abbate M, Brusegan V, Bontempelli M, Ruggenenti P (2002) Rituximab for idiopathic membranous nephropathy. Lancet 360(9337):923–924
Fervenza FC, Abraham RS, Erickson SB, Irazabal MV, Eirin A, Specks U, Nachman PH, Bergstralh EJ, Leung N, Cosio FG, Hogan MC, Dillon JJ, Hickson LJ, Li X, Cattran DC, Mayo Nephrology Collaborative Group (2010) Rituximab therapy in idiopathic membranous nephropathy: a 2-year study. Clin J Am Soc Nephrol 5(12):2188–2198
Beck LH Jr, Fervenza FC, Beck DM, Bonegio RG, Malik FA, Erickson SB, Cosio FG, Cattran DC, Salant DJ (2011) Rituximab-induced depletion of anti-PLA2R autoantibodies predicts response in membranous nephropathy. J Am Soc Nephrol 22(8):1543–1550
Michel PA, Dahan K, Ancel PY, Plaisier E, Mojaat R, De Seigneux S, Daugas E, Matignon M, Mesnard L, Karras A, François H, Pardon A, Caudwell V, Debiec H, Ronco P (2011) Rituximab treatment for membranous nephropathy: a French clinical and serological retrospective study of 28 patients. Nephron Extra 1(1):251–261
Souqiyyeh MZ, Shaheen FA, Alsuwaida A, Alghonaim M, Alwakeel J, Mosa D, Akhtar F, Rahman E, Husain M, Roujouleh H, Siddiqi N, Bukhari I, Sadaqa N, Mushtaque F, Awn NM, Shariya F, Alfi A, Amin M, Ahmad M, Rowaie FA, Almueilo S, Kechrid MC, Karkar A (2015) Rituximab as a rescue therapy in patients with glomerulonephritis. Saudi J Kidney Dis Transpl 26(1):47–55
Roccatello D, Sciascia S, Di Simone D, Solfietti L, Naretto C, Fenoglio R, Baldovino S, Menegatti E (2016) New insights into immune mechanisms underlying response to Rituximab in patients with membranous nephropathy: a prospective study and a review of the literature. Autoimmun Rev 15(6):529–538
Moroni G, Depetri F, Del Vecchio L, Gallelli B, Raffiotta F, Giglio E, Brunini F, D’Amico M, Longhi S, Radice A, Messa P, Sinico RA (2017) Low-dose rituximab is poorly effective in patients with primary membranous nephropathy. Nephrol Dial Transplant 32(10):1691–1696
Wang X, Cui Z, Zhang YM, Qu Z, Wang F, Meng LQ, Cheng XY, Liu G, Zhou FD, Zhao MH (2018) Rituximab for non-responsive idiopathic membranous nephropathy in a Chinese cohort. Nephrol Dial Transplant 33(9):1558–1563
Bagchi S, Subbiah AK, Bhowmik D, Mahajan S, Yadav RK, Kalaivani M, Singh G, Dinda A, Agarwal Kumar S (2018) Low-dose Rituximab therapy in resistant idiopathic membranous nephropathy: single-center experience. Clin Kidney J 11(3):337–341
Seitz-Polski B, Dahan K, Debiec H, Rousseau A, Andreani M, Zaghrini C, Ticchioni M, Rosenthal A, Benzaken S, Bernard G, Lambeau G, Ronco P, Esnault VLM (2019) High-dose rituximab and early remission in PLA2R1-related membranous nephropathy. Clin J Am Soc Nephrol 14(8):1173–1182
Dahan K, Debiec H, Plaisier E, Cachanado M, Rousseau A, Wakselman L, Michel PA, Mihout F, Dussol B, Matignon M, Mousson C, Simon T, Ronco P, GEMRITUX Study Group (2017) Rituximab for severe membranous nephropathy: a 6-month trial with extended follow-up. J Am Soc Nephrol 28(1):348–358
Fervenza FC, Appel GB, Barbour SJ, Rovin BH, Lafayette RA, Aslam N, Jefferson JA, Gipson PE, Rizk DV, Sedor JR, Simon JF, McCarthy ET, Brenchley P, Sethi S, Avila-Casado C, Beanlands H, Lieske JC, Philibert D, Li T, Thomas LF, Green DF, Juncos LA, Beara-Lasic L, Blumenthal SS, Sussman AN, Erickson SB, Hladunewich M, Canetta PA, Hebert LA, Leung N, Radhakrishnan J, Reich HN, Parikh SV, Gipson DS, Lee DK, da Costa BR, Jüni P, Cattran DC, MENTOR Investigators (2019) Rituximab or cyclosporine in the treatment of membranous nephropathy. N Engl J Med. 381(1):36–46
Praga M, Barrio V, Juárez GF, Luño J, Grupo Español de Estudio de la NefropatíaMembranosa (2007) Tacrolimus monotherapy in membranous nephropathy: a randomized controlled trial. Kidney Int 71(9):924–930
Waldman M, Beck LH Jr, Braun M, Wilkins K, Balow JE, Austin HA 3rd (2016) Membranous nephropathy: pilot study of a novel regimen combining cyclosporine and Rituximab. Kidney Int Rep 1(2):73–84
Rovin BH, Teng YKO, Ginzler EM, Arriens C, Caster DJ, Romero-Diaz J, Gibson K, Kaplan J, Lisk L, Navarra S, Parikh SV, Randhawa S, Solomons N, Huizinga RB (2021) Efficacy and safety of voclosporin versus placebo for lupus nephritis (AURORA 1): a double-blind, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 397(10289):2070–2080
Berg AL, Nilsson-Ehle P, Arnadottir M (1999) Beneficial effects of ACTH on the serum lipoprotein profile and glomerular function in patients with membranous nephropathy. Kidney Int 56(4):1534–1543
Ponticelli C, Passerini P, Salvadori M, Manno C, Viola BF, Pasquali S, Mandolfo S, Messa P (2006) A randomized pilot trial comparing methylprednisolone plus a cytotoxic agent versus synthetic adrenocorticotropic hormone in idiopathic membranous nephropathy. Am J Kidney Dis 47(2):233–240
Hladunewich MA, Cattran D, Beck LH, Odutayo A, Sethi S, Ayalon R, Leung N, Reich H, Fervenza FC (2014) A pilot study to determine the dose and effectiveness of adrenocorticotrophic hormone (H.P. Acthar® Gel) in nephrotic syndrome due to idiopathic membranous nephropathy. Nephrol Dial Transplant 29(8):1570–1577
van de Logt AE, Beerenhout CH, Brink HS, van de Kerkhof JJ, Wetzels JF, Hofstra JM (2015) ynthetic ACTH in high risk patients with idiopathic membranous nephropathy: a prospective, open label cohort study. PLoS One 10(11):e0142033
Kittanamongkolchai W, Cheungpasitporn W, Zand L (2016) Efficacy and safety of adrenocorticotropic hormone treatment in glomerular diseases: a systematic review and meta-analysis. Clin Kidney J 9(3):387–396
Branten AJ, du Buf-Vereijken PW, Vervloet M, Wetzels JF (2007) Mycophenolate mofetil in idiopathic membranous nephropathy: a clinical trial with comparison to a historic control group treated with cyclophosphamide. Am J Kidney Dis 50(2):248–256
Dussol B, Morange S, Burtey S, Indreies M, Cassuto E, Mourad G, Villar E, Pouteil-Noble C, Karaaslan H, Sichez H, Lasseur C, Delmas Y, Nogier MB, Fathallah M, Loundou A, Mayor V, Berland Y (2008) Mycophenolate mofetil monotherapy in membranous nephropathy: a 1-year randomized controlled trial. Am J Kidney Dis 52(4):699–705
Choi JY, Kim DK, Kim YW, Yoo TH, Lee JP, Chung HC, Cho KH, An WS, Lee DH, Jung HY, Cho JH, Kim CD, Kim YL, Park SH (2018) The effect of mycophenolate mofetil versus cyclosporine as combination therapy with low dose corticosteroids in high-risk patients with idiopathic membranous nephropathy: a multicenter randomized trial. J Korean Med Sci 33(9):e74
Barrett C, Willcocks LC, Jones RB, Tarzi RM, Henderson RB, Cai G, Gisbert SI, Belson AS, Savage CO (2020) Effect of belimumab on proteinuria and anti-phospholipase A2 receptor autoantibody in primary membranous nephropathy. Nephrol Dial Transplant 35(4):599–606
Podestà MA, Ruggiero B, Remuzzi G, Ruggenenti P (2020) Ofatumumab for multirelapsing membranous nephropathy complicated by rituximab-induced serum-sickness. BMJ Case Rep 13(1):e232896
Podestà MA, Gennarini A, Portalupi V, Rota S, Alessio MG, Remuzzi G, Ruggenenti P (2020) Accelerating the depletion of circulating anti-phospholipase A2 receptor antibodies in patients with severe membranous nephropathy: preliminary findings with double filtration plasmapheresis and ofatumumab. Nephron 144(1):30–35
Sethi S, Kumar S, Lim K, Jordan SC (2020) Obinutuzumab is effective for the treatment of refractory membranous nephropathy. Kidney Int Rep 5(9):1515–1518
Klomjit N, Fervenza FC, Zand L (2020) Successful treatment of patients with refractory PLA2R-associated membranous nephropathy with obinutuzumab: a report of 3 cases. Am J Kidney Dis 76(6):883–888
Hartono C, Chung M, Kuo SF, Seshan SV, Muthukumar T (2014) Bortezomib therapy for nephrotic syndrome due to idiopathic membranous nephropathy. J Nephrol 27(1):103–106
Salhi S, Ribes D, Colombat M, Fortenfant F, Faguer S (2021) Bortezomib plus dexamethasone for rituximab-resistant PLA2R+ membranous nephropathy. Kidney Int 100(3):708–709
Barbari A, Chehadi R, KfouryAssouf H, Kamel G, Jaafar M, Abdallah A, Rizk S, Masri M (2017) Bortezomib as a novel approach to early recurrent membranous glomerulonephritis after kidney transplant refractory to combined conventional rituximab therapy. Exp Clin Transplant 15(3):350–354
Cheng YJ, Cheng XY, Zhang YM, Wang F, Wang X, Meng LQ, Liu G, Cui Z, Zhao MH (2022) Effects of hydroxychloroquine on proteinuria in membranous nephropathy. J Nephrol 35(4):1145–1157. https://doi.org/10.1007/s40620-021-01182-z (Epub 2021 Nov 30)
Howman A, Chapman TL, Langdon MM, Ferguson C, Adu D, Feehally J, Gaskin GJ, Jayne DR, O’Donoghue D, Boulton-Jones M, Mathieson P (2013) Immunosuppression for progressive membranous nephropathy: a UK randomised controlled trial. Lancet 381(9868):744–751
Ponticelli C, Glassock RJ (2013) Treatment of membranous nephropathy in patients with renal insufficiency: what regimen to choose? J Nephrol 26(3):427–429. https://doi.org/10.5301/jn.5000289
Hanset N, Esteve E, Plaisier E, Johanet C, Michel PA, Boffa JJ, Fievet P, Mesnard L, Morelle J, Ronco P, Dahan K (2019) Rituximab in patients with phospholipase A2 receptor-associated membranous nephropathy and severe CKD. Kidney Int Rep 5(3):331–338
Ramachandran R, Prabakaran R, Priya G, Nayak S, Kumar P, Kumar A, Kumar V, Agrawal N, Rathi M, Kohli HS, Nada R (2022) Immunosuppressive therapy in primary membranous nephropathy with compromised renal function. Nephron 146(2):138–145
Funding
None received.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that he has no conflict of interest.
Ethical approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Claudio, P. Primary membranous nephropathy: an endless story. J Nephrol 36, 563–574 (2023). https://doi.org/10.1007/s40620-022-01461-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40620-022-01461-3