
Abstract
Purpose of Review
Sclerostin, encoded by the gene Sost, is a regulatory glycoprotein produced by mature osteocytes in bone. Findings in animals and humans revealed that Sost/sclerostin deficiency results in increased bone density, and neutralizing antibodies to this protein are being investigated for treatment of postmenopausal osteoporosis. While it is clear that sclerostin is a major regulator of skeletal homeostasis, the specific mechanisms that control its expression are not completely understood.
Recent Findings
Growing evidence suggest that epigenetic phenomena such as histone modification, DNA methylation, or microRNAs influence Sost/sclerostin expression under physiologic and pathologic conditions. Furthermore, these epigenetic mechanisms control Sost/sclerostin production in a time- and cell-context manner. Together with previous literature, these new findings indicate that Sost/sclerostin regulation is complex and requires coordination of multiple mechanisms.
Summary
This review summarizes the current knowledge on the epigenetic regulation of Sost/sclerostin expression and discusses future research needed to unravel the mechanisms by which Sost/sclerostin expression is controlled in a cell-, time-, and space-specific manner.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bone remodeling is a lifelong process that repairs bone damage and maintains mineral homeostasis. This process removes old bone and creates new bone in a balanced manner and involves multiple and coordinated cellular and molecular events [1, 2]. Advances during the last two decades revealed that bone remodeling is orchestrated by osteocytes, the most abundant cells in bone, which regulate the bone-resorbing activity of osteoclasts and bone-forming activity of osteoblasts by mechanisms involving cell-to-cell contact and the release of soluble factors [3, 4].
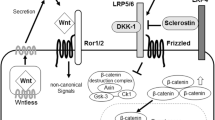
Osteocytes are major producers of antagonists of the Wnt/β-catenin signaling, a pathway that promotes bone formation by stimulating the maturation and survival of cells of the osteoblastic lineage and inhibits osteoclastogenesis by increasing the production of osteoprotegerin (Opg) in osteoblasts and osteocytes [5•]. Sclerostin, the translatable product of the gene Sost, is a potent Wnt signaling antagonist secreted by osteocytes. This secreted glycoprotein achieves its inhibitory function by binding to the Wnt co-receptors Lrp 4/5/6 thus interfering with the formation of Wnt ligand-LRP receptor complexes and thereby antagonizing downstream signaling [6•, 7, 8, 9]. Consistent with a central role of sclerostin in the regulation of bone homeostasis, mutations in the Sost gene in humans resulting in either the absence of the protein or its secretion are associated with high bone mass conditions and exaggerated bone formation, including sclerosteosis, van Buchem disease, and craniodiaphyseal dysplasia [5•, 10, 11]. Similarly, mice with genetic deletion of the gene Sost display increased bone mass and bone formation [12]. In contrast, overexpression of Sost/sclerostin decreases bone mass and reduces the bone-forming activity of osteoblasts [13,14,15]. Further, mice carrying mutations resulting in a deficient binding of sclerostin to Lrp5 also exhibit increased bone mass [16, 17]. In concert, all these results provided the basis to target the anti-osteoanabolic actions of sclerostin as a therapeutic approach for patients with decreased bone formation and low bone mass. Monoclonal neutralizing antibodies to sclerostin were generated, and beneficial skeletal outcomes have been observed in animal studies and clinical trials [18, 19]. As a result, an application for approval of a sclerostin-targeted therapy was submitted to the FDA in 2016.
The extensive Sost/sclerostin-related work generated over the last years has significantly expanded our understating of sclerostin skeletal functions and bone remodeling. However, the pathways that control Sost/sclerostin expression and the mechanisms that allow the regulation of this gene in a cell-, time-, and site-specific manner remain unclear. Growing evidence suggests that epigenetic mechanisms that alter DNA accessibility and chromatin structure dictate Sost/sclerostin patterns of expression. In this review, we summarize the current knowledge on the epigenetic regulation of Sost/sclerostin and propose future research needed to understand the mechanisms controlling Sost expression.
Epigenetic Regulation of Gene Expression
All cells in an organism have the same DNA sequence, but they differ in their gene expression patterns, which allow the cells to specialize and perform different functions. Epigenetic factors play a critical role in the establishment of cell-specific gene expression patterns and the dynamic regulation of gene expression during development and in adult tissues through reversible modifications in the chromatin, without changing DNA sequence [20]. In addition, through specific changes in gene expression patterns, epigenetic mechanisms enable temporal and spatial control of gene activity to adapt cells and organisms to changing conditions of the local microenvironment and/or the external environment.
The genome is organized in nucleosomes, consisting of 146 base pairs of DNA wrapped around a histone octamer protein core, formed by an H3-H4 tetramer and two H2A-H2B dimers. Nucleosomes fold into more complex structures that determine the accessibility of the DNA to the transcription machinery [21, 22]. DNA regions to be transcribed are in a looser chromatin conformation; thus, transcription factors and the RNA polymerase can access the target genes. Posttranslational modifications of the histone tails are essential determinants of chromatin packaging and govern activation/inactivation of genes to further influence cellular behavior. These modifications include methylation, acetylation, phosphorylation, sumoylation, biotinylation, and ubiquitylation [23]. Some modifications, such as acetylation of histones H3 and H4 and mono- or tri-methylation of lysine 4 in H3 (H3K4me1 and H3K4me3), are associated with active transcription. On the other hand, modifications such as methylation of H3 at lysines 9 and 27, H3K9me3, and H3K27me3 are related to repression of gene repression.
A variety of enzymes contribute to the covalent modifications of histones [24, 25]. Five families are involved in the incorporation of methyl groups. The SET domain contains proteins of the methyltransferase superfamily including Dot1-like proteins and human protein arginine methyltransferases (PRMT family), methylates, lysines, and arginines. Further, the amine oxidases and JumonjiC (JmjC) domain contain iron-dependent dioxygenases that contribute to histone demethylation [26, 27]. Histone acetyltransferases (HATs) and histone deacetylases (HDACs) are responsible for the acetylation and deacetylation of histones, respectively [25, 28]. Sirtuins, class III HDACs, are of particular interest since they not only regulate chromatin structure and gene expression but also DNA repair, senescence, cell differentiation, and stress cellular responses [29,30,31]. In addition to the histone-modifying proteins, other remodeling complexes can modify the chromatin package and state. These protein complexes are composed of an ATPase-dependent component to remove histones from DNA, thereby restructuring nucleosomes. Based on distinct domain structures, there are four well-characterized families of mammalian chromatin-remodeling ATPases, which are SWItch/Sucrose Non-Fermenting (SWI/SNF), Imitation SWItch (ISWI), Nucleosome Remodeling and Deacetylation (NuRD)/Mi-2/chromodomain, helicase, DNA binding (CHD), and INOsitol requiring 80/Sick With Rat8 ts (INO80/SWR1) [22, 32].
One of the most widely studied epigenetic marks is DNA methylation and specifically the addition of a methyl group to cytosines that are in the 5′ position of a guanine (i.e., CpG dinucleotides). Most cytosines in CpG sites are methylated (about 80%). The distribution of CpG in the human genome is uneven, and there are regions in the genome with a high density of CpG sites, the so-called CpG islands [33]. In the human genome, CpG islands are common in the promoter regions of many genes. The methylation of cytosines in gene promoters is frequently associated to gene repression [34]. It is noteworthy that several epigenome-wide association studies have revealed phenotype-associated differentially methylated sites in regulatory DNA regions distant from gene promoters. This finding suggests that the methylation of CpGs in enhancers and other regulatory regions, and not only in the proximal promoters, play an important role in the epigenetic regulation of gene transcription [35]. 5-Methylcytosine (5mC) is a stable epigenetic mark that is potentially transmissible to daughter cells. DNA methyltransferases (DNMTs) are responsible for the transfer of a methyl group from the methyl donor, S-adenosyl-l-methionine (SAM), to the 5-position of cytosines. DNMT1 is responsible for the maintenance of the 5mC after cell division, whereas DNMT3A/DNMT3B are involved in the de novo methylation of cytosines [36].
Cytosine hydroxymethylation is a recently identified type of DNA modification; however, its exact biological role is still unclear [37, 38]. 5-hydroxymethylcytosine (5hmC) is formed through oxidation of 5mC by the Ten-Eleven Translocation (TET) family of proteins. It has been shown that in addition to 5hmC, TET proteins can generate 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) [39, 40]. 5caC is specifically recognized and excised by thymine-DNA glycosylase (TDG), which then results in an unmethylated cytosine. This observation has led some investigators to consider that 5hmC is merely an intermediate in DNA demethylation. However, many studies suggest that 5hmC is indeed a stable epigenetic mark directly implicated in the regulation of gene expression by influencing genome structure and function [41, 42]. Supporting this notion, chromatin regulators, such as Mbd3, only bind to 5hmC and not to 5mC, thus regulating the expression of genes with 5hmC marks. Further, UHRF1, a factor involved in the maintenance of DNA methylation marks, binds to 5hmC and 5mC with similar affinity. Moreover, 5-hmC is frequently found in gene bodies and associated with active gene expression [43,44,45]. However, unlike the extensive evidence supporting a negative relationship between DNA methylation levels and gene expression, and the well-established cooperative interactions between DNA methylation and histone modifications and other epigenetic marks [33, 46,47,48], there is still limited information about the relationship between 5hmC and gene expression or its crosstalk with other epigenetic marks.
Non-coding RNAs (ncRNAs) represent other epigenetic mechanism and have gained widespread attention in recent years. They can be classified in small ncRNAs, smaller than 200 nucleotides, and long ncRNAs (lncRNAs), larger than 200 nucleotides. Among small ncRNAs, microRNAs (miRNAs) are single-stranded RNA molecules of ∼22 nucleotides in length that interact with mRNA targets posttranscriptionally to regulate gene translation [49, 50]. miRNAs are involved in many biological processes, including cell development and differentiation, immunity, and disease development and progression. miRNAs repress gene expression by blocking mRNA translation and, in some cases, inducing mRNA degradation [51]. Coding sequences for miRNAs are distributed throughout the genome in introns, exons, and intergenic regions. The biogenesis of miRNAs starts with the transcription of pri-miRNAs, which are capped with 7-methylguanosine and bear a poly-(A) tail. After transcription, the enzyme complex Drosha-DeGiorgio Critical Region 8 binds and cleaves pri-miRNAs to obtain pre-miRNAs. pre-miRNAs are then exported from the nucleus by Exportin 5. Once in the cytoplasm, Dicer, a Rnase III, processes the pre-miRNA into 22 nt duplex miRNA and then it is denatured into a single strand miRNA. This miRNA is loaded onto the RNA-induced silencing complex (RISC), which is formed by Dicer, TRBP, Ago2, and GW182. The resultant complex miRNA-RISC recognizes the mRNA targets, resulting in either the degradation of the target mRNA or blockade of the mRNA-dependent synthesis of polypeptide chains [52, 53].
LncRNAs are poorly conserved among species, but accumulating evidence suggests that this type of regulatory RNAs plays an important role in a diversity of biological processes [54]. They can be classified according to their relation to protein coding genes into five categories: sense, antisense, bidirectional, intronic, or intergenic. This group of RNAs can act as regulators of gene expression at different levels. They can interact at the transcription level by favoring or repressing the binding of transcription factors. Further, they can also act at the posttranscriptional level influencing the degradation, splicing, translation, or transportation of mRNA. Moreover, they can modulate miRNAs pathways, thereby acting as regulator of regulators [55, 56].
Sost/Sclerostin Expression and Regulation
The expression of Sost/sclerostin appears to be osteocyte-specific in bone. This is supported by immunohistochemical evidence showing sclerostin staining in the body of osteocytes, osteocytic lacunae, and canaliculae but not in osteoblasts or lining cells covering quiescent surfaces of the bone [9, 57, 58]. In vitro, Sost mRNA expression is undetectable in primary osteoblasts, but its expression progressively increases as osteoblasts mature and acquire the osteocytic cellular and molecular signature [58, 59]. Consistent with this, high levels of sclerostin are found in mature osteocytes surrounded by mineral but rarely detected in newly embedded osteocytes [57, 58]. These findings indicate that changes in the Sost regulatory machinery occur during the acquisition of the osteocytic phenotype and enable the expression of this gene only in latter stages of osteoblast differentiation. Further, the expression of Sost/sclerostin is also regulated in a site-specific manner. For instance, mechanical loading reduces the expression of Sost/sclerostin only in osteocytes located close to high bone formation surfaces thus coordinating regional and local osteogenesis [13]. Moreover, Sost/sclerostin production is also controlled in a timely manner, as demonstrated by the increases in sclerostin levels with age or the transient inhibition of its expression by parathyroid hormone (PTH) [60, 61].
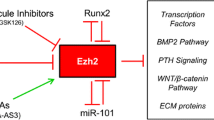
The Sost gene is organized into two exons and two major regulatory regions. Originally identified in Van Buchem’s patients, the Sost gene contains a 52-kb distal enhancer element (ECR5) located ∼35 kb downstream of the transcription start site (TSS) [11]. More recently, it was demonstrated that the Sost proximal promoter (∼1.4 kb upstream the TSS of the Sost gene) also controls the expression of this gene [62•]. These two regulatory regions contain response elements for a number of hormones and transcription factors that modulate Sost/sclerostin expression in bone [6•]. Thus, Sost/sclerostin production is complex and involves multiple mechanisms that allow dynamic regulation of its expression.
Regulation of Sost Expression by HDACs
There are few data about the role of posttranslational modifications of histones in bone metabolism and in osteocyte function in particular. Nevertheless, several studies indicate that different chromatin modifications contribute to the maintenance of bone mass; however, thus far, deacetylation of histone tails has received most attention. Deacetylation of lysine side chains in histones is involved in skeletal development and maintenance of bone mass [63, 64]. With some exceptions, in vitro data supports that HDACs inhibit bone formation and stimulate bone resorption, and animal and clinical studies using HDAC inhibitors resulted in complex effects in the bone [63, 64]. Some of these effects are mediated by direct actions on the Sost gene. Baertschi, Keller, and colleagues provided the first evidence supporting that HDACs participate in the regulation of Sost/sclerostin [65•]. Using UMR106, a rat osteosarcoma cell line that expresses Sost/sclerostin, they found that silencing of class I HDACs 1, 2, and 3 inhibits the expression of Sost, suggesting a role of these HDACs in the regulation of constitutive expression of this gene. Further, HDAC5 knockout mice exhibit elevated Sost mRNA levels and increased sclerostin-positive osteocytes [66•], demonstrating that HDAC5 negatively regulates Sost/sclerostin in osteocytes. Mechanistic studies revealed that HDAC5 binds to Mef2C, a major regulator of Sost expression, and inhibits its function [66•]. Moreover, HDAC5 also mediates the regulation of Sost/sclerostin by PTH, a known inhibitor of its expression, by mechanisms that involved Mef2c and interactions with response elements located in the ECR5 regulatory region [66•, 67, 68].
The sirtuin family includes seven proteins (sirtuin1–7) with a highly conserved NAD-binding catalytic domain. Sirtuin (Sirt)1, Sirt6, and Sirt7 are predominantly nuclear proteins, whereas other Sirts are located in the cytosol or the mitochondria [69]. Sirt1 is the most conserved and most studied mammalian sirtuin. Sirt1 has protein-deacetylase activity and has been shown to remove acetyl residues from H3K9Ac, H4K16Ac, and H1K26Ac [70]. Because histone acetylation tends to associate with relaxed chromatin and active gene transcription, Sirt-mediated deacetylation of histones at gene promoters contributes to inhibit the expression of a variety of genes. Sirt1 haplo-insufficient mice exhibit reduced bone formation and low bone mass, whereas increased Sirt1 activity promotes the differentiation of MSCs towards the osteoblastic phenotype and increases bone mass in mice, at least in part by enhancing Wnt/β-catenin signaling [71,72,73]. The effects of Sirt1 on skeletal homeostasis may be mediated in part through Sost/sclerostin regulation. In fact, Sirt1 has an inhibitory effect on Sost expression in mouse osteoblastic cells, the osteocytic cell line MLO-Y4, and human osteoblasts. The molecular mechanisms have not been fully elucidated but may be related to the deacetylation of histone 3 at lysine 9 (H3K9) at the SOST promoter [74•].
Together, these data suggest that different histone deacetylases could have opposite effects on Sost/sclerostin expression. Thus, future research is required to elucidate the effects of individual HDACs on the regulation of this gene. Similarly, the role of other chromatin modifications (methylation, sumoylation, …) on Sost transcription is understudied and demands further investigation.
Regulation of Sost Expression by DNA Methylation
Advances in the last 20 years have demonstrated that DNA methylation is key in the differentiation programs and establishment of gene expression patterns of several bone cells, including osteoblasts, osteocytes, osteoclasts, and their progenitors [35, 62, 75,76,77,78,79,80]. Although the role of this epigenetic mark on bone homeostasis is now starting to be revealed, novel findings associate methylation patterns of genes with BMD levels in postmenopausal women [81•], suggesting that aberrant DNA methylation patterns may underlie the pathophysiology of common skeletal diseases such as osteoporosis and osteoarthritis [77].
The Sost gene has two CpG-rich regions, one located in the proximal promoter and the other one located in body of the gene, in the exon 1. CpG islands are rare in gene bodies and usually are heavily methylated [62•]. Methylation in these areas is usually associated with genomic stability and active transcription, indicating that methylation in gene bodies has different functions than in promoter sequences. Consistent with this, the CpG island region present in the body of the Sost gene is largely methylated in both osteoblastic and non-osteoblastic cells [62•].
Much of the work on DNA methylation has focused on CpG-rich regions located at promoter regions. Unmethylated promoters are usually associated with nucleosome-free regions at the TSS, and thus, gene expression is controlled by transcription factors with binding elements present in the promoter sequence. Methylation in promoters is usually observed in long-term silenced genes and in genes that are specifically expressed in germ cells versus somatic cells or in undifferentiated cells versus differentiated cells. In the Sost gene, the CpG island present in the promoter is hypermethylated in active osteoblasts and their precursors, but it is largely hypomethylated in osteocytes [62•]. Further, bone-lining cells, cells that cover quiescent surfaces of bone, show an intermediate methylation profile between primary osteoblasts and osteocytes. These results indicate that DNA methylation negatively controls Sost/sclerostin in the osteoblastic lineage and that changes in the DNA methylation during osteoblast-osteocyte transition enable the expression of this gene exclusively in osteocytes. Consistent with this notion, pharmacologic demethylation of the Sost promoter markedly upregulates the expression of this gene in both osteoblasts and non-osteoblastic cells [62•]. Moreover, functional studies showed that the region −581/+30 of the Sost promoter gene, which contains the CpG-rich region, is critical for the regulation of the transcriptional activity of this gene. Mechanistic experiments demonstrated that CpG methylation decreases Sost promoter activity by preventing the binding of transcription factors to the proximal promoter, including Bmp2, Runx2, and Osx [62•, 82].
While it is well accepted that DNA methylation represses Sost expression, the mechanisms controlling this dynamic regulation of DNA methylation at the Sost promoter are largely unknown. Recent findings show that Bmp-2, a known regulator of Sost/sclerostin expression, induces demethylation of the CpG-rich region located in the proximal promoter [62•, 83], suggesting that changes in the levels of BMP could regulate the transition from a methylated to a non-methylated Sost promoter. Additional experiments are needed to gain insight into the specific mechanisms and stimuli that promote the demethylation of the Sost promoter during osteoblast differentiation towards osteocytes.
After the seminal studies by our group [62•], several investigators hypothesized that the dysregulation of Sost/sclerostin expression bone pathologies was associated with aberrant methylation patterns in the proximal promoter of the Sost gene. Reppe and colleagues studied the messenger RNA (mRNA) levels of Sost, serum sclerostin, and DNA methylation patterns in the Sost promoter in a cohort of osteoporotic patients and healthy subjects [81•]. Osteoporotic patients exhibited increased CpG methylation in the gene promoter region compared to the healthy subjects and reduced expression of Sost mRNA and circulating levels of sclerostin, suggesting that DNA methylation may act as a compensatory mechanism that lowers Sost/sclerostin to counteract the inhibition of Wnt/β-catenin signaling and promote bone formation. In contrast, no differences were found in the degree of methylation in the Sost promoter between osteoporotic and osteoarthritic patients in a smaller cohort of patients [84]. More recently, it was shown that chondrocytes from osteoarthritic patients exhibit a hypomethylated Sost promoter and increased Sost mRNA compared to normal subjects [85]. Altogether, these results suggest that changes in the methylation pattern of the Sost promoter could also underlie changes in the Sost/sclerostin levels observed in several bone pathologies. Future studies in larger cohorts and in patients with bone disorders should clarify the specific contribution of DNA methylation to the altered expression of Sost/sclerostin in diseased bone.
Regulation of Sost Expression by microRNAs
Information gathered during the past decade suggests that microRNAs (miRNAs) are key regulators of bone development and control bone formation and resorption in the adult skeleton [86,87,88]. Although high-throughput screening helped to identify changes in the expression of a number of miRNAs in several bone pathologies, scarce information is available regarding how these non-coding RNAs affect the expression of Sost/sclerostin.
Osx and Runx2 induce Sost expression by binding to response elements present in the proximal Sost promoter in both rodent and human cell systems [82, 89]. Besides this direct influence, it is possible that Osx modulate Sost/sclerostin levels by influencing miRNA expression. In this regard, Chen et al. have shown that Osx decreases the levels of miRNA-204/211, which has an inhibitory effect on Sost/sclerostin levels [90]. Consistent with this finding, transfection of this miRNA binds to the 3′-UTR of Sost mRNA, reduces the activity of Sost 3′-UTR luciferase reporter vectors, and decreases the endogenous levels of Sost/sclerostin in UMR106 cells [91•]. Further, many miRNAs have been shown to modulate the activity of Osx and Runx2 during osteoblastogenesis, including miRNA-23, miRNA-30, miRNA-31, miRNA-34, miRNA-93, miRNA-103, miRNA-125, miRNA-133, miRNA-135, miRNA-137, miRNA-145, miRNA-204, miRNA-205, miRNA-211, miRNA-214, miRNA-217, miRNA-218, miRNA-335, miRNA-338, miRNA-433, miRNA-637, and miRNA-3077 [78, 79, 92, 93]. Given the stimulatory effect of Runx2 and Osx on Sost expression, we cannot discard the possibility that some of these miRNAs may secondarily regulate Sost/sclerostin levels. However, the precise role and the relative importance of these miRNAs in the regulation of Sost have not been elucidated yet. miR-218 expression is induced during osteoblast differentiation and promotes the commitment and differentiation of osteoblast precursors by activating the Wnt signaling pathway. This is related, at least in part, to the downregulation of several Wnt inhibitors, including Sost, Dkk2, and Sfrp2. Consistent with this, miR-218 transfection reduces endogenous Sost mRNA levels in MCT3 cells and decreases the activity of Sost 3′-UTR luciferase reporters [91•].
miRNAs are emerging as attractive therapeutic targets. However, the development of miRNA-based therapies to modulate Sost/sclerostin expression will require identification of miRNAs that exclusively target the Sost mRNA in a bone-specific manner in order to minimize potential side effects due to the multiple target nature of miRNAs. Given the clinical interest in targeting sclerostin for the treatment of several bone conditions, one can envision future experiments, combining in vivo and in vitro approaches, to identify miRNAs that regulate the expression of Sost/sclerostin in bone.
Conclusions and Future Directions
Sost/sclerostin has starred a remarkable bench-to-bedside journey to become a promising anabolic drug for the treatment of osteoporosis and other bone diseases. Although much is known about its biological function in the skeleton, the transcriptional regulation of this gene remains unclear. Advances during the last decade have provided relevant information on the regulation of Sost/sclerostin by epigenetic mechanisms. Repression of Sost expression in osteoblasts and non-osteoblastic cells appears to occur through the proximal promoter rather than the distal enhancer. DNA methylation locks the Sost gene in an off position, thus impeding its transcription. Demethylation of the CpG-rich region together with multiple factors including chromatin modification, miRNAs, hormones, and transcription factors turns on the expression of this gene by acting on both the ECR5 and the proximal promoter regulatory regions. This complex regulation enables the quick adjustment of Sost/sclerostin levels to spatial or temporal demands.
Future research efforts should focus on understanding the regulatory sequences that underlie the expression of Sost/sclerostin in a cell type-specific manner, the interplay between the different epigenetic marks and their influence on transcription, and whether these processes are dysregulated in bone pathologies. The development of novel techniques, including cell-labeling, RNA-seq, high-throughput sequencing, or ChIP-seq, makes feasible to investigate in detail the dynamics of epigenetic changes and how they correlate with transcriptional changes at the single-cell level.
Pharmacological modulation of epigenetic mechanisms has been shown to regulate Sost/sclerostin in in vivo models (AzadC, HDACs) [66•, 94, 95]. However, the use of these drugs and the interpretation of the results on bone mass are limited by the complexity of the physiological activities exhibited by these inhibitors. Thus, the challenge for the future will be to translate the current and future knowledge on the regulation of Sost/sclerostin into the development of new therapies that allow selective regulation of this gene in a bone-specific manner.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Allen MR, Burr DB. Bone modeling and remodeling. In: D B, M A, editors. Basic and applied bone biology. First ed: Elsevier; 2014. p. 75–90.
Seeman E. Bone modeling and remodeling. Crit Rev Eukaryot Gene Expr. 2009;19(3):219–33. doi:10.1615/CritRevEukarGeneExpr.v19.i3.40.
Delgado-Calle J, Bellido T. Osteocytes and skeletal pathophysiology. Curr Mol Biol Rep. 2015;1(4):157–67. doi:10.1016/j.bone.2016.10.007.
Bellido T. Osteocyte-driven bone remodeling. Calcif Tissue Int. 2013;94(1):25–34. doi:10.1007/s00223-013-9774-y.
• Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013;19(2):179–92. doi:10.1038/nm.3074. Excellent revision of the role of Wnt/beta-catenin signaling and Sost/sclerostin on bone homeostasis
• Delgado-Calle J, Sato AY, Bellido T. Role and mechanism of action of sclerostin in bone. Bone. 2017;96:29–37. doi:10.1016/j.bone.2016.10.007. This review summarizes the current knowledge on Sost/sclerostin regulation and function.
Van Bezooijen RL, Roelen BA, Visser A, Wee-Pals L, de Wilt E, Karperien M, et al. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med. 2004;199(6):805–14. doi:10.1084/jem.20031454.
Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003;22(23):6267–76. doi:10.1093/emboj/cdg599.
Poole KE, Van Bezooijen RL, Loveridge N, Hamersma H, Papapoulos SE, Lowik CW, et al. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005;19(13):1842–4. doi:10.1096/fj.05-4221fje.
Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet. 2001;10(5):537–43. doi:10.1093/hmg/10.5.537.
Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39(2):91–7. doi:10.1093/hmg/10.5.537.
Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D'Agostin D, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23(6):860–9. doi:10.1359/jbmr.080216.
Tu X, Rhee Y, Condon KW, Bivi N, Allen MR, Dwyer D, et al. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone. 2012;50(1):209–17. doi:10.1016/j.bone.2011.10.025.
Delgado-Calle J, Tu X, Pacheco-Costa R, McAndrews K, Edwards R, Pellegrini G, et al. Control of bone anabolism in response to mechanical loading and PTH by distinct mechanisms downstream of the PTH receptor. J Bone Miner Res. 2017;32(3):522–35. doi:10.1002/jbmr.3011.
Kramer I, Loots GG, Studer A, Keller H, Kneissel M. Parathyroid hormone (PTH)-induced bone gain is blunted in SOST overexpressing and deficient mice. J Bone Miner Res. 2010;25(2):178–89. doi:10.1359/jbmr.090730.
Kedlaya R, Veera S, Horan DJ, Moss RE, Ayturk UM, Jacobsen CM, et al. Sclerostin inhibition reverses skeletal fragility in an Lrp5-deficient mouse model of OPPG syndrome. Sci Transl Med. 2013;5(211):211ra158. doi:10.1126/scitranslmed.3006627.
Niziolek PJ, Farmer TL, Cui Y, Turner CH, Warman ML, Robling AG. High-bone-mass-producing mutations in the Wnt signaling pathway result in distinct skeletal phenotypes. Bone. 2011;49(5):1010–9. doi:10.1016/j.bone.2011.07.034.
McClung MR, Grauer A, Boonen S, Bolognese MA, Brown JP, ez-Perez A, et al. Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med. 2014;370(5):412–20. doi:10.1016/j.bone.2011.07.034.
Ominsky MS, Boyce RW, Li X, Ke HZ. Effects of sclerostin antibodies in animal models of osteoporosis. Bone. 2017;96:63–75. doi:10.1016/j.bone.2016.10.019.
Schones DE, Zhao K. Genome-wide approaches to studying chromatin modifications. Nat Rev Genet. 2008;9(3):179–91. doi:10.1038/nrg2270.
Teif VB, Erdel F, Beshnova DA, Vainshtein Y, Mallm J-P, Rippe K. Taking into account nucleosomes for predicting gene expression. Methods. 2013;62(1):26–38. doi:10.1016/j.ymeth.2013.03.011.
Lim PS, Li J, Holloway AF, Rao S. Epigenetic regulation of inducible gene expression in the immune system. Immunology. 2013;139(3):285–93. doi:10.1111/imm.12100.
Tessarz P, Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol. 2014;15(11):703–8. doi:10.1038/nrm3890.
Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17(8):487–500. doi:10.1038/nrg.2016.59.
Ha M, Ng DW-K, Li W-H, Chen ZJ. Coordinated histone modifications are associated with gene expression variation within and between species. Genome Res. 2011;21(4):590–8. doi:10.1101/gr.116467.110.
Jin B, Li Y, Robertson KD. DNA methylation: superior or subordinate in the epigenetic hierarchy? Genes & cancer. 2011;2(6):607–17. doi:10.1177/1947601910393957.
Gibney ER, Nolan CM. Epigenetics and gene expression. Heredity. 2010;105(1):4–13. doi:10.1038/hdy.2010.54.
Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol. 2014;6(4):a018713. doi:10.1101/cshperspect.a018713.
Roth M, Chen WY. Sorting out functions of sirtuins in cancer. Oncogene. 2014;33(13):1609–20. doi:10.1038/onc.2013.120.
Rodriguez RM, Fernandez AF, Fraga MF. Role of sirtuins in stem cell differentiation. Genes & cancer. 2013;4(3–4):105–11. doi:10.1177/1947601913479798.
Crujeiras AB, Parra D, Goyenechea E, Martínez JA. Sirtuin gene expression in human mononuclear cells is modulated by caloric restriction. Eur J Clin Investig. 2008;38(9):672–8. doi:10.1111/j.1365-2362.2008.01998.x.
Brownlee PM, Meisenberg C, Downs JA. The SWI/SNF chromatin remodelling complex: its role in maintaining genome stability and preventing tumourigenesis. DNA Repair. 2015;32:127–33. doi:10.1016/j.dnarep.2015.04.023.
Bird A. DNA methylation patterns and epigenetic memory DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi:10.1101/gad.947102.
Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4(2):143–53. doi:10.1038/nrc1279.
del Real A, Pérez-Campo FM, Fernández AF, Sañudo C, Ibarbia CG, Pérez-Núñez MI, et al. Differential analysis of genome-wide methylation and gene expression in mesenchymal stem cells of patients with fractures and osteoarthritis. Epigenetics. 2017;12(2):113–22. doi:10.1080/15592294.2016.1271854.
Chen T, Ueda Y, Dodge JE, Wang Z, Li E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol Cell Biol. 2003;23(16):5594–605. doi:10.1128/MCB.23.16.5594-5605.2003.
Richa R, Sinha RP. Hydroxymethylation of DNA: an epigenetic marker. EXCLI J. 2014;13:592–610.
Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science (New York, NY). 2009;324(5929):929–30. doi:10.1126/science.1169786.
He Y-F, Li B-Z, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science (New York, NY). 2011;333:1303–7.
Tan L, Shi YG. Tet family proteins and 5-hydroxymethylcytosine in development and disease. Development. 2012;139(11):1895–902. doi:10.1242/dev.070771.
Yildirim O, Li R, Hung J-H, Chen PB, Dong X, Ee L-S, et al. Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell. 2011;147(7):1498–510. doi:10.1016/j.cell.2011.11.054.
Frauer C, Hoffmann T, Bultmann S, Casa V, Cardoso MC, Antes I, et al. Recognition of 5-hydroxymethylcytosine by the Uhrf1 SRA domain. PLoS One. 2011;6(6):e21306. doi:10.1371/journal.pone.0021306.
Cheng Y, Xie N, Jin P, Wang T. DNA methylation and hydroxymethylation in stem cells. Cell Biochem Funct. 2015;33(4):161–73. doi:10.1002/cbf.3101.
Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PAC, Rappsilber J, et al. Tet1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473(7347):343–8. doi:10.1038/nature10066.
Peper JS, Dahl RE. Genome-wide mapping of DNA hydroxymethylation in osteoarthritic chondrocytes. Arthritis Rheumatol. 2015;67(8):2129–40. doi:10.1002/art.39179.
Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115. doi:10.1186/gb-2013-14-10-r115.
Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8(4):286–98. doi:10.1038/nrg2005.
Shen L, Song CX, He C, Zhang Y. Mechanism and function of oxidative reversal of DNA and RNA methylation. Annu Rev Biochem. 2014;83:585–614. doi:10.1146/annurev-biochem-060713-035513.
Barrett LW, Fletcher S, Wilton SD. Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements. Cell Mol Life Sci. 2012;69(21):3613–34. doi:10.1007/s00018-012-0990-9.
Mondal T, Kanduri C. Maintenance of epigenetic information: a noncoding RNA perspective. Chromosom Res. 2013;21(6–7):615–25. doi:10.1007/s10577-013-9385-5.
Huang Y, Shen XJ, Zou Q, Wang SP, Tang SM, Zhang GZ. Biological functions of microRNAs: a review. J Physiol Biochem. 2011;67(1):129–39. doi:10.1007/s13105-010-0050-6.
Blahna MT, Hata A. Regulation of miRNA biogenesis as an integrated component of growth factor signaling. Curr Opin Cell Biol. 2013;25(2):233–40. doi:10.1016/j.ceb.2012.12.005.
Romero-Cordoba SL, Salido-Guadarrama I, Rodriguez-Dorantes M, Hidalgo-Miranda A. miRNA biogenesis: biological impact in the development of cancer. Cancer biology & therapy. 2014;15(11):1444–55. doi:10.4161/15384047.2014.955442.
Weikard R, Demasius W, Kuehn C. Mining long noncoding RNA in livestock. Anim Genet. 2017;48(1):3–18. doi:10.1111/age.12493.
Angrand P-O, Vennin C, Le Bourhis X, Adriaenssens E. The role of long non-coding RNAs in genome formatting and expression. Front Genet. 2015;6:165. doi:10.3389/fgene.2015.00165.
Peschansky VJ, Wahlestedt C. Non-coding RNAs as direct and indirect modulators of epigenetic regulation. Epigenetics. 2014;9(1):3–12. doi:10.4161/epi.27473.
Delgado-Calle J, Arozamena J, Garcia-Renedo R, Garcia-Ibarbia C, Pascual-Carra MA, Gonzalez-Macias J, et al. Osteocyte deficiency in hip fractures. Calcif Tissue Int. 2011;89(4):327–34. doi:10.1007/s00223-011-9522-0.
Irie K, Ejiri S, Sakakura Y, Shibui T, Yajima T. Matrix mineralization as a trigger for osteocyte maturation. J Histochem Cytochem 2008;56(6):561–567. doi: 10.1369/jhc.2008.950527.
Bonewald LF. The amazing osteocyte. J Bone Miner Res. 2011;26(2):229–38. doi:10.1002/jbmr.320.
Bellido T. Downregulation of SOST/sclerostin by PTH: a novel mechanism of hormonal control of bone formation mediated by osteocytes. J Musculoskelet Neuronal Interact. 2006;6(4):358–9.
Keller H, Kneissel M. SOST is a target gene for PTH in bone. Bone. 2005;37(2):148–58. doi:10.1016/j.bone.2005.03.018.
• Delgado-Calle J, Sanudo C, Bolado A, Fernandez AF, Arozamena J, Pascual-Carra MA, et al. DNA methylation contributes to the regulation of sclerostin expression in human osteocytes. J Bone Miner Res. 2012;27(4):926–37. doi:10.1002/jbmr.1491. This paper describes for the first time the regulation of Sost/sclerostin expression by DNA methylation in its proximal promoter
Bradley EW, Carpio LR, van Wijnen AJ, McGee-Lawrence ME, Westendorf JJ. Histone deacetylases in bone development and skeletal disorders. Physiol Rev. 2015;95(4):1359–81. doi:10.1152/physrev.00004.2015.
Cantley MD, Zannettino AC, Bartold PM, Fairlie DP, Haynes DR. Histone deacetylases (HDAC) in physiological and pathological bone remodelling. Bone. 2017;95:162–74. doi:10.1016/j.bone.2016.11.028.
• Baertschi S, Baur N, Lueders-Lefevre V, Voshol J, Keller H. Class I and IIa histone deacetylases have opposite effects on sclerostin gene regulation. J Biol Chem. 2014;289(36):24995–5009. doi:10.1074/jbc.M114.564997. This article is the first to show the role of HDACs in the constitutive expression of Sost/sclerostin
• Wein MN, Spatz J, Nishimori S, Doench J, Root D, Babij P, et al. HDAC5 controls MEF2C-driven sclerostin expression in osteocytes. J Bone Miner Res. 2015;30(3):400–11. doi:10.1002/jbmr.2381. This article shows the role of HDAC5 on Sost/sclerostin expression through the ECR5 regulatory region
St John HC, Hansen SJ, Pike JW. Analysis of SOST expression using large minigenes reveals the MEF2C binding site in the evolutionarily conserved region (ECR5) enhancer mediates forskolin, but not 1,25-dihydroxyvitamin D or TGFbeta responsiveness. J Steroid Biochem Mol Biol. 2016;164:277–80. doi:10.1016/j.jsbmb.2015.09.005.
Leupin O, Kramer I, Collette NM, Loots GG, Natt F, Kneissel M, et al. Control of the SOST bone enhancer by PTH using MEF2 transcription factors. J Bone Miner Res. 2007;22(12):1957–67. doi:10.1359/jbmr.070804.
Mei Z, Zhang X, Yi J, Huang J, He J, Tao Y. Sirtuins in metabolism, DNA repair and cancer. J Exp Clin Cancer Res. 2016;35(1):182. doi:10.1186/s13046-016-0461-5.
Carafa V, Rotili D, Forgione M, Cuomo F, Serretiello E, Hailu GS, et al. Sirtuin functions and modulation: from chemistry to the clinic. Clin Epigenetics. 2016;8:61. doi:10.1186/s13148-016-0224-3.
Simic P, Zainabadi K, Bell E, Sykes DB, Saez B, Lotinun S, et al. SIRT1 regulates differentiation of mesenchymal stem cells by deacetylating β-catenin. EMBO Molecular Medicine. 2013;5(3):430–40. doi:10.1002/emmm.201201606.
Iyer S, Han L, Bartell SM, Kim HN, Gubrij I, de CR, et al. Sirtuin1 (Sirt1) promotes cortical bone formation by preventing beta-catenin sequestration by FoxO transcription factors in osteoblast progenitors. J Biol Chem. 2014;289(35):24069–78. doi:10.1074/jbc.M114.561803.
Zhou Y, Song T, Peng J, Zhou Z, Wei H, Zhou R, et al. SIRT1 suppresses adipogenesis by activating Wnt/beta-catenin signaling in vivo and in vitro. Oncotarget. 2016;7(47):77707–20. doi:10.18632/oncotarget.12774.
• Cohen-Kfir E, Artsi H, Levin A, Abramowitz E, Bajayo A, Gurt I, et al. Sirt1 is a regulator of bone mass and a repressor of Sost encoding for sclerostin, a bone formation inhibitor. Endocrinology. 2011;152(12):4514–24. doi:10.1210/en.2011-1128. This paper shows the regulation of sost/sclerostin by Sirt1
Delgado-Calle J, Sanudo C, Sanchez-Verde L, Garcia-Renedo RJ, Arozamena J, Riancho JA. Epigenetic regulation of alkaline phosphatase in human cells of the osteoblastic lineage. Bone. 2011;49(4):830–8. doi:10.1016/j.bone.2011.06.006.
Delgado-Calle J, Sanudo C, Fernandez AF, Garcia-Renedo R, Fraga MF, Riancho JA. Role of DNA methylation in the regulation of the RANKL-OPG system in human bone. Epigenetics. 2012;7(1):83–91. doi:10.4161/epi.7.1.18753.
Delgado-Calle J, Riancho JA. The role of DNA methylation in common skeletal disorders. Biology (Basel). 2012;1(3):698–713. doi:10.3390/biology1030698.
Delgado-Calle J, Garmilla P, Riancho JA. Do epigenetic marks govern bone mass and homeostasis? Curr Genomics. 2012;13(3):252–63. doi:10.2174/138920212800543129.
Vrtacnik P, Marc J, Ostanek B. Epigenetic mechanisms in bone. Clin Chem Lab Med. 2014;52(5):589–608. doi:10.1515/cclm-2013-0770.
Gordon JA, Montecino MA, Aqeilan RI, Stein JL, Stein GS, Lian JB. Epigenetic pathways regulating bone homeostasis: potential targeting for intervention of skeletal disorders. Curr Osteoporos Rep. 2014;12(4):496–506. doi:10.1007/s11914-014-0240-1.
• Reppe S, Noer A, Grimholt RM, Halldorsson BV, Medina-Gomez C, Gautvik VT, et al. Methylation of bone SOST, its mRNA, and serum sclerostin levels correlate strongly with fracture risk in postmenopausal women. J Bone Miner Res. 2015;30(2):249–56. doi:10.1002/jbmr.2342. This article shows aberrant DNA methylation at the Sost proximal promoter in osteoporotic patients
Perez-Campo FM, Santurtun A, Garcia-Ibarbia C, Pascual MA, Valero C, Garces C, et al. Osterix and RUNX2 are transcriptional regulators of sclerostin in human bone. Calcif Tissue Int. 2016;99(3):302–9. doi:10.1007/s00223-016-0144-4.
Delgado-Calle J, Arozamena J, Perez-Lopez J, Bolado-Carrancio A, Sanudo C, Agudo G, et al. Role of BMPs in the regulation of sclerostin as revealed by an epigenetic modifier of human bone cells. Mol Cell Endocrinol. 2013;369(1–2):27–34. doi:10.1016/j.mce.2013.02.002.
Delgado-Calle J, Fernandez AF, Sainz J, Zarrabeitia MT, Sanudo C, Garcia-Renedo R, et al. Genome-wide profiling of bone reveals differentially methylated regions in osteoporosis and osteoarthritis. Arthritis Rheum. 2013;65(1):197–205. doi:10.1002/art.37753.
Papathanasiou I, Kostopoulou F, Malizos KN, Tsezou A. DNA methylation regulates sclerostin (SOST) expression in osteoarthritic chondrocytes by bone morphogenetic protein 2 (BMP-2) induced changes in Smads binding affinity to the CpG region of SOST promoter. Arthritis Res Ther. 2015;17:160. doi:10.1186/s13075-015-0674-6.
Peng S, Gao D, Gao C, Wei P, Niu M, Shuai C. MicroRNAs regulate signaling pathways in osteogenic differentiation of mesenchymal stem cells (review). Mol Med Rep. 2016;14(1):623–9. doi:10.3892/mmr.2016.5335.
Xie Y, Zhang L, Gao Y, Ge W, Tang P. The multiple roles of microrna-223 in regulating bone metabolism. Molecules. 2015;20(10):19433–48. doi:10.3390/molecules201019433.
Papaioannou G. MiRNAs in bone development. Current Genomics. 2015;16(6):427–34. doi:10.2174/1389202916666150817202425.
Yang F, Tang W, So S, De Crombrugghe B, Zhang C. Sclerostin is a direct target of osteoblast-specific transcription factor osterix. Biochem Biophys Res Commun. 2010;400(4):684–8. doi:10.1016/j.bbrc.2010.08.128.
Chen Q, Liu W, Sinha KM, Yasuda H, de Crombrugghe B. Identification and characterization of microRNAs controlled by the osteoblast-specific transcription factor Osterix. PLoS One. 2013;8(3):e58104. doi:10.1371/journal.pone.0058104.
• Hassan MQ, Maeda Y, Taipaleenmaki H, Zhang W, Jafferji M, Gordon JA, et al. miR-218 directs a Wnt signaling circuit to promote differentiation of osteoblasts and osteomimicry of metastatic cancer cells. J Biol Chem. 2012;287(50):42084–92. doi:10.1074/jbc.M112.377515. This paper shows downregulation of Sost/sclerostin expression by miR-218
Lian JB, Stein GS, Van Wijnen AJ, Stein JL, Hassan MQ, Gaur T, et al. MicroRNA control of bone formation and homeostasis. Nat Rev Endocrinol. 2012;894:212–27. doi:10.1038/nrendo.2011.234.
Seeliger C, Er B, van Griensven M. miRNAs related to skeletal diseases. Stem Cells Dev. 2016;25(71):1261–8. doi:10.1089/scd.2016.0133.
Perez-Campo FM, May T, Zauers J, Sanudo C, Delgado-Calle J, Arozamena J, et al. Generation and characterization of two immortalized human osteoblastic cell lines useful for epigenetic studies. J Bone Miner Metab. 2017;35(2):150–60. doi:10.1007/s00774-016-0753-z.
Wein MN, Liang Y, Goransson O, Sundberg TB, Wang J, Williams EA, et al. SIKs control osteocyte responses to parathyroid hormone. Nat Commun. 2016;7:13176. doi:10.1038/ncomms13176.
Acknowledgements
Research reported in this manuscript was supported by the American Society of Hematology Scholar Award and the International Myeloma Foundation Brian D. Novis Junior Research Grant to J.D.C. and by a grant from Instituto de Salud Carlos III (PI12/615), through a program potentially co-funded by FEDER Funds from the European Union to J.A.R.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Álvaro del Real and Jesus Delgado-Calle each declare no potential conflicts of interest.
José A. Riancho reports grants from Instituto de Salud Carlos III during the conduct of the study, grants from Amgen, and other from Amgen, outside the submitted work.
Human and Animal Rights and Informed Consent
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Additional information
This article is part of the Topical Collection on Molecular Biology of Skeletal Development
Rights and permissions
About this article

Cite this article
del Real, Á., Riancho, J.A. & Delgado-Calle, J. Epigenetic Regulation of Sost/sclerostin Expression. Curr Mol Bio Rep 3, 85–93 (2017). https://doi.org/10.1007/s40610-017-0063-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40610-017-0063-9