
Abstract
Purpose
Papillary thyroid carcinoma (PTC) is characterized by epithelial malignancy and is the most prevalent thyroid neoplasm with the best overall prognosis. Notably, recently published studies have indicated remarkably high expression of dipeptidyl peptidase 4 (DPP4) in PTC. However, the underlying molecular mechanism and regulatory factors of PTC progression remain unknown. Therefore, the current study aimed to elucidate the effects of DPP4 gene silencing on PTC and further investigated whether the mechanism of PTC progression is related to the mitogen-activated protein kinase (MAPK) pathway.
Methods
Herein, microarray-based gene expression profiling of PTC was conducted to identify the differentially expressed genes between tumor thyroid tissue and normal thyroid tissue as well as the underlying signaling pathway involved in PTC pathogenesis. Moreover, protein quantification was performed to assess the protein expression of DPP4 in PTC tissues collected from 65 patients. In addition, DPP4 was silenced in PTC cell lines (GLAG-66 and TPC-1) through siRNA-mediated DPP4 knockdown or sitagliptin (inhibitor of DPP4)-mediated inhibition to assess the effects of DPP4 on the MAPK pathway and cellular processes, including proliferation, apoptosis, and epithelial-to-mesenchymal transition (EMT).
Results
Intriguingly, our data revealed markedly high expression of DPP4 in PTC tissues. However, in GLAG-66 and TPC-1 cells, the silencing of DPP4 resulted in significantly reduced expression of ERK1/2, JNK1, P38 MAPK, VEGF, FGFR-1, TGF-β1, Snail, HIF-1α, N-cadherin, and Bcl-2 along with reduced phosphorylation of ERK1/2, JNK1, and P38 MAPK, whereas the expression of E-cadherin and Bax was increased. Furthermore, DPP4 silencing was found to hinder cell proliferation and potentiate cell apoptosis.
Conclusion
Collectively, the present study demonstrated that DPP4 gene silencing inhibits PTC cell proliferation and EMT and promotes cell apoptosis via suppression of the MAPK pathway, thus highlighting a possible regulatory pathway in PTC progression.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Papillary thyroid carcinoma (PTC), one of the most prevalent endocrine malignancies, has been reported to account for approximately 80–85% of all thyroid malignancies [1]. The incidence of PTC is higher in women between the ages of 20 and 55 years [2]. However, a small percentage of patients can develop local recurrence or distant metastasis, which may lead to death, indicating the existence of more aggressive variants of this malignancy [3]. Notably, over the past few decades, a remarkable increase in PTC-related morbidity has been reported, whereas the mortality rate has remained relatively stable [4]. The general prognosis of PTC patients is attributed to an overall favorable 5-year survival rate of 90% [5]. However, the significant increase in the incidence of thyroid cancer suggests that intensive study to identify novel molecular targets for the development of an effective therapeutic strategy for PTC is required [6]. Importantly, genetic alterations have been reported to play a crucial role in the progression of PTC, and PTC is often characterized by rearrangements in the rearranged during transfection (RET) gene or point mutations in the RAS or BRAF proto-oncogenes; these observations suggest that either of these factors can potentially trigger the activation of mitogen-activated protein kinase (MAPK) [7].
Of note, DPP4 (also known as DPPIV/CD26) is an exopeptidase that plays a crucial role in protein regulation, and significant increases in its expression have been reported in a wide array of organ systems and tissue types. Moreover, DPP4 has been indicated to affect various kinds of substrates that further play vital roles in metabolism, glucose regulation, inflammation, cell migration, and cell differentiation [8]. Further studies have demonstrated the presence of the DPP4 protein on the surface of a variety of cell types [9]. Interestingly, a recently published gene expression profiling study revealed remarkably high expression of the DPP4 protein in immune system and epithelial cells in patients with papillary thyroid carcinoma [10]. Another study reported a relatively high level of DPP4 expression in PTC tissues compared to normal tissues, suggesting that DPP4 is a potential candidate molecular marker of PTC [11, 12]. Moreover, a correlation has also been observed between increased expression of the DPP4 protein and improved survival of all cancer cells [13]. On the other hand, DPP4 knockdown has been indicated to suppress MAPK pathway activity in atherosclerotic lesions [14]. The MAPK pathway represents a ubiquitous signal transduction pathway and plays a pivotal role in the regulation of cell viability, apoptosis, and invasion; however, it has been reported to be altered in human disease [15]. Notably, recently published studies have indicated that the long noncoding RNA (lncRNA) SLC26A4-AS1 inactivates the MAPK pathway in TPC-1 cells, which reduces cell migration, proliferation, invasion, and epithelial-to-mesenchymal transition (EMT) and promotes apoptosis in PTC [16]. Based on these findings described above, the present study was designed to evaluate the role of the DPP4 gene in the regulation of PTC cells through the MAPK pathway. Thus, we attempted to identify a potential biomarker that could aid in the development of an effective therapy to treat PTC.
Materials and methods
Ethics statement
This study was conducted with the approval of the ethics committee of the Institutional Review Board of Affiliated Dongguan People's Hospital, Southern Medical University (Dongguan People's Hospital), and it was performed in accordance with the Declaration of Helsinki. All the participants, or their parents or guardians, signed an informed consent document before enrollment.
Microarray analysis
For the microarray analysis, PTC-related datasets (GSE33630, GSE3467, GSE3678, and GSE66783), which were retrieved from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/), were used to screen differentially expressed genes, and the detailed information of these datasets is shown in Table 1. Then, the R language affy package [17] was applied for background correction and standardized pretreatment of the data from the PTC-related datasets. The limma package [18] was used for differential gene analysis with adj. p val < 0.05 and |LogFoldChange|> 2 as the screening criteria, followed by volcano map plotting. Then, Venn online software was used to calculate and draw custom Venn diagrams (https://bioinformatics.psb.ugent.be/webtools/Venn/) to compare the differentially expressed genes obtained from the aforementioned four datasets and to identify gene intersections and potential differentially expressed genes. Thereafter, DigSee (https://210.107.182.61/geneSearch/), an engine based on searching the MEDLINE abstract to obtain evidence for retrieving PTC-related genes [19], was used to identify PTC-related genes. Finally, the PTC-related genes and differentially expressed genes were analyzed using the String database (https://string-db.org/), and the gene interaction network was constructed by Cytoscape 3.6.0 software [20].
Study subjects
Sixty-five PTC patients from the Affiliated Dongguan People's Hospital, Southern Medical University (Dongguan People's Hospital) were selected for the current study between June 2015 and December 2016, and PTC tissues and adjacent normal tissues (control group) were collected from the participants. Specimens (larger than 5 mm × 5 mm) were pathologically diagnosed with PTC and divided into two groups; one group was stored at -80℃ within 20 minutes of collection while the other group was fixed in 10% formalin. Thereafter, the tumors were categorized according to the 8th edition of the tumor-nodes-metastasis (TNM) classification criteria for thyroid cancer as follows: stage I, n = 18; stage II, n = 20; stage III, n = 17; and stage IV, n = 10 [21]. There were 29 males and 36 females aged 21–58 years, and the median age was 43 years. Twenty-five patients had lymph node metastasis (LNM), and 40 patients had non-LNM. The diagnosis of all the patients was confirmed based on clinical data and postoperative pathological diagnosis. Then, the samples were fixed in 10% formalin, embedded in paraffin, cut into 4-μm serial sections, and finally subjected to immunohistochemical staining. Patients were included if they met the following criteria: (1) patients diagnosed with PTC by cytology or histology; (2) patients diagnosed with PTC and suitable for surgical resection based on clinical evaluation; (3) patients capable of undergoing resection; (4) patients with adjacent normal tissue specimens that were pathologically confirmed to be tumor-free; and (5) patients receiving neither radiotherapy nor chemotherapy before the operation [22]. However, patients were excluded from the study if they had any severe complications [22].
Immunohistochemical staining
The prepared paraffin sections from skin tissues were incubated in a 60 °C oven for 30 min, dewaxed with xylene I and II (15 min each), and routinely hydrated (100, 95, 85, and 75%; 5 min each); then, the sections were subjected to microwave antigen retrieval with 1 mM Tris–EDTA (pH = 9.0) for 15 min. After that, the sections were cooled to room temperature and washed twice with phosphate-buffered saline (PBS) (5 min each). Then, the endoperoxidase activity was removed by immersion in 3% H2O2 methanol for 10 min at room temperature. Thereafter, the sections were blocked with 50 μL of 10% goat serum (C-0005, Shanghai Haoran Biological Technology Co., Ltd, Shanghai, China) for 30 min at 37 ℃ and incubated with a primary rabbit polyclonal antibody against CD26/DPP IV (ab28340, 1:2000, Abcam Inc, Cambridge, UK) overnight at 4 ℃. Then, the samples were incubated with a goat anti-mouse secondary IgG antibody (ab6785, 1:1000, Abcam, Cambridge, UK) at 37 °C for 30 min followed by incubation with a working solution of horseradish peroxidase (HRP)-labeled Streptomyces ovalbumin (ab7403, 1:1000, Abcam Inc, Cambridge, UK) at 37 °C for 15 min and washed three times with PBS (5 min each). Subsequently, the sections were stained with freshly prepared 3,3-diaminobenzidine (DAB) solution for 10 min in the dark, followed by microscopic observation. Following complete development, the reaction was halted by washing with tap water for 10 min. The sections were then counterstained with hematoxylin for 5 min, dehydrated with gradient alcohol, cleared with xylene, and sealed with neutral gum. Blue or light blue sections indicated negative results, and yellow–brown particles in the sections indicated positive results. In addition, PBS was used as the primary antibody solution for the negative control (NC). The number of positive cells in three randomly selected nonoverlapping visual fields from each section was calculated in a double-blind manner using image analysis software (Nikon Instech Co. Ltd., Tokyo, Japan). The formula applied was as follows: the percentage of positive cells = number of positive cells/total cells × 100%. Staining intensity was scored according to the staining results of tumor cells (A): 0 points indicated no staining, 1 point indicated buff staining, 2 points indicated tan staining, and 3 points indicated brown staining. Positive expression of DPP4 was scored according to the ratio of positive cells to all cells (B): 0 points indicated negative cells, 1 point indicated ≤ 10% positive cells, 2 points indicated 11–50% positive cells, 3 points indicated 51–75% positive cells, and 4 points indicated > 75% positive cells. The accumulated points of each field equaled A + B: scores below 3 points were denoted as negative (−), 3–5 points as positive (+), 5–7 points as strongly positive, and scores ≥ 7 points as highly strong positive (+++).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cells using TRIzol reagent (16096020, Thermo Fisher Scientific Co., Ltd, New York, USA). The concentration and purity of the extracted total RNA were subsequently evaluated using an ultraviolet spectrophotometer, and samples with A260/A280 = 1.8 ~ 2.0 were used in the experiments. Then, the 28S and 18S rRNA bands were visualized by 1% agarose gel electrophoresis, which demonstrated that the extracted RNA was intact. Thereafter, 5 µg of total RNA was reverse transcribed into complementary DNA (cDNA) according to the manufacturer’s instructions of the RT-qPCR kits (Applied Biosystems Co., Foster City, CA, USA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as the internal reference for the relative expression of DPP4, extracellular signal-regulated kinase (ERK)1, ERK2, c-Jun N-terminal kinase (JNK)1, P38 MAPK, vascular endothelial growth factor (VEGF), fibroblast growth factor receptor 1 (FGFR-1), B-cell lymphoma 2 (Bcl-2), Bax, Snail, hypoxia-inducible factor 1α (HIF-1α), epithelial (E)-cadherin and neural (N)-cadherin. The primers were synthesized by Shanghai Generay Biotech Co., Ltd., and the sequences are presented in Table 2. The relative expression levels of the target genes were calculated based on the \({2}^{{ - \Delta \Delta C_{{\text{t}}} }}\) method [23].
Western blot analysis
Approximately 100 mg of fresh specimens was collected and lysed with protein lysis buffer supplemented with radioimmunoprecipitation assay (RIPA) lysis solution (P0013B, Beyotime Biotechnology Co., Shanghai, China) and a protease inhibitor. The specimens were rapidly ground with liquid nitrogen at a low temperature and lysed on ice for 30 min. The concentration of the proteins in the supernatant obtained by centrifugation (4 ℃, 25,764g for 15 min) was determined using a bicinchoninic acid (BCA) kit (20201ES76, Shanghai Yeasen Biotechnology Co., Ltd, Shanghai, China). Then, 30 μg of the total protein was added to a loading buffer containing β-mercaptoethanol and incubated in a 100 ℃ water bath for 5 min for albuminous degeneration. The proteins were subsequently transferred to a membrane following separation using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Then, the membrane was blocked with 5% skim milk for 1 h and then probed overnight at 4 °C with the following primary antibodies: rabbit polyclonal antibody against CD26 (ab28340, 1:2000), rabbit polyclonal antibody against JNK1 (ab47337, 1:200), mouse anti-human ERK1/2 antibody (ab54230, 1:1000), mouse anti-human P38 MAPK antibody (ab31828, 1:1000), rabbit polyclonal antibody against Snail (ab82846, 1:300), mouse anti-human HIF-1α antibody (ab113642, 1:1500), and mouse anti-human N-cadherin antibody (ab76057, 1:1000). All the aforementioned antibodies were purchased from Abcam Inc. (Cambridge, UK). Following three washes (5 min each) with Tris-buffered saline with Tween (TBST), the membrane was reprobed with horseradish peroxidase a (HRP)-conjugated secondary goat anti-rabbit immunoglobulin G (IgG) antibody (ab6721, 1:2000, Abcam Inc, Cambridge, UK) and rabbit anti-mouse IgG antibody (ab6734, 1:1000, Abcam Inc, Cambridge, UK) with continuous shaking at room temperature for 1 h. The membrane was then washed with PBS three times (5 min each) at room temperature and subsequently visualized using an enhanced chemiluminescence (ECL) solution (prepared by a mixture of liquid A and liquid B in equal volumes; Pierce, Waltham, Thermo Fisher Scientific, Massachusetts, USA) at room temperature for 1 min. The band intensities were quantified using the Bio-Rad Image Analysis System (Bio-Rad Inc, Hercules, California, USA) and Quantity One v4.6.2 software. The ratio of the gray value of the target band to that of GAPDH was representative of the relative protein expression.
Cell culture and grouping
The DPP4 gene was found to be highly expressed in the PTC cell line GLAG-66 (Shenzhen Otwo Biotech Inc, Shenzhen, China) and in the human TC cell line TPC-1 (Shanghai Zishi Biotech Co., Ltd). Furthermore, the GLAG-66 cell line was cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum, 100 U/mL penicillin, and 100 µg/mL streptomycin, while the TPC-1 cell line was cultured in Roswell Park Memorial Institute (RPMI)-1640 medium (31800022, Invitrogen Inc. New York, USA) in a 5% CO2 incubator (thromo3111, Jinan Beisheng Medical Instrument Co., Ltd, Shandong, China) at 37 °C. The cells were divided into a blank group (nontransfected cells), NC group (cells transfected with a NC small interfering RNA [siRNA] sequence, which was synthesized by Shanghai GenePharma Co., Ltd), DPP4-siRNA group (cells transfected with siRNA targeting DPP4, which was synthesized by Shanghai GenePharma Co., Ltd), and DPP4-sitagliptin group (cells treated with 1 mM sitagliptin, an inhibitor of DPP4). The sequences are listed in Table 3. Briefly, 24 h before transfection, GLAG-66 and TPC-1 cells in the logarithmic phase of growth were harvested and seeded into a 6-well plate at a density of 5 × 104 cells per well. When the cell density reached 30–50%, the cells were transfected using the Lipofectamine 2000 reagent (American Invitrogen Co., Ltd) according to the manufacturer’s instructions. Serum-free Opti-minimum essential medium (MEM) (250 μL, Gibco, Grand Island, New York, USA) was used to dilute 100 pmol of DPP4-siRNA or the NC (final concentration added to the cells was 50 nM), while the remaining serum-free medium Opti-MEM (250 μL) was used to dilute 5 μL of Lipofectamine 2000; then, these two solutions were separately mixed and incubated at room temperature for 5 min. Thereafter, the two aforementioned solutions were mixed, incubated at room temperature for 20 min, and added to the cell culture plate. The cells were then cultured with high-glucose DMEM after incubation in 5% CO2 for 6–8 h. Moreover, the GLAG-66 cells were cultured in high-glucose DMEM, and the TPC-1 cells were cultured in RPMI-1640 medium. The following experiments were conducted after incubation for 48 h.
Cell counting kit-8 (CCK-8) assay
Cell proliferation was measured using a CCK-8 assay kit (GM-040101-5, Dojindo, Molecular Technologies Inc., Gaithersburg, MD, USA) according to the manufacturer’s instructions. Three groups of cells in the logarithmic phase of growth following 48 h of transfection were harvested, washed with PBS, and digested using trypsin. After washing with PBS, the cells were suspended and seeded in a 96-well plate at a density of 5 × 103 cells per well. Six duplicate wells were set for each group, and 5% fetal bovine serum was added to the cells, which were incubated in an incubator in 5% CO2 at 37 °C. The wells were then cultured and examined at 24, 48, 72, and 96 h, and 10 μL of CCK-8 solution was added to each well before the examination. After incubation at 37 °C for 2 h, the optical density (OD), which indicated cell proliferation, of each well was measured at a wavelength of 450 mm using a spectrophotometer (UV-1800A, Shanghai Macy Instrument Co., Ltd, Shanghai, China). A growth curve was plotted with time as the abscissa and OD value as the ordinate. The experiment was independently repeated three times.
Flow cytometry
The method for propidium iodide (PI) single staining was as follows: After 48 h of cell transfection, the culture supernatant was removed, and the cells were washed once with PBS, digested with trypsin, collected, and centrifuged at 179 g and 4 °C for 5 min; then, the supernatant was discarded. Next, the cell pellet was washed twice with precooled PBS and harvested via centrifugation at 1000 rpm/minute for 5 min. The cells were then treated with 70% precooled ethanol and fixed at 4 °C overnight. Following PBS washing and centrifugation at 179 g for 5 min, 10 μL of RNase enzyme was added to the cells and incubated at 37 ℃ for 5 min. Then, the cells were stained with 1% PI (40710ES03, Shanghai Qcbio Science and Technologies Co., Ltd., Shanghai, China) in the dark for 30 min. The specimens were then analyzed with a flow cytometer (FACSCalibur, BD, FL, NJ, USA) to determine the red fluorescence at an excitation wavelength of 488 nm to analyze the cell cycle. In the scatter diagram, the lower left quadrant (Q4) indicates live cells, the lower right quadrant (Q3) indicates early apoptotic cells, the upper right quadrant (Q2) represents late apoptotic cells, and the upper left quadrant (Q1) represents necrotic cells. The results were then analyzed using ModFit software. The experiments were independently repeated three times.
The Annexin V-fluorescein isothiocyanate (FITC)/PI double staining method was used to assess cell apoptosis. Briefly, GLAG-66 and TPC-1 cells were seeded in a 6-well plate at a density of 2 × 105 cells per well and divided into the blank group, the NC group and the DPP4-siRNA group. Following 48 h of transfection, the cells were digested with EDTA-free trypsin, harvested, and centrifuged at 4 °C and 179g for 5 min, and the supernatant was removed. Precooled PBS was subsequently used to wash the cells, and the supernatant was separated via centrifugation at 179g for 5 min. Cell apoptosis was examined using an Annexin-V-FITC/PI apoptosis assay kit (CA1020, Beijing Solarbio Science and Technology Co., Ltd., Beijing, China). The cells were washed with binding buffer and added to a mixture of Annexin V-FITC and binding buffer at a ratio of 1:40. After resuspension, oscillation, mixing, and incubation at room temperature for 30 min, the cells were added to the PI mixture, and the binding buffer was added at a ratio of 1:40, followed by mixing and incubating at room temperature for 15 min. Cell apoptosis was measured by flow cytometry. The following controls were used to set up compensation and quadrants: unstained cells, cells stained with Annexin V-FITC alone (no PI), and cells stained with PI alone (no Annexin V-FITC). Then, the data from each sample tube were collected and analyzed by CellQuest3.0 software to calculate the percentage of apoptotic cells. The apoptotic rate (%) was expressed as the percentage of the total number of early apoptotic cells (Q3) to the total number of late apoptotic cells (Q2). The experiment was independently conducted three times.
Statistical analysis
All the data were processed using SPSS 21.0 statistical software (IBM Corp. Armonk, NY, USA). Measurement data are expressed as the mean ± standard deviation. Data obeying normal distribution and homogeneity of variance between the two groups were compared using a t test, while comparisons among multiple groups were conducted by one-way analysis of variance (ANOVA), and a nonparametric (Mann–Whitney U) test was applied for data with skewed distribution and defect variances. Enumeration data are expressed as percentages or ratios, and comparisons were performed using x2 text. A value of p < 0.05 indicated a statistically significant difference.
Results
Bioinformatics analysis predicting the differentially expressed genes in PTC
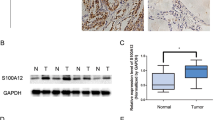
For bioinformatics analysis, the R language affy package and limma package were used to analyze the PTC-related datasets with the screening criteria of adj. p. val < 0.05 and |LogFoldChange|> 2. There were 132, 64, 92, and 1123 differentially expressed genes screened from the GSE33630, GSE3467, GSE3678, and GSE66783 datasets, respectively. Subsequently, the first 100 differentially expressed genes from each dataset were plotted on a Venn diagram (Fig. 1a), which revealed 11 intersections, including KCNJ2, PROS1, IGFBPL1, LRRK2, TPO, GABRB2, TFF3, LRP1B, PKHD1L1, CDH16, and DPP4. The aforementioned genes were then subjected to a follow-up analysis as candidate genes. PTC-related genes were researched in DigSee software, and the first 20 genes (TP53, TG, BRAF, RET, LGALS3, CEACAM5, ERBB2, BRCA1, CCND1, HRAS, KRT19, MET, VIM, TP63, VEGFA, KRAS, CDH1, TPO, KRT7, and MUC1) were regarded as disease-related genes. Furthermore, the String database was used to analyze the interaction between differentially expressed genes and disease-related genes followed by the construction of the gene interaction network. Intriguingly, we found that in the gene interaction network, the disease-related genes showed close interactions, and DPP4 was the differentially expressed gene that closely interacted with the other genes (Fig. 1b), emphasizing that the differential expression of DPP4 was likely to influence PTC. Thereafter, a volcano plot was constructed that depicted the first 50 differentially expressed genes obtained from the GSE3467 (Fig. 1c) and GSE33630 datasets (Fig. 1d). However, DPP4 was more highly expressed in PTC tissues than in normal control tissues. Moreover, in the GSE3678 and GSE66783 datasets (as shown in Fig. 1e, f), the expression of DPP4 was found to be significantly elevated in the PTC tissues. Accordingly, previously published studies have also reported the activation of the MAPK pathway in PTC [24, 25], while another study has demonstrated that DPP4 plays a pivotal role in activating the MAPK pathway [26]. Additionally, the DPP4 inhibitor has been shown to suppress the MAPK pathway [14]. Thus, based on these findings described above, we speculated that differentially expressed DPP4 could influence the MAPK pathway.

mRNA expression profiles in PTC. a Venn diagram of the first 100 differentially expressed genes in the PTC-related GSE33630, GSE3467, GSE3678, and GSE66783 datasets, revealing 11 intersected genes; b interaction network of differentially expressed genes related to PTC and disease-related genes; the orange diamond refers to differentially expressed genes, and the blue round refers to disease-related genes; genes irrelevant to the other genes are not shown in this network; c volcano plot of the first 50 differentially expressed genes in the GSE3467 dataset; d volcano plot of the first 50 differentially expressed genes in the GSE33630 dataset; In panels c and d, the abscissa refers to sample number and ordinate refers to differentially expressed genes; the histogram in the upper right represents color gradation, and each rectangle refers to a sample value; e expression of DPP4 in the GSE3678 dataset; f expression of DPP4 in the GSE66783 dataset
High DPP4 protein expression in PTC tissues exhibits a positive correlation with patient tumor size, tumor staging, and LNM
Furthermore, the positive expression of the DPP4 protein in PTC tissues and adjacent normal tissues was determined by immunohistochemical staining. Intriguingly, our results demonstrated prominently enhanced DPP4 protein expression in the cell membrane, and its positive expression was reflected by yellowish-brown or brown granules; these granules were predominantly observed in the upper region of the membrane of follicular cells, forming a yellow line along the cavity surface of papilla or follicles. However, the basement membrane was found to be negative (Fig. 2). Moreover, the positive expression rate of the DPP4 protein was 7.69% in adjacent normal tissues and 81.54% in PTC tissues, indicating that the positive expression rate of the DPP4 protein was elevated in PTC tissues compared to adjacent normal tissues (p < 0.05).

PTC tissues exhibit increased positive expression of the DPP4 protein, which is positively correlated with patient tumor size, tumor staging, and LNM. a Representative view of DPP4 positive expression in PTC tissues and adjacent normal tissues (n = 65) analyzed by immunohistochemistry (scale bar = 25 μm); b Quantitation of the positive expression of the DPP4 protein in PTC tissues and adjacent normal tissues. *p < 0.05 compared with adjacent normal tissues
Further analysis of the relationship between the positive expression of the DPP4 protein and the clinicopathological characteristics of PTC patients (Table 4) revealed that the expression of the DPP4 protein was not related to sex or age (p > 0.05) but was related to tumor size, tumor staging, and the presence or absence of LNM (p < 0.05). Thus, larger tumor size reflected a higher tumor staging and a higher positive expression rate of the DPP4 protein. Moreover, a higher positive expression rate of the DPP4 protein was observed among the patients with LNM than among those without LNM (p < 0.05). Taken together, these results suggested that the expression of DPP4 was positively related to patient tumor size, tumor staging, and lymph node metastasis.
DPP4 promotes the expression of MAPK-related genes in PTC tissues
Thereafter, the mRNA expression of DPP4 and MAPK pathway-related factors (ERK1, ERK2, JNK1, and P38 MAPK) in PTC tissues and adjacent normal tissues was examined by RT-qPCR. Importantly, our data indicated that compared with adjacent normal tissues, PTC tissues exhibited significantly increased mRNA expression of DPP4, ERK1, ERK2, JNK1, and P38 MAPK (p < 0.05) (Fig. 3a).

DPP4 increases the expression of MAPK pathway-related factors in PTC tissues. a mRNA expression of DPP4, ERK1, ERK2, JNK1 and P38 MAPK in PTC tissues and adjacent normal tissues (n = 65); b representative Western blots of the DPP4 protein and its quantification in PTC tissues and adjacent normal tissues (n = 3; a representative sample from three repeated experiments was selected), normalized to GAPDH. c Representative Western blots of the DPP4, ERK1, ERK2, JNK1, and P38 MAPK proteins and their quantification in PTC tissues and adjacent normal tissues (n = 3; a representative sample from three repeated experiments was selected), normalized to GAPDH. *p < 0.05 compared with adjacent normal tissues
Moreover, the protein expression of DPP4 and the expression of MAPK pathway-related factors (p-ERK1/2, p-JNK1, p-P38 MAPK, ERK1/2, JNK1, and P38 MAPK) in PTC tissues and adjacent normal tissues were verified by Western blot analysis. Notably, the results revealed lower levels of DPP4 protein expression in the adjacent normal tissues than in the PTC tissues (p < 0.05) (Fig. 3b).
The protein expression of DPP4, ERK1/2, JNK1, and P38 MAPK, along with the level of phosphorylated ERK1/2, JNK1, and P38 MAPK, was significantly higher in the PTC tissues than in adjacent normal tissues (p < 0.05) (Fig. 3c). Thus, DPP4 is correlated with MAPK pathway-related genes. Hence, we hypothesized a role of the DPP4 gene in promoting the phosphorylation of MAPK pathway-related proteins and activating the MAPK pathway in PTC tissues, which could be the key mechanism of PTC progression. Subsequently, a cell transfection experiment was carried out.
DPP4 silencing decreases the mRNA expression of MAPK pathway-, cell proliferation- and EMT-related genes while enhancing that of cell apoptosis-related genes in vitro
Furthermore, RT-qPCR and Western blot analyses were conducted to examine the mRNA and protein expression, respectively, of DPP4, MAPK pathway-related factors (ERK1/2, JNK1, and P38 MAPK), cell proliferation-related factors (VEGF, FGFR-1, and TGF-β1), EMT-related factors (Snail, HIF-1α, E-cadherin, and N-cadherin), and cell apoptosis-related factors (Bcl-2, and Bax) in GLAG-66 and TPC-1 cell lines. As shown in Fig. 4, the results obtained from the GLAG-66 cell line were consistent with those obtained from the TPC-1 cell line. Specifically, compared with the blank and NC groups, the DPP4-siRNA group exhibited decreased mRNA and protein expression of DPP4, ERK1, ERK2, JNK1, P38 MAPK, VEGF, FGFR-1, TGF-β1, Snail, HIF-1α, N-cadherin, and Bcl-2 and decreased phosphorylation of ERK1/2, JNK1, and P38 MAPK (p < 0.05), whereas the expression of E-cadherin and Bax was increased (p < 0.05). On the other hand, no difference was observed regarding the aforementioned factors between the DPP4-sitagliptin group and the DPP4-siRNA group, the blank group, and the NC group (p > 0.05). Taken together, the results reported above showed that DPP4 silencing could diminish the expression of MAPK pathway-, cell proliferation- and EMT-related factors, whereas cell apoptosis-related genes were upregulated, in vitro.

DPP4 silencing decreases the mRNA expression of DPP4, ERK1, ERK2, JNK1, P38 MAPK, VEGF, FGFR-1, TGF-β1, Snail, HIF-1α, N-cadherin, and Bcl-2 but increases that of E-cadherin and Bax in PTC cells (GLAG-66 and TPC-1). *p < 0.05 compared with the blank group and the NC group
DPP4 silencing inhibits the protein expression of MAPK pathway-, cell proliferation- and EMT-related genes while increasing the protein expression of cell apoptosis-related genes in vitro
The protein expression of DPP4, MAPK pathway-related factors (ERK1/2, JNK1, and P38 MAPK), cell proliferation-related factors (VEGF, FGFR-1, and TGF-β1), EMT-related factors (Snail, HIF-1α, E-cadherin, and N-cadherin), and cell apoptosis-related factors (Bcl-2 and Bax) was detected by Western blot analysis. The results obtained from the GLAG-66 cell line were consistent with those obtained from the TPC-1 cell line (Fig. 5). Importantly, a significant decrease in the protein expression of DPP4, ERK1/2, JNK1, P38 MAPK, VEGF, FGFR-1, TGF-β1, Snail, HIF-1α, N-cadherin, and Bcl-2 as well as the phosphorylation of ERK1/2, JNK1, and P38 MAPK (p < 0.05) was observed. However, markedly increased expression of E-cadherin and Bax protein was observed in the DPP4-siRNA group compared to the blank group and the NC group (p < 0.05). However, no significant difference was detected regarding the aforementioned factors between the DPP4-sitagliptin group and the DPP4-siRNA group, the blank group and the NC group (p > 0.05). Collectively, these results described above suggest that DPP4 silencing could suppress the protein expression of MAPK pathway-, cell proliferation-, and EMT-related factors but potentiate the protein expression of cell apoptosis-related genes in vitro.

DPP4 silencing inhibits the protein expression of DPP4, ERK1/2, JNK1, P38 MAPK, VEGF, FGFR-1, TGF-β1, Snail, HIF-1α, N-cadherin, and Bcl-2 and the phosphorylation of ERK1/2, JNK1, and P38 MAPK while increasing the expression of E-cadherin and Bax in PTC cells (GLAG-66 and TPC-1). a Representative Western blots of the DPP4, ERK1/2, JNK1, P38 MAPK, VEGF, FGFR-1, TGF-β1, Snail, HIF-1α, N-cadherin, Bcl-2, E-cadherin, and Bax proteins and their quantification in GLAG-66 cells with DPP4 silencing, normalized to GAPDH; b Representative Western blots of the DPP4, ERK1/2, JNK1, P38 MAPK, VEGF, FGFR-1, TGF-β1, Snail, HIF-1α, N-cadherin, Bcl-2, E-cadherin and Bax proteins and their quantification in TPC-1 cells with DPP4 silencing, normalized to GAPDH. *p < 0.05 compared with the blank and NC groups
DPP4 silencing inhibits PTC cell proliferation in vitro
Furthermore, the CCK-8 assay was employed to evaluate PTC cell proliferation. Importantly, we found that there was no significant difference (p > 0.05) in cell proliferation after 24 h (Fig. 6). However, the proliferation of PTC cells was diminished over time in the DPP4-siRNA group compared to the blank group and the NC group (p < 0.05). Moreover, no notable difference was observed in terms of cell proliferation between the DPP4-sitagliptin group and the DPP4-siRNA group, the blank group, and the NC group (p > 0.05). Thus, the results described above showed that DPP4 silencing impaired PTC cell proliferation in vitro.

DPP4 silencing attenuates the proliferation of PTC cells in vitro. a GLAG-66 cell proliferation was measured by CCK-8 assay following treatment with DPP4-siRNA or DPP4-sitagliptin; b TPC-1 cell proliferation was measured by CCK-8 assay following treatment with DPP4-siRNA or DPP4-sitagliptin; *p < 0.05 compared with the blank group and the NC group
DPP4 silencing arrests more PTC cells at the G0/G1 phase but fewer cells at the S phase in vitro
Then, the PI single staining results showed (Fig. 7) that compared with the blank group and the NC group, the DPP4-siRNA group exhibited more G0/G1 phase-arrested cells and fewer S phase-arrested cells (p < 0.05). However, no significant difference was detected regarding cell cycle distribution between the DPP4-sitagliptin group and the DPP4-siRNA group, blank group and NC group (p > 0.05). Collectively, these results further supported the evidence that DPP4 silencing could inhibit PTC cell proliferation in vitro.

DPP4 silencing arrests more PTC cells at the G0/G1 phase but fewer cells at the S phase in vitro. a GLAG-66 cell cycle distribution was measured by flow cytometry following treatment with DPP4-siRNA or DPP4-sitagliptin; b statistical results of panel a; c TPC-1 cell cycle distribution was measured by flow cytometry following treatment with DPP4-siRNA or DPP4-sitagliptin; d statistical results of Panel C. *p < 0.05 compared with the blank and NC groups
DPP4 silencing promotes PTC cell apoptosis in vitro
Furthermore, the Annexin staining results indicated (Fig. 8) that after transfection, the total early- and late-stage apoptosis rates of the GLAG-66 cells and TPC-1 cells were (4.58 ± 0.53)%, (4.77 ± 0.48)%, (4.18 ± 0.42)%, and (4.37 ± 0.45)%, respectively, in the NC group and the blank group, which were much lower than those in the DPP4-siRNA group [(12.13 ± 1.14)% and (11.73 ± 1.19)%] (p < 0.05). On the other hand, no prominent difference was observed regarding the cell apoptosis rate between the DPP4-sitagliptin group and the DPP4-siRNA group, the NC group, and the blank group (p > 0.05). Thus, these results collectively suggest that DPP4 silencing stimulated the apoptosis of PTC cells in vitro.

DPP4 silencing increases the apoptosis of PTC cells in vitro. a Representative data of apoptotic GLAG-66 cells and their quantification following treatment with DPP4-siRNA or DPP4-sitagliptin; b statistical results of panel a; c representative data of apoptotic TPC-1 cells and their quantification following treatment with DPP4-siRNA or DPP4-sitagliptin; d statistical results of panel c. In the scatter diagram, the lower left quadrant (Q4) shows the live cells, the lower right quadrant (Q3) shows the early apoptotic cells, the upper right quadrant (Q2) shows the late apoptotic cells and the upper left quadrant (Q1) shows the necrotic cells. *p < 0.05 compared with the blank and NC groups
Discussion
PTC represents the most common type of thyroid cancer and includes several subtypes, including the follicular subtype [27]. Notably, recent studies have indicated that high expression of DPP4 is attributed to better survival of cancer cells, thus emphasizing the crucial role of DPP4 as a potential biomarker for cancer prognosis [13]. The present study was conducted to investigate the effect of DPP4 gene silencing on PTC cell proliferation, apoptosis, and EMT via the MAPK pathway. Intriguingly, our results illustrated that DPP4 gene silencing could potentially hinder MAPK pathway activation, which suppressed PTC cell proliferation and EMT and promoted apoptosis, thus ultimately delaying the progression of PTC.
Following a comprehensive set of experimental investigations, our results revealed that the mRNA and protein expression of DPP4 and MAPK pathway-related factors was remarkably enhanced in PTC tissues. Similarly, a previously published study demonstrated the upregulated positive expression of the DPP4 protein in PTC tissues, and its high expression was found to be associated with extrathyroidal extension (p < 0.001), BRAF mutation (p < 0.001), and advanced tumor stage (p = 0.007) in PTC [28]. Moreover, Lee et al. reported that DPP4 could suppress various carcinoma phenotypes of thyroid cancer cells via regulation of the TGFβ pathway. Certain findings associated with DPP4 gene silencing were characterized by pharmacological inhibition, emphasizing that the biological role of DPP4 in thyroid cancer was at least partially regulated by certain activities [28]. However, the current study remained focused on the mechanism by which DPP4 gene silencing regulates the MAPK pathway, affecting the proliferation, apoptosis, and EMT of PTC cells. Therefore, we attempted to explore the effect of DPP4 knockdown on PTC cell proliferation, cell cycle progression, and apoptosis based on comprehensive bioinformatics analysis and an array of human and cellular experiments. Hence, the focus of the present study, namely the downstream signaling pathway and the MAPK pathway-related factors at the cellular level, was the main difference between this paper and Lee's article.
In addition, the results from the current study revealed that the expression of MAPK pathway-related factors was enhanced in PTC tissues, and the DPP4 gene was found to enhance the activity of the MAPK pathway in PTC tissues by increasing the expression of pathway-related genes. Consistently, recent evidence has demonstrated that inactivation of the MAPK pathway by miRNAs results in the tumor suppression of PTC, thus highlighting the crucial role of MAPK in the tumorigenesis of various tumors, including PTC [29]. Moreover, MAPK signaling components represent crucial factors in the context of converting extracellular stimuli into a wide range of cellular responses and are reported to be upregulated in human tumors; these findings form the basis for developing novel therapies aimed at the inhibition of these factors [30]. Accordingly, previously reported experimental data have indicated that sitagliptin (a DPP4 inhibitor) was able to inhibit the progression of a certain disease by suppressing the MAPK pathway [14]. In addition, numerous reports have suggested that inhibition of DPP4 influences ERK-mediated gene expression [31], and the expression of TGF-β can be suppressed by a DPP-4 inhibitor [32]. Interestingly, the positive correlation of DPP4 expression with MAPK pathway activation has also been highlighted in oxidative stress [33]. Hence, these studies described above further support our finding that the DPP4 protein affects the regulation of the MAPK pathway.
E-cadherin (an EMT-related factor) has been characterized as a cell adhesion molecule that plays a critical role in cell-to-cell adhesion [34]. Moreover, previously published studies have demonstrated that E-cadherin inhibits cell growth by inducing apoptosis [35]. Interestingly, DPP4 has also been indicated to inhibit EMT [36]. However, inhibition of DPP4 has been reported to induce suppression of EMT in human bronchial epithelial cells [37]. Another study illustrated that knockdown of Snail (an EMT-inducible transcription factor) can reduce ATP-binding cassette (ABC) transporter levels in doxorubicin-treated DPP4-deficient 4T1 cells [38]. Vildagliptin and sitagliptin (DPP4 inhibitors) have been found to reduce HIF-1α levels in the context of testicular torsion/detorsion-induced injury by antioxidative stress mechanisms [39]. The abovementioned findings suggested the role of DPP4 in the inhibition of EMT in PTC cells.
Furthermore, our data indicated a remarkable reduction in cell proliferation, whereas cell apoptosis was elevated, in the presence of DPP4 silencing or inhibitor treatment. Notably, ubiquitous expression of DPP4 has been identified in a wide variety of cells, including endothelial cells and epithelial cells, and its diverse biological activities have been shown to influence protease activity, regulate intracellular signal transduction, and control cell migration and proliferation [40]. Furthermore, a recent study investigated the interaction between DPP4 and apoptosis mediators or inhibitors, and their data demonstrated the upregulation of the pro-apoptotic factor Bax and the downregulation of the anti-apoptotic factor Bcl-2 following high expression of DPP4 [41]. However, inhibition of DPP4 by miR-29a has been reported to contribute to the suppression of proliferation, migration, and invasion in PTC cells [42].
In summary, the present study demonstrated that silencing the DPP4 gene could potentially inhibit activation of the MAPK pathway to prevent PTC cell proliferation and EMT while enhancing cell apoptosis (Fig. 9). These findings emphasize the significance of DPP4 gene silencing as a promising strategy for PTC. Importantly, the present study provides a unique perspective on research for developing novel PTC therapies. Although our study remained focused on evaluating the DPP4 gene in PTC, it is important to note that DPP4 may not be the sole factor relevant to PTC therapy. Therefore, further research is required to explore the mechanism that involves not only the DPP4 gene but also other accelerating genes to verify the conclusion of the present study and to facilitate the development of treatments for PTC.

Graphical representation of the underlying regulatory pathway and function of DPP4 in PTC. DPP4 was upregulated in PTC tissues. DPP4 activated the MAPK pathway and then promoted the proliferation and EMT, while inhibiting the apoptosis, of PTC cells
References
LiVolsi VA (2011) Papillary thyroid carcinoma: an update. Mod Pathol 24(Suppl 2):S1-9
Chen W, Zheng R, Baade PD et al (2016) Cancer statistics in China, 2015. CA Cancer J Clin 66:115–132
Toniato A, Boschin I, Casara D et al (2008) Papillary thyroid carcinoma: factors influencing recurrence and survival. Ann Surg Oncol 15:1518–1522
Stern S, Dagan E, Alon EE et al (2016) Papillary thyroid carcinoma treatment—is the pendulum shifting? Harefuah 155:511–515
Liu FC, Lin HT, Lin SF et al (2017) Nationwide cohort study on the epidemiology and survival outcomes of thyroid cancer. Oncotarget 8:78429–78451
McLeod DS, Sawka AM, Cooper DS (2013) Controversies in primary treatment of low-risk papillary thyroid cancer. Lancet 381:1046–1057
Abdullah MI, Junit SM, Ng KL et al (2019) Papillary thyroid cancer: genetic alterations and molecular biomarker investigations. Int J Med Sci 16:450–460
Glorie LL, Verhulst A, Matheeussen V et al (2012) DPP4 inhibition improves functional outcome after renal ischemia-reperfusion injury. Am J Physiol Renal Physiol 303:F681-688
Sato Y, Kamada T, Yamauchi A (2014) The role of dipeptidyl peptidase 4 (DPP4) in the preservation of renal function: DPP4 involvement in hemoglobin expression. J Endocrinol 223:133–142
Ozog J, Jarzab M, Pawlaczek A et al (2006) Expression of DPP4 gene in papillary thyroid carcinoma. Endokrynol Pol 57(Suppl A):12–17
Huang Y, Prasad M, Lemon WJ et al (2001) Gene expression in papillary thyroid carcinoma reveals highly consistent profiles. Proc Natl Acad Sci USA 98:15044–15049
Gubala E, Wiench M, Oczko-Wojciechowska M et al (2005) Gene expression analysis by DNA microarray in papillary thyroid cancer. Endokrynol Pol 56:752–757
Javidroozi M, Zucker S, Chen WT (2012) Plasma seprase and DPP4 levels as markers of disease and prognosis in cancer. Dis Markers 32:309–320
Zeng Y, Li C, Guan M et al (2014) The DPP-4 inhibitor sitagliptin attenuates the progress of atherosclerosis in apolipoprotein-E-knockout mice via AMPK- and MAPK-dependent mechanisms. Cardiovasc Diabetol 13:32
Lee S, Rauch J, Kolch W (2020) Targeting MAPK signaling in cancer: mechanisms of drug resistance and sensitivity. Int J Mol Sci 21:1102
Wang DP, Tang XZ, Liang QK et al (2020) Overexpression of long noncoding RNA SLC26A4-AS1 inhibits the epithelial-mesenchymal transition via the MAPK pathway in papillary thyroid carcinoma. J Cell Physiol 235:2403–2413
Gautier L, Cope L, Bolstad BM et al (2004) affy—analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20:307–315
Smyth GK (2004) Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3:1–25
Kim J, So S, Lee HJ et al (2013) DigSee: disease gene search engine with evidence sentences (version cancer). Nucleic Acids Res 41:W510-517
Shannon P, Markiel A, Ozier O et al (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504
Kim K, Kim JH, Park IS et al (2018) The updated AJCC/TNM staging system for papillary thyroid cancer (8th edition): from the perspective of genomic analysis. World J Surg 42:3624–3631
Song HM, Luo Y, Li DF et al (2015) MicroRNA-96 plays an oncogenic role by targeting FOXO1 and regulating AKT/FOXO1/Bim pathway in papillary thyroid carcinoma cells. Int J Clin Exp Pathol 8:9889–9900
Ayuk SM, Abrahamse H, Houreld NN (2016) The role of photobiomodulation on gene expression of cell adhesion molecules in diabetic wounded fibroblasts in vitro. J Photochem Photobiol B 161:368–374
Occhi G, Barollo S, Regazzo D et al (2015) A constitutive active MAPK/ERK pathway due to BRAFV600E positively regulates AHR pathway in PTC. Oncotarget 6:32104–32114
Romitti M, Wajner SM, Ceolin L et al (2016) MAPK and SHH pathways modulate type 3 deiodinase expression in papillary thyroid carcinoma. Endocr Relat Cancer 23:135–146
Wronkowitz N, Gorgens SW, Romacho T et al (2014) Soluble DPP4 induces inflammation and proliferation of human smooth muscle cells via protease-activated receptor 2. Biochim Biophys Acta 1842:1613–1621
Cancer Genome Atlas Research N (2014) Integrated genomic characterization of papillary thyroid carcinoma. Cell 159:676–690
Hong S, Yu S, Li J et al (2016) MiR-20b displays tumor-suppressor functions in papillary thyroid carcinoma by regulating the MAPK/ERK signaling pathway. Thyroid 26:1733–1743
Lee JJ, Wang TY, Liu CL et al (2017) Dipeptidyl peptidase IV as a prognostic marker and therapeutic target in papillary thyroid carcinoma. J Clin Endocrinol Metab 102:2930–2940
Wagner EF, Nebreda AR (2009) Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer 9:537–549
Ta NN, Li Y, Schuyler CA et al (2010) DPP-4 (CD26) inhibitor alogliptin inhibits TLR4-mediated ERK activation and ERK-dependent MMP-1 expression by U937 histiocytes. Atherosclerosis 213:429–435
Wang WJ, Chang CH, Sun MF et al (2014) DPP-4 inhibitor attenuates toxic effects of indoxyl sulfate on kidney tubular cells. PLoS ONE 9:e93447
Beckers PAJ, Gielis JF, Van Schil PE et al (2017) Lung ischemia reperfusion injury: the therapeutic role of dipeptidyl peptidase 4 inhibition. Ann Transl Med 5:129
Wang X, Zhang J, Fan M et al (2009) The expression of E-cadherin at the invasive tumor front of oral squamous cell carcinoma: immunohistochemical and RT-PCR analysis with clinicopathological correlation. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 107:547–554
Lowy AM, Knight J, Groden J (2002) Restoration of E-cadherin/beta-catenin expression in pancreatic cancer cells inhibits growth by induction of apoptosis. Surgery 132:141–148
Huang CN, Wang CJ, Yang YS et al (2016) Hibiscus sabdariffa polyphenols prevent palmitate-induced renal epithelial mesenchymal transition by alleviating dipeptidyl peptidase-4-mediated insulin resistance. Food Funct 7:475–482
Sun J, Chu S, Lu M et al (2020) The roles of dipeptidyl peptidase-4 and its inhibitors in the regulation of airway epithelial-mesenchymal transition. Exp Lung Res 46:163–173
Li S, Fan Y, Kumagai A et al (2020) Deficiency in dipeptidyl peptidase-4 promotes chemoresistance through the CXCL12/CXCR4/mTOR/TGFbeta signaling pathway in breast cancer cells. Int J Mol Sci 21:805
Abdelzaher WY, Rofaeil RR, Ali DME et al (2020) Protective effect of dipeptidyl peptidase-4 inhibitors in testicular torsion/detorsion in rats: a possible role of HIF-1alpha and nitric oxide. Naunyn Schmiedebergs Arch Pharmacol 393:603–614
Shi S, Kanasaki K, Koya D (2016) Linagliptin but not Sitagliptin inhibited transforming growth factor-beta2-induced endothelial DPP-4 activity and the endothelial-mesenchymal transition. Biochem Biophys Res Commun 471:184–190
El-Sherbeeny NA, Nader MA (2016) The protective effect of vildagliptin in chronic experimental cyclosporine A-induced hepatotoxicity. Can J Physiol Pharmacol 94:251–256
Wang Y, Han J, Lv Y et al (2019) miR-29a inhibits proliferation, invasion, and migration of papillary thyroid cancer by targeting DPP4. OncoTargets Ther 12:4225–4233
Acknowledgements
We would like to give our sincere appreciation to the reviewers for their helpful comments on this article.
Funding
None.
Author information
Authors and Affiliations
Contributions
XH, SC, CX, ZL, ZW, and ZY designed the study. XH, SC, and CX collected the data, carried out the data analyses and produced the initial draft of the manuscript. ZL, ZW, and ZY contributed to drafting the manuscript. All the authors have read and approved the final submitted manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Ethical approval
The study was approved by the ethics committee of the Institutional Review Board of Affiliated Dongguan People's Hospital, Southern Medical University (Dongguan People's Hospital) and performed in strict accordance with the Declaration of Helsinki.
Informed consent
All the participants, or their parents or guardians, provided informed consent before enrollment.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hu, X., Chen, S., Xie, C. et al. DPP4 gene silencing inhibits proliferation and epithelial-mesenchymal transition of papillary thyroid carcinoma cells through suppression of the MAPK pathway. J Endocrinol Invest 44, 1609–1623 (2021). https://doi.org/10.1007/s40618-020-01455-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-020-01455-7