
Abstract
Purpose of Review
Cadmium has been recognized as a potential risk factor for cardiovascular disease (CVD). We present a review of cadmium toxicity, its effect on cellular activities, and a summary of reported association between environmental cadmium exposure and CVD. We also discuss the possible therapeutic benefit of cadmium chelation.
Recent Findings
Experimental data suggest that cadmium affects several signaling pathways which may lead to endothelial dysfunction and vascular tissue damage, promoting atherosclerosis. This is further supported by epidemiological studies that have shown an association of even low-level cadmium exposure with an increased risk of clinical cardiovascular events. The Trial to Assess Chelation Therapy (TACT) provided inferential evidence for the cardiovascular benefit of treating toxic metal burden. However, at the present time, there is no direct evidence, but suggestive findings from clinical trials indicating that removal of cadmium from body stores may be associated with improved cardiovascular outcomes.
Summary
An evolving body of evidence supports environmental cadmium exposure as a pro-atherosclerosis risk factor in CVD; however, the mechanisms for the proatherogenic effect of cadmium are still not completely understood. Further studies in translational toxicology are needed to fill the knowledge gaps regarding the molecular mechanisms of cadmium toxicity and the promotion of atherosclerosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cadmium is a non-essential metal that has been recognized as an environmental and industrial pollutant with diverse toxic effects on human health [1, 2]. The world production of cadmium mainly comes from the by-product extraction of primary ores of lead, copper, and principally zinc. Cadmium extraction markedly increased during the twentieth century because of the widespread use of this metal in consumer products, including rechargeable nickel-cadmium batteries, jewelry and toys, pigments for plastic, ceramics and glassware, stabilizers for plastics, and protective electroplating of metal surfaces [1, 3,4,5]. Cadmium is also a common constituent in fertilizers derived from phosphate rock [6]. According to the US Geological Survey, the global cadmium production in 2017 was approximately 23,000 tons [7].
This widespread use of cadmium has resulted in many sources of exposure from which it can be inhaled or ingested [1, 3]. Industrial emissions of cadmium arise from non-ferrous metal production, iron and steel production, fossil fuel combustion, recycling and incineration of metal and electronic waste [8]. During production and use of cadmium-containing materials, contaminated aerosols and small particles are dispersed by wind, polluting soil, and water. The deposition, accumulation, and mobilization of cadmium in soil, freshwater, and ocean waters lead to contamination of foods. In the USA, dietary intake is the primary exposure route in non-smokers [1]. High levels of cadmium are found in mollusks and crustaceans, and in organ meats such as liver and kidney [9,10,11]. Plants can selectively take up cadmium via root cells through micronutrient transporters such as zinc-regulated transporter, iron-regulated transporter, and natural resistance-associated macrophage protein [12]. Cadmium concentrations vary among crop genotypes, being particularly high in grains, starchy root vegetables, and leafy greens such as lettuce, spinach, and tobacco. The high concentration of cadmium in tobacco leaves results in an additional source of exposure in smokers and consumers of smokeless tobacco products, who have a twofold higher body burden than non-smokers [13]. About 1 to 3 μg of cadmium is absorbed from smoking one pack of cigarettes per day [1]. Public health policies and regulations may have been able to reduce cadmium exposure in the USA, likely related to the decline in smoking rate [14]. Yet chronic cadmium exposure remains a significant hazard due to both ongoing and past exposures.
Following ingestion or inhalation, cadmium is transported in blood plasma bound to albumin and other proteins and delivered to the liver and kidney, where it accumulates with a half-life of approximately 10 years and 30 years, respectively [1]. Cadmium is taken up in the liver and induces the synthesis of metallothionein [3]. Metallothioneins are low-molecular-weight, cysteine-rich metal-binding proteins that play an important role in the retention of cadmium in various tissues [15]. Cadmium binds to metallothionein and is released into the circulation then delivered to target cells and tissues where deleterious effects may ensue, inducing mitochondrial damage and cell death, inflammation, and fibrosis [3]. The cadmium-metallothionein complex is taken up by the kidney, where it undergoes glomerular filtration and accumulates in the proximal renal tubules. The level of cadmium in the urine is proportional to the level of cadmium in the kidney [16]. Therefore, urine cadmium concentration serves as a biomarker for the cumulative body burden of cadmium [17].
Cadmium Levels in the US Population
Many of the studies available on cadmium exposure and cardiovascular outcomes use data from the National Health and Nutrition Examination Survey (NHANES), a series of representative samples of non-institutionalized US adults. Data from NHANES surveys between 1999 and 2016 show that the geometric mean (95% CI) of blood cadmium levels ranged from 0.41 (0.37–0.44) μg/L in 1999–2000 to 0.23 (0.22–0.25) μg/L in 2016. Blood cadmium levels were lower among males than among females and are higher after 20 years of age [18].
Cadmium Levels and Cardiovascular Disease
Several observational studies conducted in the NHANES population have shown an association between cadmium exposure and the risk for atherosclerotic disease and cardiovascular mortality [19,20,20, 22,23,23, 26,27,28,29,30] (Table 1). In NHANES 1999–2000, blood cadmium levels were strongly associated with increased prevalence of peripheral arterial disease (PAD). The adjusted odds ratio (OR) for PAD comparing the 4th vs. 1st quartile of blood cadmium was 2.42 (95% CI 1.13–5.15) [19]. This study also showed that after adjustment for cadmium in current smokers, the odds ratio for PAD comparing smokers to never smokers decreased from 4.1 to 1.8 suggesting that at least part of the adverse vascular effect of smoking may be mediated by the content of cadmium in cigarettes [19]. Similarly, an environment-wide association study using datasets from NHANES 1999–2004 was used to evaluate environmental factors associated with PAD [20]. The study showed that smoking-associated factors, including urinary cadmium, had a significant association with PAD. In fact, urinary cadmium was found to mediate most of the harm contributed by smoking [20]. In a small, exploratory cross-sectional study of 43 patients with coronary artery disease and PAD, toxic metals were measured before and after edetate disodium (EDTA)-based chelation. The study showed that, within this group of patients with CAD, the severity of PAD was associated with an increase in urinary cadmium level in both spontaneous and post-chelation urine (Fig. 1) [21].

Urinary cadmium in coronary artery disease patients with and without PAD. Individual p-values for urinary cadmium levels within groups of patients with CAD without PAD, CAD, and PAD but no critical limb ischemia, and CAD with critical limb ischemia was significantly higher with increased severity of disease
Other cross-sectional analyses of NHANES have also reported that elevated blood or urine cadmium level are associated with increased risk of myocardial infarction (MI) in women (NHANES 1988-1994; 4th vs. 1st quartile OR 95% CI: 1.80 (1.06–3.04)) but not statistically in men (OR 95% CI: 1.26 (0.71–2.26)) [22], stroke (NHANES 1999–2006; 4th vs. 1st quartile OR 95% CI: 1.38 (1.14–1.67) and heart failure (NHANES 1999-2006, 4th vs. 1st quartile OR 95% CI: 1.48 (1.17–1.87) (Table 1) [26]. Furthermore, in NHANES 1999–2004, blood and urine cadmium levels remained a risk factor for cardiovascular mortality (80th vs. 20th percentile: OR 95% CI: 1.74 (1.07–2.83)) [28]. Similarly, in another prospective investigation of NHANES III 1988–1994 participants, urinary cadmium was associated with cardiac mortality in men but not in women. The multivariable-adjusted hazard ratios for cardiovascular and coronary heart disease mortality associated with a 2-fold increase in creatinine-corrected urinary cadmium in men were 1.21 (1.07–1.36)) and 1.36 (1.11–1.66) respectively vs. 0.93 (0.84–1.04) and 0.82 (0.76–0.89) in women [23]. Combining data from NHANES III 1988–1994 and NHANES 1999–2004, including 15,421 adults ≥ 40 years old, it was estimated that 8.4% (95% CI: 4.3–36.4) deaths/100 000 person-years) of the absolute reduction in cardiovascular deaths between the two periods was attributed to reductions in cadmium burden, reflecting 31.0% lower blood cadmium levels in 1999–2004 compared with 1988–1994. This relationship held after adjustment for cardiovascular risk factors, sociodemographic factors, and lead exposure [31].
In summary, in the US population, as represented in the NHANES surveys, cadmium levels have been associated with risk of PAD, MI, stroke, heart failure, and cardiovascular mortality. Some of these studies suggest that there may be sex differences in cardiovascular outcomes related to cadmium exposure [22, 23]. Sex differences on cadmium-related cardiovascular endpoints may be due to confounding factors, random sampling variability, or biological factors. For instance, women had higher urine cadmium concentrations than men regardless of smoking status. Iron deficiency in women may lead to greater intestinal absorption of cadmium which may result in higher urinary and blood cadmium levels in women compared with men [24, 25]. Observations from in vivo and in vitro studies have found that cadmium may also possess estrogen-like activity, which may influence total body burden of cadmium in women [28]. An interesting speculation on the aforementioned studies is that sex differences in cadmium pharmacokinetics may contribute to the known sex differences in cardiovascular outcomes among men and women.
The Strong Heart Study is an ongoing multi-tribal study of 3348 American Indian adults in the American West. Higher baseline urinary cadmium levels measured in 1989–1991 were associated with increased incident cardiovascular disease (CVD) and CVD mortality, including coronary heart disease, stroke, and heart failure (Table 1). The subgroup with diabetes showed a stronger, statistically significant association between cadmium and CVD endpoints, when compared with those without diabetes [29]. Moreover, prospective analyses of the Strong Heart Study cohort further added to the evidence that cadmium is a risk factor for new-onset peripheral arterial disease, independent of smoking [30].
The evidence reviewed, therefore, supports the conclusion that cadmium has an adverse impact on health outcomes, particularly cardiovascular [14]. Overall, based on basic and epidemiological evidence cadmium may be an untargeted risk factor for the development of CVD.
Cadmium Toxicity at the Cellular Level
Ion channels on the cell membrane are the major route for cadmium entry into the cell. Ion channels are expressed in nearly all vascular cells, including endothelial cells, smooth muscle cells (SMCs), and fibroblasts. These ion channels play an important role in the regulation of physiological processes such as cell membrane potential and vasomotor function; when disrupted, they contribute to pathophysiological conditions of the vessel wall such as atherosclerosis [32]. Normal, healthy vascular homeostasis involves maintenance of the balance between vasodilation and vasoconstriction which is accomplished in part by calcium and potassium ion channels on the cell membrane of SMCs. Voltage-dependent calcium channels (VDCC) on the cell membrane of SMCs become activated when depolarized, allowing influx of extracellular calcium and initiate smooth muscle contraction. In a negative feedback loop, calcium-sensitive potassium channels along with voltage-gated potassium channels are activated hyperpolarizing the cell membrane promoting potassium efflux and leading to smooth muscle vasodilation [33]. Ion channel dysfunction and alterations in the ion influx and efflux mechanism contributes to vascular remodeling and stiffening and has been linked to hypertension [33]. In addition, alterations in the cell to cell communication and ion channel signaling between vascular endothelial cells and surrounding SMCs in the arteriole walls are also important in the development of atherosclerosis. Due to similarities in the chemical properties of cadmium and calcium, cadmium can mimic calcium entry through the cell membrane VDCC leading to cadmium influx and alterations in calcium homeostasis [34, 35]. This was exemplified in rat hepatocyte models exposed to cadmium in the absence or presence of different calcium channel blockers or a calcium agonist. The uptake of cadmium was inhibited by a calcium channel blocker suggesting calcium channels are the mode of cadmium entry into the cell [36].
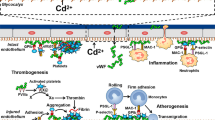
Thus, cadmium-induced calcium overload is one of the proposed mechanisms causing direct vasoconstriction [37]. Cadmium interference in calcium signaling distorts other intracellular calcium-dependent processes including inositol triphosphate (IP3) production and activation of calmodulin-dependent enzyme (CaMK) and protein kinase C (PKC). For example, cadmium activates PKC, a protein kinase involved in the sensitization of contraction to calcium by stimulation of mitogen-activated protein kinase (MAPK) [32]. In primary cultures of vascular smooth muscle cells of spontaneously hypertensive rats exposed to cadmium, there was greater increase in expression of MAPK in VSMs when compared with normotensive rat controls. In addition, PKC inhibitor reduced the effect of cadmium on PKC expression [38]. Cadmium also exerts its calcium mimetic effect on another calcium-sensitive enzyme, CaMK, which mediates apoptotic cell death [39]. The role of cadmium-induced CaMK in vascular cells is still poorly understood; however, we know that chronic apoptosis of vascular SMCs accelerates atherogenesis [40]. Cadmium also affects another specific calmodulin-dependent enzyme in endothelial cells, nitric oxide synthase (eNOS), which plays an important role in nitric oxide (NO) production and regulation of vasoactivity [41]. In an in vitro study, low levels of cadmium (0.01–1 μM) blocked endothelial NO phosphorylation, reduced endothelial cell NO production, and disrupted endothelial vasodilation [42]. The vascular effect of NO was examined in the rats poisoned with hypertensive doses of cadmium. Rat mesenteric arteries exposed to hypertensive doses of cadmium demonstrated a decrease in vascular reactivity with evidence of decreased serum NO concentration [43]. In an experimental study of the Ca2+ ATPase activity of the sarcoplasmic reticulum in rabbits, cadmium altered calcium homeostasis functioning as a potent inhibitor of the Ca2+ pump [44]. Cadmium-induced calcium pump inhibition causes an acute transient elevation of calcium concentration in the cytosol, but chronic exposure to cadmium leads to reduced calcium pools in the endoplasmic reticulum and a diminished response to evoked calcium signaling [45]. Another target of cadmium toxicity is the vascular endothelial (VE) cadherin-dependent cell adhesion molecules which maintain the integrity of endothelial cell to cell contact and vascular permeability [46]. In vitro studies have shown cadmium can displace calcium from cadherin-binding sites thereby interfering with its adhesive function [46]. Crosstalk between the various signaling pathways highlights the complexity and multifactorial mechanisms of cadmium-induced toxicity. Cadmium can also be toxic by inducing redox-active species, which may contribute to lipid peroxidation and oxidative DNA damage. In a study investigating the effects of cadmium on lipid levels, rats chronically exposed to 50 to 100 parts per million cadmium through drinking water for 7 weeks showed a significant increase in total serum cholesterol and triglycerides [47]. Subsequently, there was a 2-fold increase in oxidized LDL levels which mediate the transformation of macrophages to foam cells, key players in the pathogenesis of atherosclerosis [47, 48]. Furthermore, there is evidence of a reduction in the activity of antioxidant glutathione and enzymes, such as paraoxonase 1 (PON1), catalase, and superoxide dismutase, as well as depletion of radical scavengers with exposure to cadmium [49, 50]. PON1 is thought to contribute to the anti-inflammatory and antioxidant effect of high-density lipoproteins. In a prospective cohort study, low level of PON1 activity was associated with a higher prevalence of CVD [51, 52]. In the experimental rat model chronically exposed to cadmium, there was a significant decrease in PON1 activity [47]. Oxidative stress can lead to apoptosis of vascular smooth muscle cells, endothelial cells, and macrophages, which is a mechanism implicated in atherogenesis [53, 54]. Nuclear factor kappa B (NF-kB) is a redox-sensitive transcription factor important in the regulation of apoptosis [55]. NF-kB initiates transcription of target genes such as the inhibitors of apoptosis (IAPs) which are critical in cell survival as they bind to caspases and inhibit apoptosis [56]. The effect of cadmium on cell death signaling pathway was examined in rat kidney epithelial cells exposed to 20μM Cd for 5 h. The study showed that the activity of NF-kB was significantly reduced, downregulating of the target gene products IAPs and enhancing cadmium-induced apoptosis [56].Induction of vascular SMC and endothelial cell death by cadmium suppression of NF-kB activity may play a central role in the promotion of atherosclerosis [56,57,58]. Hence, cadmium exposure can result in endothelial and vascular SMCs dysfunction by interfering with the NO and intracellular calcium signaling pathways, increasing oxidative stress and apoptosis (Fig. 2), all of which could promote atherosclerosis. A full understanding of cadmium toxicity at the cellular level would provide further insight into possible mechanisms of cadmium-induced CVD.

Mechanism of cadmium toxicity. Stimulatory (“+”) and inhibitory (“−”) effects of cadmium on second messenger signaling pathways leading to decreased nitric oxide, increased superoxide anions (ROS), cellular dysfunction, and necrotic cell death. G, GTP binding; IP3, inositol-1,4,5 triphosphate; CAM, Ca2+-calmodulin; PKC, protein kinase C; NO, nitric oxide; NAD(P)H, nicotinamide adenine dinucleotide phosphate oxidase
Discussion
The above studies, ranging from basic science, to small-scale clinical studies, to large-scale epidemiologic studies representative of the US population, form a robust body of evidence indicating that cadmium, even at relatively low levels of exposure, is vasculotoxic and may be a risk factor for atherosclerosis. Preventive measures should address reduction of cadmium exposure in the general population to minimize atherosclerotic risk. Yet its protracted biological half-life, low rate of excretion, and preferential storage in soft issues, as described above, emphasize the need for a biomarker or surrogate measure of cadmium body burden.
Biomarkers are objective measurements that serve as indicators of disease processes or unintended environmental exposure. As discussed earlier, urine cadmium concentration has served as a biomarker for long-term cadmium exposure in epidemiologic research. Studies have shown that urine cadmium is a temporally stable biomarker as evidenced by its reproducibility when repeat samples are collected years apart [59, 60]. Yet, factors that influence renal physiology greatly impact the variability of urine cadmium. One such factor of urinary biomarkers is the degree of urine dilution, which is typically adjusted for by urine creatinine or specific gravity. However, residual influence of diuresis may persist even after correction, which may influence variations in urinary cadmium [61]. Moreover, there is a positive correlation between urine cadmium and the glomerular filtration rate. In the presence of a normal, efficient renal function, there is faster elimination of cadmium in the urine [62]. However, cadmium can adversely impair glomerular and renal tubular function and may increase urinary excretion of low-molecular-weight proteins [61]. In patients with renal disease and diabetes, due to damage to the proximal renal tubules, inhibition of renal tubular reabsorption of low molecular weight proteins such as retinol-binding protein results in increased co-excretion of these proteins and cadmium in the urine [63]. Blood cadmium can also be used to assess exposure to cadmium. It has been recognized, however, to reflect recent exposure rather than chronic exposure, although a compartment in blood cadmium also reflects long-term cadmium exposure [11, 64]. For instance, recent smoking is associated with markedly higher blood cadmium concentration vs. urine cadmium. Similarly, a decline in blood cadmium concentration after smoking cessation most likely occurs within days and weeks, although former smokers still have higher blood cadmium levels compared with never smokers [65].
The use of metallothionein as a biomarker of chronic cadmium exposure has been entertained in prior studies [66,67,68]. Metallothionein plays an important role in the cadmium retention in tissues, mainly the liver and kidney. As briefly discussed earlier, cadmium stimulates synthesis of metallothionein enhancing metallothionein gene transcription leading to an increase in apo-metallothionein that tightly binds to cadmium cations in the cytosol. It is a specific metal-binding protein, with particularly high affinity towards cadmium, which makes it an attractive option as a biomarker, and thus, the level of cadmium in the body should be proportional to the level of saturated metal-binding ligands of metallothionein. However, metallothionein’s major role is to regulate zinc and copper metabolism. Zinc status influences the expression of metallothionein and the ability to bind cadmium [69]. For instance, zinc losses through the urine in the presence of diabetes could influence metallothionein expression and potentially explain higher cadmium toxicity in the presence of diabetes [70]. The evidence available suggesting the potential use of metallothionein as a cadmium biomarker has largely been from environmental biomonitoring studies in the marine environment [71, 72]. It is unclear if metallothionein protein is associated with CVD. Further studies are needed to fully understand the use of metallothionein as a potential biomarker in humans, and its association with cadmium toxicity in CVD.
Other Mechanisms of Toxicity
Epigenetics has been recognized as an additional layer in the understanding of cadmium toxicity [73]. To the best of our knowledge, few studies have investigated the relationship between cadmium epigenetic effects and CVD. This novel area of research suggests potential participation of DNA methylation, post-translation histone modifications, and non-coding RNA or microRNA in CVD pathogenesis including atherosclerosis [74]. As previously discussed, cadmium is a well-known blocker of voltage-dependent calcium channels influencing vascular cell dysfunction. It is plausible that cadmium toxicity may itself cause changes in the gene expression of voltage-dependent calcium channels, which then leads to smooth muscle proliferation in atherosclerosis. Epigenome-wide association study of 2,325 adults 45–74 years of age who participated in the Strong Heart Study in 1989–1991 suggests that smoking-related cadmium exposure may be partly associated with changes in DNA methylation at specific sites; however, the functional consequence of the differential methylation at these sites remains to be understood [75]. Experimental and observation data has also linked dysregulation in microRNA expression with exposure to cadmium [76, 77]. Studies focused on the nephrotoxic effects of cadmium, have shown that deregulation of microRNA may play a role in the pathophysiology of cadmium-induced kidney damage [11]. It is unclear if cadmium is involved in the alteration of key regulators of gene expression in atherosclerosis, such as microRNA-126, microRNA-145, and microRNA-155 [78, 79]. The discovery of its role in microRNA dysregulation in atherosclerosis may shed light on the mechanism of cadmium toxicity and perhaps provide novel treatment pathways for CVD.
Interventional Studies
Despite the epidemiological and experimental evidence, there is not yet outcome data from clinical trials showing that a reduction of cadmium body burden is associated with improved CV outcomes. In fact, chelation to treat low-level metal intoxication remains a controversial therapy in clinical toxicology and cardiology. Chelation involves binding of cationic metallic and nonmetallic ions and their mobilization from physiological tissue. Various chelating agents have been used for the treatment of cadmium toxicity such as dimercaprol (BAL), dimercaptopropanesulfonic acid (DMPS), dimercaptosuccinic acid (DMSA), and edetate disodium or edetate calcium disodium (EDTA), though no specific therapy has been approved for clinical use due to limited clinical research. EDTA is the most widely accepted chelating agent that can form complexes with cadmium, which are then excreted in the urine. In fact, the edetates are potent cadmium chelators. A single infusion of edetate disodium will increase cadmium excretion by about 700% in a few hours [80, 81].
In the mid-twentieth century, EDTA was used to treat symptoms of CVD in small, uncontrolled studies [82, 83]. Since then, subsequent case reports and case studies and 3 small clinical trials, studying an aggregate of 269 subjects, followed. Each of these studies had methodological problems, but principally, in aggregate, the studies were too small to exclude a small to moderate effect of therapy [84,85,86].
The first properly powered clinical trial of edetate disodium–based chelation took place from 2002 to 2012. Funded by the National Institutes of Health, The Trial to Assess Chelation Therapy (TACT) was designed as a double-blind, placebo-controlled, randomized trial designed to determine the safety and efficacy of edetate disodium–based chelation in patients with a prior MI [87,88,89].
There were 1708 patients enrolled in the clinical trial across 134 sites in the USA and Canada between 2003 and 2010. The primary endpoint for the study was a composite of all-cause mortality, MI, stroke, coronary revascularization, and hospitalization for angina at median follow-up of 55 months.
Edetate chelation reduced the primary composite endpoint by 18%: hazard ratio (HR), 0.82; 95% confidence interval (CI), 0.69–0.99, p = 0.035 (Fig. 3); with a 5-year number needed to treat (NNT) of 18 patients to prevent an event [88]. In the prespecified group of patients with diabetes (n = 633), there was a 41 % reduction of the primary endpoint by edetate disodium–based chelation (primary endpoint: EDTA chelation vs. placebo: 25% vs. 38%; HR = 0.59; 95% CI, 0.44–0.79; p = 0.0002) (Fig. 4). The NNT in 5 years to prevent a single event in diabetic patients was 6.5. In this subgroup of participants, the rate of the major secondary endpoints was also markedly reduced by EDTA treatment. Death from any cause was reduced by 43% (10% vs 16%; HR = 0.57; 95% CI, 0.36–0.88; p = 0.011) with a 5-year NNT of 12 (Fig. 5). Furthermore, there was a 52% relative reduction in the risk of recurrent MI (HR = 0.48; 95% CI 0.26–0.88; p = 0.015) and a 32% relative reduction in the risk of coronary revascularization (HR = 0.68; 95% CI, 0.47–0.99; p = 0.042) [89]. TACT provided the strongest inferential evidence thus far for the potential benefit of toxic metal chelation to reduce major adverse cardiovascular events. However, metal levels were not measured in this study, and it was unclear if clinical outcomes were linked to a reduction in cadmium or/and other toxic metals that are removed by EDTA or by other mechanisms (e.g., improving endothelial function and oxidative stress, calcium removal/homeostasis, etc.). An ongoing replicative study (TACT2) enrolling post-myocardial infarction patients with diabetes will investigate the possible mechanisms of benefit including the relationship of cadmium chelation with outcomes.

Primary endpoint in the overall patient population

Primary endpoint in patients with diabetes mellitus

Mortality in patients with diabetes mellitus by infusion group
A post hoc, non-prespecified analysis of 162 TACT patients with diabetes and PAD demonstrated an even greater benefit from edetate disodium–based chelation compared with placebo. Patients exhibited a 48% relative risk reduction (p = 0.0069) of the TACT primary endpoint [90].
Given the evidence of high levels of cadmium in patients with severe PAD and critical limb ischemia (CLI) and the beneficial effects observed in TACT, an open-label study was performed to evaluate the safety and efficacy of EDTA-based chelation as a therapeutic option in patients with diabetes and CLI [90]. This recently published unblinded pilot study demonstrated a potential signal of benefit and preliminary evidence of safety. The study enrolled 10 patients with diabetes and moderate to severe infra-popliteal chronic limb ischemia, each receiving up to 50 edetate disodium–based infusions. All patients had coronary artery disease, previous lower extremity revascularization, ischemic limb pain, and 70% had non-healing ulcers or dry gangrene. There were no adverse events related to therapy. At 1-year follow-up, there were no major adverse cardiovascular events. No limb amputations occurred in patients who completed more than 20 infusions. The patients who completed at least 40 infusions showed complete wound healing and improvements in overall quality of life as ascertained by PAD Questionnaire and SF-36 [91]. Still, the conclusion that a reduction in cadmium led to clinical benefit is confounded by concomitant increased excretion of other cations such as lead and calcium and by the absence of a control group in this pilot study [80]. Further studies are needed with measurements of blood or urinary cadmium concentrations before and after chelation treatments and a placebo control in order to conclude that there is a causal relationship between the use of a cadmium-avid chelator and improved outcomes in patients with severe vascular disease.
Conclusion
Cadmium has significant impact on human disease and may be a modifiable risk factor for the development and progression of atherosclerosis as supported by epidemiological studies and the TACT trial. However, there are still several limitations in understanding the role of cadmium in CVD. The knowledge gaps regarding the mechanisms of cadmium toxicity and promotion of atherosclerosis underscore the need for additional translational toxicology research. Identification of other molecular targets of cadmium and epigenetic mechanisms in cadmium-induced gene alterations are necessary for translation into clinically useful therapeutics. In addition, early markers of cadmium cardiovascular toxicity as well as the interaction of cadmium with other cardiovascular risk factors including levels of other toxic and non-toxic metals merit further research. TACT2, currently enrolling, will provide further light on the potential benefits of cadmium removal therapy to improve cardiovascular outcomes. TACT2 will maintain a biorepository to investigate the mechanistic role of cadmium chelation. Additionally, a trial (TACT3a) to further assesses the safety and efficacy of EDTA-based chelation in patients with PAD is currently underway.
Abbreviations
- Cd:
-
Cadmium
- CVD:
-
Cardiovascular disease
- TACT:
-
Trial to assess chelation therapy
- MI:
-
Myocardial infarction
- Ca2+ :
-
Calcium
- SMCs:
-
Smooth muscle cells
- VDCC:
-
Voltage-dependent calcium channels
- IP3:
-
Inositol triphosphate
- PKC:
-
Protein kinase C
- CAMK:
-
Calmodulin dependent enzyme
- MAPK:
-
Mitogen-activated protein kinase
- NO:
-
Nitric oxide
- PON1:
-
Paraoxonase 1
- NF-kB:
-
Nuclear factor kappa B
- NHANES:
-
National Health and Nutrition Examination Survey
- PAD:
-
Peripheral arterial disease
- RNA:
-
Ribonucleic acid
- EDTA:
-
Edetate disodium
- BAL:
-
Dimercapral
- DMPS:
-
Dimercaptopropanesulfonic acid
- DMSA:
-
Dimercaptosuccinic acid
- LDL:
-
Low-density lipoprotein
- OR:
-
Odds ratio
- HR:
-
Hazard ratio
- CI:
-
Confidence interval
- NNT:
-
Number needed to treat
- CLI:
-
Critical limb ischemia
References
Agency for Toxic Substances and Disease Registry. Toxicological profile for cadmium; Agency for Toxic Substances and Disease Registry, Public Health Service, U.S. Department of Health and Human Services: Atlanta, GA, USA, 2012; pp. 1–487.
International Agency for Research on Cancer (IARC) [Accessed July 2013]; Beryllium, cadmium, mercury, and exposures in the glass manufacturing industry. 1993 Available online at: http://monographs.iarc.fr/ENG/Monographs/vol58/
Nordberg GF, Nogawa K, Nordberg M, Friberg L. Handbook on the toxicology of metals: cadmium. Amsterdam, Elsevier; 2007. p. 445–86.
Staessen JA, Vyncke G, Lauwerys RR, Roles HA, Celis HG, Claeys F, et al. Transfer of cadmium from a sandy acidic soil to man: a population study. Environ Res. 1992;58(1):25–34.
Grant CA, Sheppard SC. Fertilizer Impacts on Cadmium availability in agricultural soils and crops. Hum Ecol Risk Assess. 2008;14:210–28.
Roberts TL. Cadmium and phosphorous fertilizers: the issues and the science. Procedia Engineering. 2014;83:52–9.
U.S. Geological Survey, 2018, Mineral resources data system: commodity statistics and information available online at https://minerals.usgs.gov/minerals/pubs/commodity/cadmium/. (Accessed May 14, 2018)
Pacyna JM, Pacyna EG. An assessment of global and regional emissions of trace metals to the atmosphere from anthropogenic sources worldwide. Environ Rev. 2001;9(4):269–98.
Satarug S, Baker JR, Urbenjapol S, Haswell EM, Reilly PE, Williams DJ, et al. A global perspective on cadmium pollution and toxicity in non-occupationally exposed population. Toxicol Lett. 2003;137:65–83.
Miranda M, Lopez Alonso ML, Castillo C, Hernandez J, Benedito JL. Cadmium levels in liver, kidney and meat in calves from Asturias (North Spain). Eur Food Res Technol. 2001;212:426–30.
Jarup L, Akesson A. Current status of cadmium as an environmental health Problem. Toxicol Appl Pharmacol. 2009;238(3):201–8.
Asgher M, Khan MIR, Anjum NA, Khan NA. Minimising toxicity of cadmium in plants-role of plant growth regulators. Protoplasma. 2015;252(2):399–413.
Pappas RS, Polzin GM, Zhang L, Watson CH, Paschal DC, Ashley DL. Cadmium, lead, and thallium in mainstream tobacco smoke particulate. Food Chem Toxicol. 2006;44:714–23.
Tellez-Plaza M, Navas-Acien A, Caldwell KL, Menke A, Muntner P, Guallar E. Reduction in cadmium exposure in the United States population, 1988–2008: the contribution of declining smoking rates. Environ Health Perspect. 2012;120:204–9.
Klaassen CD, Lui J, Choudhuri S. Metallothionein: an intracellular protein to protect against cadmium toxicity. Annu Rev Pharmacol Toxicol. 1999;39:267–94.
Kawada T, Shinmyo RR, Suzuki S. Changes in urinary cadmium excretion among pigment workers with improvement of the work environment. Ind Health. 1993;31(4):165–70.
Friberg L. Cadmium and the kidney. Environ Health Perspect. 1984;54:1–11.
Fourth National Report on Human Exposure to Environmental Chemicals, Updated Tables, Centers for Disease Control and Prevention National Center for Environmental Health. March 2018: http://www.cdc.gov/exposurereport
Navas-Acien A, Selvin E, Sharrett AR, Calderon-Aranda E, Silbergeld E, Guallar E. Lead, cadmium, smoking, and increased risk of peripheral arterial disease. Circulation. 2004;109:3196–201.
Zhuang X, Ni A, Liao L, Guo Y, Dai W, Jiang Y, et al. Environment-Wide Association study to identify novel factors associated with peripheral arterial disease: evidence from the National Health and Nutrition Examination Survey (1999–2004). Atherosclerosis. 2018;269:172–7.
Ujueta F, Arenas IA, Diaz D, Yates T, Beasley R, Navas-Acien A, et al. Cadmium level and severity of peripheral artery disease in patients with coronary artery disease. Eur J Prev Cardiol. 2018;26(13):1456–8.
Everett CJ, Frithsen IL. Association of urinary cadmium and myocardial infarction. Environ Res. 2008;106(2):284–6.
Menke A, Muntner P, Silbergeld EK, Platz EA, Guallar E. Cadmium levels. in urine and mortality among U.S. adults. Environ Health Perspect. 2009;117:190–6.
Vahter M, Akesson A, Lidén C, Ceccatelli S, Berglund M. Gender differences in the disposition and toxicity of metals. Environ Res. 2007;104(1):85–95.
Zacharski LR, Ornstein DL, Woloshin S, Schwartz LM. Association of age, sex, and race with body iron stores in adults: analysis of NHANES III data. AMJ. 2000;140(1):98–104.
Peters JL, Perlstein TS, Perry MJ, McNeely E, Weuve J. Cadmium exposure in association with history of stroke and heart failure. Environ Res. 2010;110(2):199–206.
Agarwal S, Zaman T, Tuzcu EM, Kapadia SR. Heavy metals and cardiovascular disease: results from the National Health and Nutrition Examination Survey (NHANES) 1999–2006. Angiology. 2011;62:422–9.
Tellez-Plaza M, Navas-Acien A, Menke A, Crainiceanu CM, Pastor-Barriuso R, Guallar E. Cadmium exposure and all-cause and cardiovascular mortality in the U.S. General Population. Environ Health Perspect. 2012;120:1017–22.
Tellez-Plaza M, Guallar E, Howard BV, Umans JG, Francesconi KA, Goessler W, et al. Cadmium exposure and incident cardiovascular disease. Epidemiology. 2013;24:421–9.
Tellez-Plaza M, Guallar E, Fabsitz RR, Howard BV, Umans JG, Framcesconi KA, et al. Cadmium exposure and incident peripheral arterial disease. Circ Cardiovasc Qual Outcomes. 2013;6(6):626–33.
Ruiz-Hernandez A, Navas-Acien A, Pastor-Barriuso R, Crainiceanu CM, Redon J, Guallar E, et al. Declining exposures to lead and cadmium contribute to explaining the reduction of cardiovascular mortality in the US population, 1988-2004. Int J Epidemiol. 2017;46(6):1903–12.
Prozialeck WC, Edwards JR, Nebert DW, Woods JM, Barchowsky A, Atchison WD. The vascular system as a target of metal toxicity. Toxicol Sci. 2008;102:207–18.
Chen J, Wen J, Wang N, Wang C, Xu Q, Yang Y. Ion channels and vascular diseases. Arterioscler Thromb Vasc Biol. 2019;39:146–56.
Hinkle PM, Kinsella PA, Osterhoudt KC. Cadmium uptake and toxicity via voltage-sensitive calcium channels. J Biol Chem. 1987;262(34):16333–7.
Bridges CC, Zalups RK. Molecular and ionic mimicry and the transport of toxic metals. Toxicol Appl Pharmacol. 2005;204(3):274–308.
Blazka ME, Shaikh ZA. Differences in cadmium and mercury uptakes by hepatocytes: role of calcium channels. Toxicol Appl Pharmacol. 1991;110(2):355–63.
Varoni MV, Palomba D, Demontis MP, Gianorso S, Pais GL, Anania V. Role of the brain renin-angiotensin system in blood pressure regulation. Vet Res Commun. 2007;31(Suppl. 1):343–6.
Washington B, Williams S, Armstrong P, Mtshali C, Robinson JT, Myles EL. Cadmium toxicity on arterioles vascular smooth muscle cells of spontaneously hypertensive rats. Int J Environ Res Public Health. 2006;3(4):323–8.
Suzuki Y, Chao SH, Zysk JR, Cheung WY. Stimulation of calmodulin by cadmium ion. Arch Toxicol. 1985;57:205–11.
Clarke MCH, Littlewood TD, Figg N, Maguire JJ, Davenport AP, Goddard M, et al. Chronic apoptosis of vascular smooth muscle cells accelerates atherosclerosis and promotes calcification and medial degeneration. Circ Res. 2008;102:1529–38.
Greif DM, Sacks DB, Michel T. Calmodulin phosphorylation and modulation of endothelial nitric oxide synthase catalysis. PNAS. 2004;101(5):1165–70.
Moncada S. Nitric oxide: Discovery and impact on clinical medicine. J R Soc Med. 1999;92:164–9.
Skoczynska A, Martynowicz H. The impact of subchronic cadmium poisoning on the vascular effect of nitric oxide in rats. Hum Exp Toxicol. 2005 Jul;24(7):353–61.
Hechtenberg S, Beyermann D. Inhibition of sarcoplasmic reticulum Ca (2+)-.ATPase activity by cadmium, lead and mercury. Enzyme. 1991;45:109–15.
Biagioli M, Pifferi S, Ragghianti M, Bucci S, Rizzuto R, Pinton P. Endoplasmic reticulum stress and alteration in calcium homeostasis are involved in cadmium induced apoptosis. Cell Calcium. 2008;43:184–95.
Prozialeck WC. Evidence that E-cadherin may be a target for cadmium toxicity in epithelial cells. Toxicol Appl Pharmacol. 2000;164(3):231–49.
Afolabi OK, Oyewo EB, Adekunle AS, Adedosu OT, Adedeji AL. Impaired lipid levels and inflammatory response in rats exposed to cadmium. EXCLI J. 2012;11:677–87 Published 2012 Sep 27.
Witztum JL, Steinberg D. Role of oxidized low density lipoprotein in atherogenesis. J Clin Invest. 1991;88(6):1785–92.
Ikediobi CO, Badisa VL, Ayuk-Takem LT, Latinwo LM, West J. Response of antioxidant enzymes and redox metabolites to cadmium-induced oxidative stress in CRL-1439 normal rat liver cells. Int J Mol Med. 2004;14:87–92.
Waisberg M, Joseph P, Hale B, Beyersmann D. Molecular and cellular mechanisms of cadmium carcinogenesis. Toxicology. 2003;192:95–117.
Bhattacharyya T, Nicholls SJ, Topol EJ, Zhang R, Yang X, Schmitt D, et al. Relationship of paraoxonase 1 (PON1) gene polymorphisms and functional activity with systemic oxidative stress and cardiovascular risk. JAMA. 2008;299(11):1265–76.
Pollack AZ, Sjaarda L, Ahrens KA, Mumford SL, Browne RW, Wactawski-Wende J, et al. Association of cadmium, lead and mercury with paraoxonase 1 Activity in women. PLoS One. 2014;9(3):e92152.
Isner JM, Kearney M, Bortman S, Passeri J. Apoptosis in human atherosclerosis and restenosis. Circulation. 1995;91:2703–11.
Messner B, Knoflach M, Seubert A, Ritsch A, Pfaller K, Henderson B. Cadmium is a novel and independent risk factor for early atherosclerosis mechanisms and in vivo relevance. Arterioscler Thromb Vasc Biol. 2009;29(9):1392–8.
Perkins ND. Integrating cell-signaling pathways with NF- kappa B and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62.
Xie J, Shaikh ZA. Cadmium-induced apoptosis in rat kidney epithelial cells involves decrease in nuclear factor-kappa B activity. Toxicol Sci. 2006;91:299–308.
Shumilla JA, Wetterhahn KE, Barchowsky A. Inhibition of NF-kappa B binding to DNA by chromium, cadmium, mercury, zinc, and arsenite in vitro: evidence of a thiol mechanism. Arch Biochem Biophys. 1998;349(2):356–62.
Hart BA, Lee CH, Shukla GS, Shukla A, Osier M, Eneman JD, et al. Characterization of cadmium-induced apoptosis in rat lung epithelial cells: evidence for the participation of oxidant stress. Toxicology. 1999;133:43–58.
Vacchi-Suzzi C, Porucznik CA, Cox KJ, Zhao Y, Ahn H, Harrington JM, et al. Temporal variability of urinary cadmium in spot urine samples and first morning voids. J Expos Sci Environ Epidemiol. 2016;27:160–6. https://doi.org/10.1038/jes.2016.2.
Meliker JR, Vacchi-Suzzi C, Harrington J, Levine K, Lui L-Y, Bauer DC, et al. Temporal stability of urinary cadmium in samples collected several years apart in a population of older persons. Int J Hyg Environ Health. 2019;222:230–4.
Chaumont A, Voisin C, Deumer G, Haufroid V, Annesi-Maesano I, Roles H, et al. Associations of urinary cadmium with age and urinary proteins: further evidence of physiological variations unrelated to metal accumulation and toxicity. Environ Health Perspect. 2013;121(9):1047–53.
Weaver VM, Kim NS, Lee BK, Parsons PJ, Spector J, Fadrowski J, et al. Differences in urine cadmium associations with kidney outcomes based on serum creatinine and cystatin C. Environ Res. 2011;111(8):1236–42.
Chaumont A, Nickmilder M, Dumont X, Lundh T, Skerfving S, Bernard A. Associations between proteins and heavy metals in urine at low environmental exposures: evidence of reverse causality. Toxicol Lett. 2012;210:345–52.
Lauwerys RR, Bernard AM, Roels HA, Buchet JP. Cadmium: exposure markers as predictors of nephrotoxic effects. Clin Chem. 1994;40(7 Pt 2):1391–4.
Adams SV, Newcomb PA. Cadmium blood and urine concentrations as measures of exposure: NHANES 1999–2010. J Expo Sci Environ Epidemiol. 2014;24:163–70.
Chung J, Nartey NO, Cherian MG. Metallothionein levels in liver and kidney of Canadians--a potential indicator of environmental exposure to cadmium. Arch Environ Health. 1986;41(5):319–23.
Hejlmajer HE, Drasch GA, Kretschmer E, Summer KH. Metallothionein,cadmium, copper and zinc in human and rat tissues. Toxicol Lett. 1987;38(3):205–11.
Bremner I, Mehra RK, Sato M. Metallothionein in blood, bile and urine. Experientia Suppl. 1987;52:507–17.
Ochi T, Otsuka F, Takahashi K, Ohsawa M. Glutathione and metallothioneins as cellular defense against cadmium toxicity in cultured Chinese hamster cells. Chem Biol Interact. 1988;65(1):1–14.
Wijesekara N, Chimienti F, Wheeler MB. Zinc, a regulator of islet function and glucose homeostasis. Diabetes Obes Metab. 2009;11(Suppl 4):202–14.
Man AK, Woo NY. Upregulation of metallothionein and glucose-6-phosphate dehydrogenase expression in silver sea bream, Sparus sarba exposed to sublethal levels of cadmium. Aquat Toxicol. 2008;89:214–21.
Shariati F, Esaili SA, Mashinchian A, Pourkazemi M. Metallothionein as potential biomarker of cadmium exposure in Persian sturgeon (Acipenser persicus). Biol Trace Elem Res. 2011 Oct;143(1):281–91.
Wang B, Li Y, Shao C, Tan Y, Cai L. Cadmium and its epigenetic effects. Curr Med Chem. 2012;19(16):2611–20.
Khalil CA. The Emerging Role of epigenetics in cardiovascular disease. Ther Adv Chronic Dis. 2014;5(4):178–87.
Domingo-Relloso A, Riffo-Campos AL, Rentero-Garrido P, Ladd-Acosta C, Fallin DM, Tang WY, et al. Cadmium, smoking, and human blood dna methylation profiles in adults from the strong heart study. Environ Health Perspect. 2020;128(6):067005. https://doi.org/10.1289/EHP6345.
Lemaire J, Van der Hauwaert C, Savary G, Dewaeles E, Perrais M, Lo Guidice JM, et al. Cadmium-induced renal cell toxicity is associated with microRNA deregulation. Int J Toxicol. 2020;39(2):103–14.
Gu S, Dai J, Qu T, He Z. Emerging Roles of MicroRNAs and Long Noncoding RNAs in Cadmium Toxicity. Biol Trace Elem Res. 2020;195:481–90.
Harris TA, Yamakuchi M, Ferlito M, Mendell JT, Lowenstein CJ. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc Natl Acad Sci U S A. 2008 Feb 5;105(5):1516–21.
Yuanyuan W, Jahantigh M, Neth P, Weber C, Schober A. MicroRNA-126, -145, and -155. Arterioscler Thromb Vasc Biol. 2013;33:449–54.
Waters RS, Bryden NA, Patterson KY, Veillon C, Anderson RA. EDTA Chelation effects on urinary losses of cadmium, calcium, chromium, cobalt, copper, lead, magnesium, and zinc. Biol Trace Elem Res. 2001;83:207–21.
Arenas IA, Navas-Acien A, Erqui I, Lamas GA. Enhanced Vasculotoxic metal excretion in post-MI patients after edetate disodium therapy. J Am Coll Cardiol. 2016;67(13 Suppl):A2125.
Clarke NE, Clarke CN, Mosher RE. The in vivo dissolution of metastatic calcium. An approach to atherosclerosis. Am J Med Sci. 1955;229:142–9.
Clarke CN, Clarke NE, Mosher RE. Treatment of angina pectoris with disodium ethylene diamine tetra acetic acid. Am J Med Sci. 1956;232:654–66.
Guldager B, Jelnes R, Jorgensen SJ, Nielsen JS, Klaerke A, Mogensen K, et al. EDTA treatment of intermittent claudication--a double-blind, placebo-controlled study. J Intern Med. 1992;231(3):261–7.
van Rij AM, Solomon C, Packer SG, Hopkins WG. Chelation Therapy for intermittent claudication. A double-blind, randomized, controlled trial. Circulation. 1994;90(3):1194–9.
Knudtson ML, Wyse DG, Galbraith PD, Brant R, Hildebrand K, Paterson D, et al. Chelation therapy for ischemic heart disease: a randomized controlled trial. JAMA. 2002;287(4):481–6.
Lamas GA, Goertz C, Boineau R, Mark DB, Rozema T, Nahin RL, et al. Design of the trial to access chelation therapy. Am Heart J. 2012;163:7–12.
Lamas GA, Goertz C, Boineau R, Mark DB, Rozema T, Nahin RL, et al. Effect of disodium EDTA chelation regimen on cardiovascular events in patients with previous myocardial infarction: the TACT randomized trial. JAMA. 2013;309:1241–50.
Escolar E, Lamas GA, Mark DB, Boineau R, Goertz C, Rosenberg Y, et al. The effect of an EDTA-based chelation regimen on patients with diabetes mellitus and prior myocardial infarction in the Trial to Assess Chelation Therapy (TACT). Circ Cardiovasc Qual Outcomes. 2014;7:15–24.
Ujueta F, Arenas IA, Escolar E, Diaz D, Boineau R, Mark DB, et al. The effect of EDTA-based chelation on patients with diabetes and peripheral artery disease in the Trial to Assess Chelation Therapy (TACT). J Diabetes Complicat. 2019;33:490–4.
Arenas IA, Ujueta F, Diaz D, Navas-Acien A, Beasley R, Yates T, et al. Limb preservation using edetate disodium-based chelation in patients with diabetes and critical limb ischemia: an open-label pilot study. Cureus. 2019;11(12):e6477.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Metals and Health
Rights and permissions
About this article

Cite this article
Diaz, D., Ujueta, F., Mansur, G. et al. Low-Level Cadmium Exposure and Atherosclerosis. Curr Envir Health Rpt 8, 42–53 (2021). https://doi.org/10.1007/s40572-021-00304-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40572-021-00304-w