
Abstract
Purpose of Review
Because of its clinical relevance, extensive work has been performed to uncover the signaling pathways modulated by parathyroid hormone (PTH). This review focuses on the recent identification of novel effectors of the anabolic effect of PTH in bone.
Recent Findings
PTH-induced activation of PKA leads to inactivation of SIK2 and nuclear translocation of HDAC4/5. This inhibits MEF2C-dependent Sost expression. However, the phenotypic characterization of the HDAC4/5 double knockout mice shows normal anabolic response to intermittent PTH, supporting the notion that HDAC/Sost-independent mechanisms must exist. Lrp6 and a member of the Usp gene family have been identified as novel targets of the transcriptional coregulator Nascent polypeptide-associated complex And Coregulator alpha (αNAC) downstream from PTH activation. We propose that the αNAC cascade is involved in the transmission of the anabolic PTH signal.
Summary
Further deciphering the signaling pathways transducing the anabolic effects of PTH will allow to develop novel therapeutically relevant molecules.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Parathyroid hormone (PTH), an 84-amino acid peptide hormone, is an important regulator of calcium homeostasis. The parathyroid gland senses low levels of circulating calcium and secretes PTH. The endocrine function of PTH is elicited by its direct effect on bone and kidneys and indirect effect on the gastrointestinal tract. High levels of PTH promote calcium and phosphorus release by activating osteoclast-mediated resorption of bone. At the level of the kidney, it acts to decrease calcium excretion and phosphorus reabsorption. The indirect effect of PTH is mediated by vitamin D activation, which in turn stimulates the absorption of dietary calcium and phosphorus from the gut [1, 2].
The profound effect of PTH on the skeleton shapes bone microarchitecture and modulates its strength. Chronic and excessive infusion of PTH has been associated with bone loss. Primary hyperparathyroidism (PHPT) is one classical example mimicked by continuous PTH administration, associated with osteopenia [3], accelerated bone loss [4], and enhanced bone turnover. However, intermittent administration of PTH at a low dosage promotes bone formation in a time interval referred to as the anabolic window [5]. This osteoanabolic effect is antagonized by the activation of bone resorption afterwards, yet the net effect is enhanced bone formation [6, 7]. While new therapeutics are in the pipeline, at the time of writing this review, the intermittent treatment with the N-terminal fragment of PTH(1–34) is the only US Food and Drug Administration (FDA)-approved osteoanabolic therapy for postmenopausal osteoporosis [8].
PTH signaling is a common factor mediating the crosstalk among osteoblasts, osteocytes, and osteoclasts. At the cellular level, the biological effect of PTH is mediated through signaling cascades and downstream targets controlling proliferation, maturation, and differentiation events. Because of its clinical relevance, extensive work has been performed to uncover the key signaling pathways modulated by PTH. In this review, we will focus on the recent work describing the effectors of the anabolic effect of PTH in bone, with emphasis on the proposed role of the transcriptional coregulator Nascent polypeptide-associated complex And Coregulator alpha (αNAC). PTH mediates phosphorylation of αNAC at residue serine 99 through a Gαs-PKA-dependent pathway [9•]. This leads to nuclear translocation of αNAC. In the nucleus of differentiated osteoblasts, αNAC associates with transcription factors, components of the basic transcriptional machinery, and other partners to regulate the expression of target genes [9•, 10, 11, 12•].
PTH1R
PTH signals in cells by activating the parathyroid hormone receptor 1 (PTH1R), a seven-transmembrane domain G protein-coupled receptor (GPCR) that belongs to the class B secretin-like GPCR family [13]. In bone, chondrocytes, osteoblasts, and osteocytes express PTH1R [14]. In mice, deletion of Pth1r either globally or in the osteoblastic lineage (osteoblasts or osteocytes) impairs proper bone development and the capacity of bone tissue to respond to PTH treatment. Systemic deletion of Pth1r results in mild cortical thickening in specific regions of long bones accompanied by a more profound decrease of trabecular bone development [15]. Osteoblast-specific inactivation with the osteocalcin-driven Cre (OC-Cre) yields a similar phenotype [16].
A mild osteopenic phenotype has also been reported when Pth1r is deleted from osteocytes postnatally using a tamoxifen-inducible Dmp1-CRE driver [17]. However, different results were reported when Pth1r was constitutively deleted from osteocytes. These animals show an increased trabecular and cortical bone volume at 3 months of age, resulting from a low bone turnover state [18, 19••]. A significant finding, regardless of the steady-state bone phenotype of mice in which Pth1r is ablated in osteocytes, is that the anabolic response to PTH is blunted in those animals [18, 19••]. These results indicate the importance of the osteocyte cell population for PTH-induced bone gain responses.
Signaling Pathways
PTH1R is coupled to different G proteins including the Gαs, the Gαq11, and the Gαq12/13 subunits which respectively primarily mediate their intracellular effects through the activation of the adenylate cyclase (ADCY), the phospholipase C (PLC), and the Ras homolog gene family, member A (RHOA) [20, 21]. Within this wide range of signaling cascades activated by PTH, it has been demonstrated that its anabolic function is mainly mediated by the activation of the Gαs-dependent accumulation of cyclic AMP (cAMP) and activation of protein kinase A (PKA) and not by Gαq-mediated PLC activation. This was initially examined using various amino-terminal fragments of PTH that differentially activate Gαs- or Gαq-mediated signaling [22]. PTH(1–34) activates both cAMP production and PLC, while PTH(1–31) only activates cAMP synthesis [23,24,25]. On the other hand, PTH(3–38) only activates the PLC pathway [24, 25]. Anabolic effects were measured upon daily injections of PTH(1–34) and PTH(1–31), but not PTH(3–38) [26, 27]. These studies established that PTH-stimulated cAMP production is sufficient for mediating the anabolic effect, but activation of the PLC pathway is insufficient. These classical biochemistry studies have now been confirmed using mouse molecular genetics approaches and osteoblast-specific postnatal inactivation of Gαs [28, 29, 30••].
The phenotype of mice with osteoblasts specifically deficient for Gαs is mimicked by a mutation leading to cytoplasmic retention of the transcriptional coregulator αNAC, suggesting that Gαs and αNAC form part of a common genetic pathway [31, 32]. Confirming this hypothesis, we found that treatment of osteoblasts with PTH(1–34) or a PKA-selective activator leads to translocation of αNAC to the nucleus. αNAC was phosphorylated by PKA at residue serine 99 in vitro. Phospho-S99-αNAC accumulates in osteoblasts exposed to PTH(1–34) but not in treated cells expressing dominant negative PKA. Nuclear accumulation is abrogated by an S99A mutation but enhanced by a phosphomimetic residue (S99D). Chromatin immunoprecipitation (ChIP) analysis showed that PTH(1–34) treatment leads to accumulation of αNAC at the osteocalcin (Bglap) promoter [9•]. These data show that αNAC is a substrate of PKA following PTH signaling. We propose that αNAC acts as a downstream effector of the anabolic action of PTH in bone. We are currently testing the physiological relevance of this proposed mechanism using site-directed mutagenesis of αNAC at S99 in knock-in mice models.
The important role of the Gαs-dependent pathway in PTH-mediated bone acquisition was also confirmed using mice expressing a constitutively active PTH1R (caPTH1R) systemically. These mice show a drastic increase of trabecular bone volume [33], but this abnormal increase of bone volume is completely reversed when Gαs is postnatally deleted from the osteoblasts [30••]. Interestingly, the same study also showed than even if the intermittent PTH (iPTH)-mediated increase of bone mass is blunted in mice lacking osteoblast expression of Gαs, PTH is still able to increase osteoblast numbers and bone formation rates [30••]. This discovery illustrates well the complexity of PTH signaling in bone cells. It also suggests than other pathways are required to work in collaboration with Gαs-dependent signalization to improve bone mass following PTH stimulation. One possibility would involve the β-arrestin 2-dependent pathway, which is a Gαs-independent cascade that has been shown to be involved in iPTH-induced bone formation [34,35,36].
The cAMP second messenger activates the PKA-dependent signaling cascade as well as a PKA-independent pathway that involves the Rap guanine nucleotide exchange factor (GEF) 3 and 4 (also called EPAC). While the physiological importance of the latter remains to be demonstrated in bone, the bone anabolic function of PKA has been confirmed using a transgenic approach [37, 38]. Forcing the expression of a constitutively active form of PKA (caPKA) in osteoblasts led to an important increase of the bone volume in long bones [38]. This phenotype is associated with an increase of bone turnover and aligns with the phenotype observed in mice expressing caPTH1R specifically in osteocytes [39, 40]. Interestingly, the increased bone volume observed when caPKA is expressed in osteoblasts is more dramatic than the increase observed when the transgene is only expressed in osteocytes [37, 38]. Here again, this difference suggests that the PKA axis is not the only one required to mediate the anabolic action of PTH on bone.
Coreceptors
Optimal activation of PTH1R by PTH involves its interaction with the LDL receptor-related protein 6 (LRP6) [41,42,43,44]. When Lrp6 expression is disrupted in mice, the anabolic action of iPTH is blunted [42•]. It has been suggested that N-cadherin regulates the amount of LRP5/LRP6 complexes at the plasma membrane [45]. This indirectly negatively impacts the abundance of PTH1R/LRP6 complexes by affecting the LRP6 pool available for an interaction with PTH1R [43]. This model is supported by ablation of the Cdh2 gene (coding for N-cadherin) in mice which increases membrane PTH1R/LRP6 complexes. This in turn promotes signaling downstream from PTH1R, enhancing bone formation following iPTH treatment [43, 46].
LRP6 availability and activity are mainly regulated via interaction with extracellular ligands and phosphorylation of its intracellular domain (reviewed in [47,48,49]). However, mechanisms regulating Lrp6 transcription remain largely unknown. Kulkarni and colleagues [50] reported that Lrp6 expression can be modulated by PTH, but the exact mechanism still needs to be determined. Recent data from our laboratory suggests that this regulation involves the transcriptional coregulator αNAC (unpublished).
In order to identify additional target genes affected by PTH-mediated αNAC phosphorylation, we performed ChIPSeq analysis against αNAC in MC3T3-E1 cells treated with vehicle or PTH(1–34). RNASeq was performed in parallel. Candidate genes that showed increased αNAC binding to their proximal promoter and increased expression following PTH(1–34) treatment were selected for further analysis. This strategy identified the Lrp6 gene as a potential new αNAC target. ChIPSeq results were validated using conventional quantitative ChIP. Deletions and point mutation experiments in transient transfection assays confirmed that the αNAC regulation of Lrp6 transcription requires a proximal promoter element; expression of the reporter was reduced by more than 50% when this element is deleted or mutated (not shown). This is the first study that characterizes transcriptional regulation of Lrp6 gene expression. It identified a novel αNAC target gene induced by PTH signaling and suggests a putative “feed-forward” mechanism to prime subsequent PTH responses.
Anabolic Action of PTH and Wnt Signaling
PTH signaling activates the different nodes of the Wingless-related integration site (Wnt) signal transduction network in osteoblastic cells. Wnt signaling plays a significant role in bone metabolism. Once activated, it fates mesenchymal stem cells (MSCs) to osteogenic commitment, regulates pre-osteoblast proliferation, differentiation, and survival, and controls osteoclastic bone resorption by augmenting osteoprotegerin (OPG) production (reviewed in [51]). Wnt proteins bind to a dual-receptor complex of the frizzled (Fzd) receptor and the LRP5 and LRP6 coreceptors, stabilizing its main downstream effector β-catenin and triggering the transcription of Wnt target genes. Recent studies have shown that the anabolic effect of PTH can be altered by the Wnt/LRP6/β-catenin axis. As discussed before, PTH-bound PTH1R engages with LRP6 and activates β-catenin in osteoblasts and osteocytes [44] and this PTH-induced activation is completely abrogated in Lrp6 deficient mice [42•]. This interaction appears to be exclusive to LRP6, as the loss of LRP5 does not affect the anabolic actions of intermittent PTH [52]. Although PTH activates β-catenin in osteoblasts [43, 53], studies using mice with β-catenin-deficient osteocytes revealed that the anabolic effects of PTH therapy in the trabecular compartment do not require osteocytic β-catenin [54].
The work of Li et al. [55] has improved our understanding of the role of T cells and other lineages in mediating PTH-induced bone anabolism. Their findings demonstrated that T cell-produced Wnt10b and its costimulatory molecule CD40 ligand (CD40L) can drive the anabolic effect of intermittent PTH on osteoblasts and bone mass [55, 56]. This information adds another level of complexity to PTH anabolism modalities and highlights the importance of investigating the interactions among osteoblasts, osteocytes, and T cells as a circular regulatory circuit of PTH anabolism. Uncertainty remains as to the identity of cytokines and Wnt modulators that play a multidirectional role mediating the connection among different PTH-responsive lineages.
An established mechanism by which PTH exerts its anabolic effect in bone is through the suppression of the sclerostin (SCL) gene (SOST). SCL is a potent Wnt antagonist, mainly secreted by osteocytes. SOST regulation by PTH remains a hot area for investigation by different research groups. Kramer and colleagues [57] reported contradictory evidence from studies in Sost transgenic and Sost-deficient mice [58, 59], raising questions about whether PTH-induced bone gain requires the downregulation of Sost or not. A recent study performed in osteocyte-specific Sost transgenic mice has resolved this discrepancy, presenting evidence that PTH can activate alternative anabolic pathways independently of Sost/SCL suppression [18].
We have reported that altered gene dosage for Gαs and αNAC in compound heterozygous mice (Gαsob+/−; αNAC+/−) results in reduced bone mass, increased numbers of osteocytes, and enhanced expression of Sost [9•]. This modulation of Sost expression levels was also observed in a different αNAC knock-in mouse model with a serine-to-alanine mutation at position 43 (S43A) that results in a decrease in nuclear αNAC [31]. Taken together, these data lead us to speculate about an important role for nuclear αNAC in Sost regulation during the transition stage from osteoblasts to osteocytes. It is likely that PTH regulates αNAC functions in osteocytes. Understanding the molecular mechanism by which αNAC regulates Sost levels in osteocytes will be insightful, and future studies will focus on the transcriptional partners of αNAC that may have a direct or indirect effect on Sost transcription.
PTH-Mediated Regulation of HDACs
In studying the regulation of Sost expression caused by PTH signaling, investigators have unveiled a role for myocyte enhancer factor 2C (MEF2C). Mice with specific inactivation of Mef2c in osteocytes have reduced Sost expression and increased bone mass [60]; targeted deletion of the Sost distal enhancer containing the MEF2C binding site yields a similar phenotype [61•]. PTH inhibits the transcription of the Mef2c gene [62, 63] and controls its binding to the upstream Sost enhancer through class IIa Histone DeAcetylases 4 and 5 (HDAC4/5) in osteocytes [64, 65]. Different classes of HDACs (I, IIa, IIb) are expressed in osteoblasts. PTH induces the nuclear translocation of HDAC5 and its binding to MEF2C, suppressing Sost expression [64]. Interestingly, this inhibitory mechanism governing Sost production has been shown to be exclusive to class IIa HDACs, whereas class I HDACs are involved in constitutive Sost expression [64].
Class IIa HDAC function is regulated via nucleo-cytoplasmic shuttling involving differential phosphorylation and protein-protein interactions, where phosphorylated class IIa HDACs are sequestered in the cytoplasm through interaction with 14-3-3 proteins [66]. Recent studies have identified the relevant kinases involved in this mechanism in osteocytes. Members of the salt inducible kinase (SIK) family phosphorylate HDAC4/5 to maintain them in the cytoplasm. Wein and colleagues [67••] have shown that PTH-induced activation of PKA leads to phosphorylation and inactivation of SIK2. This in turn permits dephosphorylation and subsequent nuclear translocation of HDAC4/5, thus inhibiting MEF2C-dependent Sost expression [67••]. The model is further supported by the demonstration that small molecule inhibitors of SIKs mimic the skeletal effects of PTH and increase bone formation and bone mass [67••]. This work is significant as it demonstrates the validity of further deciphering the signaling pathways transducing the anabolic effects of iPTH in order to develop novel therapeutically relevant molecules. Another important finding of the phenotypic characterization of the models used in the Wein et al. study is the observation that HDAC4/5 double knockout mice show normal anabolic response to intermittent PTH, supporting the notion that HDAC/SCL-independent mechanisms must exist [67••].
Basic Domain-Leucine Zipper Transcription Factors and PTH Signal Transduction
In response to PTH, the basic domain-leucine zipper (bZIP) transcription factor, cAMP-response element-binding protein (CREB), is phosphorylated [68]. It then stimulates the transcription of genes that encode for members of the AP-1 family of bZIP factors, Fos and Jun [68,69,70]. A recent study has linked the class III HDAC, Sirtuin 1 (SIRT1), to the PTH-mediated regulation of the Mmp13 promoter. The mechanism involves the direct binding of SIRT1 to cJUN and this interaction at the AP-1 site within the Mmp13 promoter blocks AP-1 activity and downregulates Mmp13 [71]. Interestingly, the authors further showed that the deacetylation of cJUN by SIRT1 is cAMP dependent [71].
We have shown that αNAC functions as a transcriptional coregulator for the AP-1 family member, cJUN [72, 73]. Protein-protein interactions were detected between αNAC and cJUN, as well as between αNAC and the TATA-binding protein (TBP), and αNAC potentiates the transcriptional activity of cJUN [72, 73]. Additional studies from our lab have also shown that αNAC can recruit HDAC corepressors at specific gene promoters regulating their expression in a promoter and cell-specific manner. Coimmunoprecipitation assays have detected complexes between the αNAC and the corepressors HDAC1 and HDAC3 in myoblasts and osteoblasts [12•]. Identifying the target promoter genes regulated by the αNAC-HDACs interaction in osteoblasts and osteocytes is of great interest. In this context, we can speculate a mechanistic model regulating Sost expression involving the interaction between αNAC and HDACs in osteocytes.
It has been shown that another bZIP transcription factor, activating transcription factor 4 (ATF4), plays a critical role in the anabolic actions of PTH in bone. ATF4 is a key regulator of osteoblast function [74,75,76]. The anabolic bone response to PTH is severely impaired in mice lacking ATF4; in these animals, PTH-stimulated osteoblast proliferation, survival, and differentiation are suppressed [77].
Our recent results show that αNAC does not exclusively function as a positive regulator of gene transcription, but rather as a docking platform for transcriptional activator or repressor complexes to control gene expression during mesenchymal differentiation [12•]. This dynamic role as an activator or repressor of gene transcription is controlled through posttranslational modification by covalent attachment of a Small Ubiquitin-like MOdifyer (SUMO). SUMOylation leads to transcriptional inhibition by providing a novel protein-protein interface, allowing interaction of the SUMOylated αNAC protein with transcriptional corepressors. The amino acid sequence of αNAC contains one copy of the composite “phospho-sumoyl switch” motif that couples sequential phosphorylation and SUMOylation [78]. We found that αNAC is selectively SUMOylated at lysine residue 127 within the motif and that SUMOylation is enhanced when a phosphomimetic mutation (serine to aspartic acid, S to D) is introduced at the nearby serine residue 132. The S132D, hyper-SUMOylated αNAC mutant, specifically interacts with the corepressor HDAC2 and enhances the inhibitory activity of the Factor Inhibiting ATF4-mediated Transcription (FIAT) on ATF4-mediated transcription from the Bglap gene promoter [11]. Considering the role of ATF4 in PTH-induced osteoblast proliferation, survival, and differentiation [77], these results suggest a further role for αNAC in the downstream events mediating the anabolic actions of PTH in bone. It has not escaped the authors that inhibition of the kinase that activates the αNAC phospho-sumoyl switch would prevent αNAC SUMOylation and its interaction with corepressors, in turn leading to increased gene expression in osteoblasts and osteocytes. This is a current intense focus of research in our laboratory.
Ubiquitination and PTH Anabolism
The recent findings on the role of HDACs in the regulation of Sost expression caused by PTH signaling is bound to spur future work on the upstream cascades regulating the stability, activity, and subcellular localization of HDACs. While it has been demonstrated that PTH signaling affects the activity of HDAC-regulating kinases [67••], it is highly probable that HDAC-ubiquitinating enzymes could affect their activity and abundance via stability and proteosomal degradation.
Protein ubiquitination is an important posttranslational modification that regulates a multitude of biological processes. These biological functions include cell cycle regulation, protein degradation, DNA repair, kinase signaling, and trafficking events. Deubiquitinating enzymes (DUBs) maintain a dynamic balance between ubiquitination and deubiquitination by removing ubiquitin moieties, thus affecting the stability and function of target proteins. Little is known about the role of the ubiquitin-specific peptidase (USP) family of enzymes in the regulation of bone mass, particularly in response to PTH stimulation. Alonso et al. [79] have reported that PTH can induce the expression of USP2, a PTH1R-specific deubiquitinating enzyme, thus modulating PTH1R sorting. Further work using MC3T3-E1 osteoblastic cells has shown that USP2 upregulation is involved in osteoblast proliferation following PTH treatment [80].
The genomic landscape encompasses about 58 different USPs, few of which have a clear assigned function and substrate. Recent studies have been oriented towards uncovering the upstream signals regulating the activity of those enzymes, their partner ligases, and their potential substrates. Our efforts to identify genes affected by PTH-mediated αNAC phosphorylation through ChIPSeq analysis against αNAC in PTH-treated MC3T3-E1 cells revealed a member of the USP family as a novel αNAC transcriptional target (unpublished). Inhibition of the expression of this USP family member using RNA knockdown appears to regulate mesenchymal cell lineage-making decisions and differentiation in response to PTH. We are pursuing this work by trying to uncover targets and partners of this USP family member using proteomics-based approaches.
Perspectives and Conclusion
Recent research focusing on the anabolic action of PTH has brought to attention the key roles of osteocytes in the transduction of the PTH signal. Future work will benefit from improved in vitro models of established osteocytic cell lines that have been recently described in the literature [65, 81•].
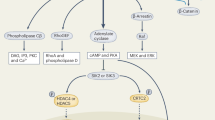
A role for class IIa HDACs has also been uncovered. Interestingly, deciphering the signaling events taking place between the ligand-receptor engagement and gene transcription responses has revealed the involvement of salt inducible kinases (Fig. 1). The demonstration that small molecule inhibitors of SIKs mimic the skeletal effects of PTH and increase bone formation and bone mass [67••] identifies potentially therapeutically relevant molecules. This may prove significant as the current formulation of iPTH is nearing the end of patent protection.

Novel pathways downstream from PTH. Optimal activation of the PTH receptor 1 (PTH1R) by PTH involves its interaction with the LDL receptor related protein 6 (LRP6). PTH1R is coupled to Gαs leading to activation of adenylate cyclase (AC), accumulation of cyclic AMP (cAMP), and activation of protein kinase A (PKA). PTH-induced activation of PKA leads to inactivation of salt inducible kinase 2 (SIK2) and nuclear translocation of Histone DeAcetylases 4 and 5 (HDAC4/5). This inhibits myocyte enhancer factor 2C (MEF2C)-dependent sclerostin gene (Sost) expression. The PTH-Gαs-PKA pathway also mediates phosphorylation of the transcriptional coregulator Nascent polypeptide-associated complex And Coregulator alpha (αNAC) at residue serine 99 (Ser99). This leads to nuclear translocation of αNAC. Lrp6 and a member of the ubiquitin-specific peptidase (Usp) gene family have been identified as novel targets of αNAC downstream from PTH activation, in addition to the characterized target osteocalcin (Bglap). This work also hints that additional members of the basic domain-leucine zipper (bZIP) family of transcription factors may interact with αNAC besides cJUN. The characterization of Lrp6 as an αNAC target gene suggests a pathway that can amplify PTH responses by increasing expression of the coreceptor molecule
The power of mouse molecular genetics revealed that anabolic response to intermittent PTH also involves HDAC/SCL-independent mechanisms, since HDAC4/5 double knockout mice show normal bone gain when treated with iPTH [67••]. Some of these implicate Wnt ligands and modulators in non-bone cell types [55, 56]. We also propose that the cascade involving αNAC and the novel targets that we recently identified are involved in the transmission of the anabolic PTH signal. We will test that hypothesis using site-specific αNAC mutations in knock-in mice strains. We predict that mice sporting a mutation of the PKA phospho-acceptor site within the αNAC sequence should exhibit a blunted response to intermittent PTH treatment. Reciprocally, mice with a phosphomimetic residue at the corresponding position could turn out to be quite informative depending on the observed phenotype. An osteopenic phenotype associated with increased osteoclastogenesis would place αNAC within the cascade mediating the effects of constant infusion with PTH and suggest an implication of the coregulator in the control of RANKL expression. Alternatively, the mice could demonstrate an exuberant response to iPTH treatment.
Through the characterization of the phenotype of knock-in mice strains in which the phosphoacceptor residue controlling the phospho-sumoyl switch within the αNAC sequence has been mutated, we will demonstrate the physiological relevance of αNAC SUMOylation. Maintaining αNAC in a hyper-SUMOylated state (phosphomimetic mutation) should prevent it from acting as a positive regulator of gene transcription. The mutation should mimic some aspects of the phenotype of mice in which αNAC is excluded from the nucleus [9•, 31]. Thus, the phosphomimetic mutant mice should exhibit decreased Bglap gene expression, and mutant bones could be osteopenic with accelerated mineralization characterized by less osteoid tissue and a reduced volume of immature, woven-type bone showing poor lamellation. Mutant bones could have poor biomechanical properties. Endocrine dysfunction with impaired energy metabolism could result from inhibited Bglap expression. Preventing SUMOylation through mutation of the acceptor lysine or mutation of the phosphoacceptor residue within the phospho-sumoyl switch to a non-phosphorylatable residue should result in increased target gene expression. Bones from these mice could show increased bone volume with favorable biomechanical properties, establishing a positive effect of reducing αNAC SUMOylation. All these mutant strains have been established in our laboratory and the analysis of their phenotypes is ongoing.
The characterization of Lrp6 as an αNAC target gene further supports the notion that αNAC is involved as an effector of the PTH signal. The results are significant as they represent the first description of a mechanism regulating Lrp6 transcription. As previously mentioned, it suggests a pathway that can amplify PTH responses by increasing expression of a coreceptor molecule (Fig. 1). Our analysis of the Lrp6 promoter could also identify additional transcription factors interacting with αNAC, besides cJUN. Interestingly, the data to date points to distinct AP-1 family members and other bZIP factors.
Finally, the identification of a USP family member as an αNAC target downstream from the PTH-PKA signal spurs future studies to uncover ubiquitin modifiers that respond to PTH and affect key bone modulators of kinases and transcription factors. Such information will expand our knowledge of the PTH-mediated mechanisms regulating osteoblast proliferation and differentiation.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Hanley DA, Watson PH, Hodsman AB, Dempster DW. Pharmacological mechanisms of therapeutics: parathyroid hormone. In: Bilezikian JP, Raisz LG, Martin TJ, editors. Principles of bone biology. 3rd ed. San Diego: Elsevier; 2008.
Levine MA. Normal mineral homeostasis. Interplay of parathyroid hormone and vitamin D. Endocr Dev. 2003;6:14–33.
Silverberg SJ, Shane E, de la Cruz L, Dempster DW, Feldman F, Seldin D, et al. Skeletal disease in primary hyperparathyroidism. J Bone Miner Res. 1989;4:283–91. doi:10.1002/jbmr.5650040302.
Grey AB, Stapleton JP, Evans MC, Reid IR. Accelerated bone loss in post-menopausal women with mild primary hyperparathyroidism. Clin Endocrinol. 1996;44:697–702.
Pleiner-Duxneuner J, Zwettler E, Paschalis E, Roschger P, Nell-Duxneuner V, Klaushofer K. Treatment of osteoporosis with parathyroid hormone and teriparatide. Calcif Tissue Int. 2009;84:159–70. doi:10.1007/s00223-009-9218-x.
Dobnig H, Turner RT. The effects of programmed administration of human parathyroid hormone fragment (1-34) on bone histomorphometry and serum chemistry in rats. Endocrinology. 1997;138:4607–12.
Qin L, Raggatt LJ, Partridge NC. Parathyroid hormone: a double-edged sword for bone metabolism. Trends Endocrinol Metab. 2004;15:60–5. doi:10.1016/j.tem.2004.01.006.
Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, et al. Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344:1434–41. doi:10.1056/NEJM200105103441904.
• Pellicelli M, Miller JA, Arabian A, Gauthier C, Akhouayri O, Wu JY, et al. The PTH-Gαs-protein kinase A cascade controls αNAC localization to regulate bone mass. Mol Cell Biol. 2014;34:1622–33. doi:10.1128/MCB.01434-13. Characterization of the PTH signal regulating nuclear translocation of αNAC
Akhouayri O, Quelo I, St-Arnaud R. Sequence-specific DNA binding by the alphaNAC coactivator is required for potentiation of c-Jun-dependent transcription of the osteocalcin gene. Mol Cell Biol. 2005;25:3452–60.
Hekmatnejad B, Akhouayri O, Jafarov T, St-Arnaud R. SUMOylated alphaNAC potentiates transcriptional repression by FIAT. J Cell Biochem. 2014;115:866–73. doi:10.1002/jcb.24729.
• Jafarov T, Alexander JW, St-Arnaud R. alphaNAC interacts with histone deacetylase corepressors to control myogenin and osteocalcin gene expression. Biochim Biophys Acta. 2012;1819:1208–16. doi:10.1016/j.bbagrm.2012.10.005. First demonstration that αNAC can act as both a positive or a negative regulator of transcription through differential interaction with HDACs
Cheloha RW, Gellman SH, Vilardaga JP, Gardella TJ. PTH receptor-1 signalling-mechanistic insights and therapeutic prospects. Nat Rev Endocrinol. 2015;11:712–24. doi:10.1038/nrendo.2015.139.
Santa Maria C, Cheng Z, Li A, Wang J, Shoback D, Tu CL, et al. Interplay between CaSR and PTH1R signaling in skeletal development and osteoanabolism. Semin Cell Dev Biol. 2016;49:11–23. doi:10.1016/j.semcdb.2015.12.004.
Lanske B, Amling M, Neff L, Guiducci J, Baron R, Kronenberg HM. Ablation of the PTHrP gene or the PTH/PTHrP receptor gene leads to distinct abnormalities in bone development. J Clin Invest. 1999;104:399–407. doi:10.1172/JCI6629.
Qiu T, Xian L, Crane J, Wen C, Hilton M, Lu W, et al. PTH receptor signaling in osteoblasts regulates endochondral vascularization in maintenance of postnatal growth plate. J Bone Miner Res. 2015;30:309–17. doi:10.1002/jbmr.2327.
Powell Jr WF, Barry KJ, Tulum I, Kobayashi T, Harris SE, Bringhurst FR, et al. Targeted ablation of the PTH/PTHrP receptor in osteocytes impairs bone structure and homeostatic calcemic responses. J Endocrinol. 2011;209:21–32. doi:10.1530/JOE-10-0308.
Delgado-Calle J, Tu X, Pacheco-Costa R, McAndrews K, Edwards R, Pellegrini GG, et al. Control of bone anabolism in response to mechanical loading and PTH by distinct mechanisms downstream of the PTH receptor. J Bone Miner Res. 2016; doi:10.1002/jbmr.3011.
•• Saini V, Marengi DA, Barry KJ, Fulzele KS, Heiden E, Liu X, et al. Parathyroid hormone (PTH)/PTH-related peptide type 1 receptor (PPR) signaling in osteocytes regulates anabolic and catabolic skeletal responses to PTH. J Biol Chem. 2013;288:20122–34. doi:10.1074/jbc.M112.441360. PTH1R signaling in osteocytes is necessary for PTH anabolic effect
Abou-Samra AB, Juppner H, Force T, Freeman MW, Kong XF, Schipani E, et al. Expression cloning of a common receptor for parathyroid hormone and parathyroid hormone-related peptide from rat osteoblast-like cells: a single receptor stimulates intracellular accumulation of both cAMP and inositol trisphosphates and increases intracellular free calcium. Proc Natl Acad Sci U S A. 1992;89:2732–6.
Singh AT, Gilchrist A, Voyno-Yasenetskaya T, Radeff-Huang JM, Stern PH. G alpha12/G alpha13 subunits of heterotrimeric G proteins mediate parathyroid hormone activation of phospholipase D in UMR-106 osteoblastic cells. Endocrinology. 2005;146:2171–5. doi:10.1210/en.2004-1283.
Iida-Klein A, Guo J, Drake MT, Kronenberg HM, Abou-Samra AB, Bringhurst FR, et al. Structural requirements of parathyroid hormone/parathyroid hormone-related peptide receptors for phospholipase C activation and regulation of phosphate uptake. Miner Electrolyte Metab. 1995;21:177–9.
Takasu H, Gardella TJ, Luck MD, Potts Jr JT, Bringhurst FR. Amino-terminal modifications of human parathyroid hormone (PTH) selectively alter phospholipase C signaling via the type 1 PTH receptor: implications for design of signal-specific PTH ligands. Biochemistry. 1999;38:13453–60.
Jouishomme H, Whitfield JF, Chakravarthy B, Durkin JP, Gagnon L, Isaacs RJ, et al. The protein kinase-C activation domain of the parathyroid hormone. Endocrinology. 1992;130:53–60.
Jouishomme H, Whitfield JF, Gagnon L, Maclean S, Isaacs R, Chakravarthy B, et al. Further definition of the protein kinase C activation domain of the parathyroid hormone. J Bone Miner Res. 1994;9:943–9. doi:10.1002/jbmr.5650090620.
Whitfield JF, Morley P, Willick GE, Ross V, Barbier JR, Isaacs RJ, et al. Stimulation of the growth of femoral trabecular bone in ovariectomized rats by the novel parathyroid hormone fragment, hPTH-(1-31)NH2 (Ostabolin). Calcif Tissue Int. 1996;58:81–7.
Armamento-Villareal R, Ziambaras K, Abbasi-Jarhomi SH, Dimarogonas A, Halstead L, Fausto A, et al. An intact N terminus is required for the anabolic action of parathyroid hormone on adult female rats. J Bone Miner Res. 1997;12:384–92. doi:10.1359/jbmr.1997.12.3.384.
Ogata N, Shinoda Y, Wettschureck N, Offermanns S, Takeda S, Nakamura K, et al. G alpha(q) signal in osteoblasts is inhibitory to the osteoanabolic action of parathyroid hormone. J Biol Chem. 2011;286:13733–40. doi:10.1074/jbc.M110.200196.
Yang D, Singh R, Divieti P, Guo J, Bouxsein ML, Bringhurst FR. Contributions of parathyroid hormone (PTH)/PTH-related peptide receptor signaling pathways to the anabolic effect of PTH on bone. Bone. 2007;40:1453–61. doi:10.1016/j.bone.2007.02.001.
•• Sinha P, Aarnisalo P, Chubb R, Poulton IJ, Guo J, Nachtrab G, et al. Loss of Gsα in the postnatal skeleton leads to low bone mass and a blunted response to anabolic parathyroid hormone therapy. J Biol Chem. 2016;291:1631–42. doi:10.1074/jbc.M115.679753. The anabolic effect of iPTH is blunted by deletion of Gsα in osteoblasts; genetic demonstration that phospholipase C activation is not required for PTH-induced bone anabolism
Meury T, Akhouayri O, Jafarov T, Mandic V, St-Arnaud R. Nuclear alpha NAC influences bone matrix mineralization and osteoblast maturation in vivo. Mol Cell Biol. 2010;30:43–53. doi:10.1128/MCB.00378-09.
Wu JY, Aarnisalo P, Bastepe M, Sinha P, Fulzele K, Selig MK, et al. Gsα enhances commitment of mesenchymal progenitors to the osteoblast lineage but restrains osteoblast differentiation in mice. J Clin Invest. 2011;121:3492–504. doi:10.1172/JCI46406.
Calvi LM, Sims NA, Hunzelman JL, Knight MC, Giovannetti A, Saxton JM, et al. Activated parathyroid hormone/parathyroid hormone-related protein receptor in osteoblastic cells differentially affects cortical and trabecular bone. J Clin Invest. 2001;107:277–86. doi:10.1172/JCI11296.
Bohinc BN, Gesty-Palmer D. Arrestins in bone. Prog Mol Biol Transl Sci. 2013;118:335–58. doi:10.1016/B978-0-12-394440-5.00013-9.
Ferrari SL, Pierroz DD, Glatt V, Goddard DS, Bianchi EN, Lin FT, et al. Bone response to intermittent parathyroid hormone is altered in mice null for {beta}-Arrestin2. Endocrinology. 2005;146:1854–62. doi:10.1210/en.2004-1282.
Gesty-Palmer D, Flannery P, Yuan L, Corsino L, Spurney R, Lefkowitz RJ, et al. A beta-arrestin-biased agonist of the parathyroid hormone receptor (PTH1R) promotes bone formation independent of G protein activation. Sci Transl Med. 2009;1:1ra. doi:10.1126/scitranslmed.3000071.
Kao RS, Abbott MJ, Louie A, O’Carroll D, Lu W, Nissenson R. Constitutive protein kinase A activity in osteocytes and late osteoblasts produces an anabolic effect on bone. Bone. 2013;55:277–87. doi:10.1016/j.bone.2013.04.001.
Tascau L, Gardner T, Anan H, Yongpravat C, Cardozo CP, Bauman WA, et al. Activation of protein kinase A in mature osteoblasts promotes a major bone anabolic response. Endocrinology. 2016;157:112–26. doi:10.1210/en.2015-1614.
O’Brien CA, Plotkin LI, Galli C, Goellner JJ, Gortazar AR, Allen MR, et al. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS One. 2008;3:e2942. doi:10.1371/journal.pone.0002942.
Rhee Y, Allen MR, Condon K, Lezcano V, Ronda AC, Galli C, et al. PTH receptor signaling in osteocytes governs periosteal bone formation and intracortical remodeling. J Bone Miner Res. 2011;26:1035–46. doi:10.1002/jbmr.304.
Li C, Wang W, Xie L, Luo X, Cao X, Wan M. Lipoprotein receptor-related protein 6 is required for parathyroid hormone-induced Sost suppression. Ann N Y Acad Sci. 2016;1364:62–73. doi:10.1111/nyas.12750.
• Li C, Xing Q, Yu B, Xie H, Wang W, Shi C, et al. Disruption of LRP6 in osteoblasts blunts the bone anabolic activity of PTH. J Bone Miner Res. 2013;28:2094–108. doi:10.1002/jbmr.1962. Genetic demonstration that LRP6 in osteoblasts is essential for the anabolic effects of PTH
Revollo L, Kading J, Jeong SY, Li J, Salazar V, Mbalaviele G, et al. N-cadherin restrains PTH activation of Lrp6/beta-catenin signaling and osteoanabolic action. J Bone Miner Res. 2015;30:274–85. doi:10.1002/jbmr.2323.
Wan M, Yang C, Li J, Wu X, Yuan H, Ma H, et al. Parathyroid hormone signaling through low-density lipoprotein-related protein 6. Genes Dev. 2008;22:2968–79. doi:10.1101/gad.1702708.
Vinyoles M, Del Valle-Perez B, Curto J, Vinas-Castells R, Alba-Castellon L, Garcia de Herreros A, et al. Multivesicular GSK3 sequestration upon Wnt signaling is controlled by p120-catenin/cadherin interaction with LRP5/6. Mol Cell. 2014;53:444–57. doi:10.1016/j.molcel.2013.12.010.
Yang H, Dong J, Xiong W, Fang Z, Guan H, Li F. N-cadherin restrains PTH repressive effects on sclerostin/SOST by regulating LRP6-PTH1R interaction. Ann N Y Acad Sci. 2016;1385:41–52. doi:10.1111/nyas.13221.
Acebron SP, Niehrs C. Beta-catenin-independent roles of Wnt/LRP6 signaling. Trends Cell Biol. 2016;26:956–67. doi:10.1016/j.tcb.2016.07.009.
Malinauskas T, Jones EY. Extracellular modulators of Wnt signalling. Curr Opin Struct Biol. 2014;29:77–84. doi:10.1016/j.sbi.2014.10.003.
Niehrs C, Shen J. Regulation of Lrp6 phosphorylation. Cell Mol Life Sci. 2010;67:2551–62. doi:10.1007/s00018-010-0329-3.
Kulkarni NH, Halladay DL, Miles RR, Gilbert LM, Frolik CA, Galvin RJ, et al. Effects of parathyroid hormone on Wnt signaling pathway in bone. J Cell Biochem. 2005;95:1178–90. doi:10.1002/jcb.20506.
Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013;19:179–92. doi:10.1038/nm.3074.
Iwaniec UT, Wronski TJ, Liu J, Rivera MF, Arzaga RR, Hansen G, et al. PTH stimulates bone formation in mice deficient in Lrp5. J Bone Miner Res. 2007;22:394–402. doi:10.1359/jbmr.061118.
Tian Y, Xu Y, Fu Q, He M. Parathyroid hormone regulates osteoblast differentiation in a Wnt/beta-catenin-dependent manner. Mol Cell Biochem. 2011;355:211–6. doi:10.1007/s11010-011-0856-8.
Kedlaya R, Kang KS, Hong JM, Bettagere V, Lim KE, Horan D, et al. Adult-onset deletion of β-catenin in 10kbDmp1-expressing cells prevents intermittent PTH-induced bone gain. Endocrinology. 2016;157:3047–57. doi:10.1210/en.2015-1587.
Li JY, Walker LD, Tyagi AM, Adams J, Weitzmann MN, Pacifici R. The sclerostin-independent bone anabolic activity of intermittent PTH treatment is mediated by T-cell-produced Wnt10b. J Bone Miner Res. 2014;29:43–54. doi:10.1002/jbmr.2044.
Robinson JW, Li JY, Walker LD, Tyagi AM, Reott MA, Yu M, et al. T cell-expressed CD40L potentiates the bone anabolic activity of intermittent PTH treatment. J Bone Miner Res. 2015;30:695–705. doi:10.1002/jbmr.2394.
Kramer I, Keller H, Leupin O, Kneissel M. Does osteocytic SOST suppression mediate PTH bone anabolism? Trends Endocrinol Metab. 2010;21:237–44. doi:10.1016/j.tem.2009.12.002.
Kramer I, Loots GG, Studer A, Keller H, Kneissel M. Parathyroid hormone (PTH)-induced bone gain is blunted in SOST overexpressing and deficient mice. J Bone Miner Res. 2010;25:178–89. doi:10.1359/jbmr.090730.
Robling AG, Kedlaya R, Ellis SN, Childress PJ, Bidwell JP, Bellido T, et al. Anabolic and catabolic regimens of human parathyroid hormone 1-34 elicit bone- and envelope-specific attenuation of skeletal effects in Sost-deficient mice. Endocrinology. 2011;152:2963–75. doi:10.1210/en.2011-0049.
Kramer I, Baertschi S, Halleux C, Keller H, Kneissel M. Mef2c deletion in osteocytes results in increased bone mass. J Bone Miner Res. 2012;27:360–73. doi:10.1002/jbmr.1492.
• Collette NM, Genetos DC, Economides AN, Xie L, Shahnazari M, Yao W, et al. Targeted deletion of Sost distal enhancer increases bone formation and bone mass. Proc Natl Acad Sci U S A. 2012;109:14092–7. doi:10.1073/pnas.1207188109. Genetic evidence that MEF2C is important for transcriptional activation of Sost in osteocytes
Leupin O, Kramer I, Collette NM, Loots GG, Natt F, Kneissel M, et al. Control of the SOST bone enhancer by PTH using MEF2 transcription factors. J Bone Miner Res. 2007;22:1957–67. doi:10.1359/jbmr.070804.
Saidak Z, Le Henaff C, Azzi S, Marty C, Marie PJ. Low-dose PTH increases osteoblast activity via decreased Mef2c/Sost in senescent osteopenic mice. J Endocrinol. 2014;223:25–33. doi:10.1530/JOE-14-0249.
Baertschi S, Baur N, Lueders-Lefevre V, Voshol J, Keller H. Class I and IIa histone deacetylases have opposite effects on sclerostin gene regulation. J Biol Chem. 2014;289:24995–5009. doi:10.1074/jbc.M114.564997.
• Wein MN, Spatz J, Nishimori S, Doench J, Root D, Babij P, et al. HDAC5 controls MEF2C-driven sclerostin expression in osteocytes. J Bone Miner Res. 2015;30:400–11. doi:10.1002/jbmr.2381. Novel osteocytic cell line that will facilitate studies of the role of osteocytes in the transduction of the PTH signal.
Parra M, Verdin E. Regulatory signal transduction pathways for class IIa histone deacetylases. Curr Opin Pharmacol. 2010;10:454–60. doi:10.1016/j.coph.2010.04.004.
•• Wein MN, Liang Y, Goransson O, Sundberg TB, Wang J, Williams EA, et al. SIKs control osteocyte responses to parathyroid hormone. Nat Commun. 2016;7:13176. doi:10.1038/ncomms13176. Identifies SIK2 as important mediator of the PTH anabolic signal; HDAC4/5 double KO mice show normal anabolic response to iPTH, suggesting additional pathways
Pearman AT, Chou WY, Bergman KD, Pulumati MR, Partridge NC. Parathyroid hormone induces c-fos promoter activity in osteoblastic cells through phosphorylated cAMP response element (CRE)-binding protein binding to the major CRE. J Biol Chem. 1996;271:25715–21.
Clohisy JC, Scott DK, Brakenhoff KD, Quinn CO, Partridge NC. Parathyroid hormone induces c-fos and c-jun messenger RNA in rat osteoblastic cells. Mol Endocrinol. 1992;6:1834–42.
McCauley LK, Koh AJ, Beecher CA, Rosol TJ. Proto-oncogene c-fos is transcriptionally regulated by parathyroid hormone (PTH) and PTH-related protein in a cyclic adenosine monophosphate-dependent manner in osteoblastic cells. Endocrinology. 1997;138:5427–33.
Fei Y, Shimizu E, McBurney MW, Partridge NC. Sirtuin 1 is a negative regulator of parathyroid hormone stimulation of matrix metalloproteinase 13 expression in osteoblastic cells: role of sirtuin 1 in the action of PTH on osteoblasts. J Biol Chem. 2015;290:8373–82. doi:10.1074/jbc.M114.602763.
Yotov WV, Moreau A, St-Arnaud R. The alpha chain of the nascent polypeptide-associated complex functions as a transcriptional coactivator. Mol Cell Biol. 1998;18:1303–11.
Moreau A, Yotov WV, Glorieux FH, St-Arnaud R. Bone-specific expression of the alpha chain of the nascent polypeptide-associated complex, a coactivator potentiating c-Jun-mediated transcription. Mol Cell Biol. 1998;18:1312–21.
Elefteriou F, Ahn JD, Takeda S, Starbuck M, Yang X, Liu X, et al. Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature. 2005;434:514–20.
Yang X, Matsuda K, Bialek P, Jacquot S, Masuoka HC, Schinke T, et al. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology. Implication for Coffin-Lowry Syndrome Cell. 2004;117:387–98.
Yoshizawa T, Hinoi E, Jung DY, Kajimura D, Ferron M, Seo J, et al. The transcription factor ATF4 regulates glucose metabolism in mice through its expression in osteoblasts. J Clin Invest. 2009;119:2807–17. doi:10.1172/JCI39366.
Yu S, Franceschi RT, Luo M, Fan J, Jiang D, Cao H, et al. Critical role of activating transcription factor 4 in the anabolic actions of parathyroid hormone in bone. PLoS One. 2009;4:e7583. doi:10.1371/journal.pone.0007583.
Yang XJ, Gregoire S. A recurrent phospho-sumoyl switch in transcriptional repression and beyond. Mol Cell. 2006;23:779–86.
Alonso V, Magyar CE, Wang B, Bisello A, Friedman PA. Ubiquitination-deubiquitination balance dictates ligand-stimulated PTHR sorting. J Bone Miner Res. 2011;26:2923–34. doi:10.1002/jbmr.494.
Shirakawa J, Harada H, Noda M, Ezura Y. PTH-induced osteoblast proliferation requires upregulation of the ubiquitin-specific peptidase 2 (Usp2) expression. Calcif Tissue Int. 2016;98:306–15. doi:10.1007/s00223-015-0083-5.
• Woo SM, Rosser J, Dusevich V, Kalajzic I, Bonewald LF. Cell line IDG-SW3 replicates osteoblast-to-late-osteocyte differentiation in vitro and accelerates bone formation in vivo. J Bone Miner Res. 2011;26:2634–46. doi:10.1002/jbmr.465. As in [65], osteocytic cell line that will facilitate studies of the role of osteocytes in the transduction of the PTH signal
Acknowledgements
Work from the authors’ laboratory is supported by CIHR grant MOP-119306 to R.St-A.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Hadla Hariri, Martin Pellicelli, and René St-Arnaud declare no potential conflicts of interest.
Human and Animal Rights and Informed Consent
All reported studies/experiments with animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (McGill University Institutional Animal Care and Use Committee and Canadian Council on Animal Care).
Additional information
This article is part of the Topical Collection on Molecular Biology of Skeletal Development
Rights and permissions
About this article

Cite this article
Hariri, H., Pellicelli, M. & St-Arnaud, R. New PTH Signals Mediating Bone Anabolism. Curr Mol Bio Rep 3, 133–141 (2017). https://doi.org/10.1007/s40610-017-0060-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40610-017-0060-z