
Abstract
Purpose of the Review
To reason that targeting chaperone-mediated autophagy (CMA) represents a promising approach for disease therapy, we will summarize advances in researches on the relationship between CMA and diseases and discuss relevant strategies for disease therapy by targeting the CMA process.
Recent Findings
CMA is a unique kind of selective autophagy in lysosomes. Under physiological conditions, CMA participates in the maintenance of cellular homeostasis by protein quality control, bioenergetics, and timely regulated specific substrate-associated cellular processes. Under pathological conditions, CMA interplays with various disease conditions. CMA makes adaptive machinery to address stress, while disease-associated proteins alter CMA which is involved in pathogeneses of diseases. As more proteins are identified as CMA substrates and regulators, dysregulation of CMA has been implicated in an increasing number of diseases, while rectifying CMA alteration may be a benefit for these diseases.
Summary
Alterations of CMA in diseases mainly including neurodegenerative diseases and many cancers raise the possibility of targeting CMA to recover cellular homeostasis as one potential strategy for therapy of relevant diseases.
Similar content being viewed by others

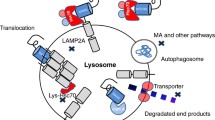
Avoid common mistakes on your manuscript.
Introduction
Chaperone-mediated autophagy (CMA), a lysosome-based catabolic process, occurs to maintain cellular homeostasis by performing catabolic lysis of excessive or unnecessary soluble cytosol proteins and can be activated in response to a variety of stress conditions to recover cellular homeostasis [1, 2]. Under physiological conditions, CMA contributes to the protein quality control by elimination of redundant and damaged proteins, participates in the cellular energetic balance by recycling amino acids, and regulates various cellular processes by degradation of specialized proteins [3]. On the other hand, a decline in CMA has been described in aged brains and neurodegenerative diseases, while the increase in CMA has been found in many cancers where CMA may play a dual role in serving as an activator or a suppressor for tumorigenesis [4]. With more proteins identified as CMA substrates and regulators, CMA has been found to be implicated in increasing disease conditions. Several lines of evidence show that reverse of altered CMA may be a benefit for disease processes, and relevant agents may serve as drug candidates for disease therapy [5]. In this review, in honor of Dr. J Fred Dice, a CMA pioneer, we first describe the basics, evolution, and relativity of CMA, and then, we present the interaction between CMA and either proteolytic pathways briefly or diseases exquisitely, summarize advances in disease therapy by targeting CMA, and discuss relevant strategies.
CMA Basics, Evolution, and Relativity
CMA is an interplayed multistep process to selectively degrade cytosolic proteins with the unique machinery, which is distinct from other two lysosome-based autophagy pathways macroautophagy (MA) and microautophagy (MI) [6]. The unique components for the CMA pathway mainly include the CMA receptor: lysosome-associated membrane protein type 2A (LAMP2A), the chief chaperone: heat shock-cognate protein of 70 kDa (Hsc70) in cytoplasm and lysosome, and substrates: KFERQ-like consensus motif-contained cytosolic proteins [7]. The unique process of CMA initiates from cytosolic Hsc70 binding to substrates through their CMA consensus motifs and guides the targeted substrates to LAMP2A in the lysosomal membrane. Once docked on the lysosomal membrane, the targeted substrates need to be unfolded before they are internalized into the lysosomal lumen in the presence of the lysosomal Hsc70 and other co-chaperone proteins including Hsc90. The substrates are degraded by proteases and hydrolases in the lysosome for the recycle of critical amino acids [7, 8]. During more than two decades until the year 2000, the selective protein degradation (the old name of CMA)-associated research was mainly performed in Dr. Dice’s laboratory where the key components mentioned above for CMA were identified [9]. Since the year 2000, the CMA-related research has been extending to multiple laboratories although most of the output still comes from the labs of Dr. Dice who passed away on January 9, 2010 [10] and of Dr. Cuervo who is a current leader in the CMA field.
Based on the literature available on CMA, we cautiously select and reason ten events as milestones in research for the evolution of the CMA process. The number 1 is to discover the first CMA substrate ribonuclease A (RNase A). In early eighties of last century, Dr. Dice’s team demonstrated that RNase A introduced into the cells with serum was degraded with a half-life of 90 h, whereas in response to serum deprivation, its rate of degradation was increased about 1.6-fold [11], and then, they showed that RNase A could be selectively degraded dependent of its N-terminal 20 amino acids by lysosomes [12]. Therefore, RNase A is the first protein identified as a substrate of CMA [13]. The number 2 is to identify KFERQ-like motif as a signature for CMA substrates. In 1988, the essential motifs related to KFERQ were identified in proteins serving as substrates of this selective degradation pathway, which is referred to as the selective pathway for degradation of cytosolic proteins by lysosomes [14]. About 30% of cytosolic proteins contain such KFERQ-like sequences [15]. The number 3 is to identify cytosolic Hsc70 as a key chaperone protein for CMA. Hsc70 can bind to KFERQ-like regions in cytosolic proteins that are targeted for lysosomal degradation in response to serum deprivation [16]. This is the first chaperone protein as well as the most common one for the CMA process. The number 4 is to identify LAMP2A as a receptor for the CMA process. LAMP2A, also named LGP96 (lysosomal glycoprotein of 96 kDa), serves as a receptor for the selective import and degradation of proteins within lysosomes [17]. The number 5 is to identify intra-lysosomal Hsc70 necessary for the CMA activity. Hsc70 is in the lumen of lysosomes and further determined to be required for the selective pathway of lysosome-mediated protein degradation [18]. The lysosomal Hsc70 is a meter for the CMA activity, and its distribution and level directly reflect the CMA activity and status [6]. The number 6 is to coin the term “chaperone-mediated autophagy” and report the CMA activity decline in aging. In the year 2000, Dr. Dice for the first time used CMA instead of “the lysosome-based selective degradation” to report the CMA decline in aging tissues and cells [9]. Although the CMA-related research and output started in the early eighties of last century, this cellular process is accepted and more popular after 2000, which partially is attributed to this new name. Also, CMA declines along with aging, indicating the potential role of newly coined cellular process in age-related diseases [9]. The number 7 is to determine cathepsin A responsible for the breakdown of LAMP2A. The dynamic change of LAMP2A from monomer to oligomer to degradation is a key step for the CMA activity. To regulate LAMP2A is a key to regulate the CMA activity. Identifying cathepsin A for LAMP2A breakdown launched the first wave of research for the local regulation of CMA [19]. The number 8 is to determine the CMA involved in the pathogenesis of Parkinson’s disease (PD). The two independent Science reports described that α-synuclein is a substrate for CMA and damages CMA to decrease degradation of malfunction neuronal survival factor MEF2D, leading to loss of dopaminergic neurons [20, 21]. This is the first time to report and confirm that the CMA is implicated in a disease. Up to now, the relationship between CMA and PD is still the hottest topic. The number 9 is to identify GFAP and EF1α as two regulators for CMA activity. GFAP can stabilize the LAMP2A-based complex, while the GTP-mediated release of EF1α from the lysosomal membrane leads to self-association of GFAP, disassembly of the LAMP2A complex, and the decrease in CMA activity [22]. Interestingly, GFAP is partially regulated by the lysosomal mTORC2/PHLPP1/Akt axis in which Akt antagonizes the function of GFAP by phosphorylation, indicating that the CMA activity may be regulated by the local and intracellular signal transduction pathway [23]. The number 10 is to identify phosphorylation of LAMP2A by p38 MAPK. This is the first time to demonstrate systematically: (1) the signal pathway from extracellular stress to CMA and (2) the regulation of LAMP2A and CMA by direct phosphorylation of LAMP2A at specific sites in response to endoplasmic reticulum (ER) stress [24]. The authors predict that this regulatory pathway may be involved in the activation of CMA by other stimuli or disease conditions because they interplay with ER stress and unfolded protein response (UPR) pathways. During the last decade, the CMA-associated research has dramatically advanced, and the bunch of high impact factor papers has been published to address other aspects of CMA, especially including several new techniques for CMA. For example, (1) the overexpression of LAMP2A can successfully correct the CMA defect in aged rodents [25], (2) CMA-based technique targets mutant huntingtin protein but not normal one for degradation [26] or other specific endogenous proteins in vitro and in vivo [27], and (3) A photoconvertible fluorescent-based technique tracks CMA to monitor its activity in basal and stress conditions [28]. However, the findings are not to elucidate mechanisms underlying the CMA process directly. Therefore, we only choose the findings highly associated with the mechanism of CMA as the evolutionary events to show a profile of CMA machinery due to the limitation of pages.
CMA is unique and distinct from other two autophagy with its unique characteristics. In our previous review [29], we described the CMA with characteristics of selectivity, saturability, and competitivity. With the progress of research in CMA, the relativity of CMA is emerging. CMA is the interplayed protein cascade process with the multiple steps [1]. The CMA relativity has been shown in the components and each step in CMA. Firstly, CMA substrates are relative, which shows that proteins with classic KFERQ-like motifs may not be suitable while proteins without such motif may be suitable substrates for CMA degradation [21, 30]. The following reasons may interpret this: (1) KFERQ-like motif is not a gold standard to identify CMA substrates. For this, the current criteria to predict CMA substrates may need to be modified via systemically identifying substrates under different conditions and analyzing profile of identified substrates; (2) three-dimension structures of proteins may hide the CMA recognition motif while modification such as phosphorylation and acetylation may make the motifs accessible for Hsc70; and (3) modifications by stress may generate a novel CMA motif in proteins without such a motif and then make them suitable to be degraded via CMA [31]. For example, acetylation can make lysine (K) to glutamine (Q)-like amino acid, leading to a new CMA substrate motif in the protein, which is accessible for recognition by the CMA chaperone protein [32]. Secondly, CMA only was previously found in the mammal cells because relevant studies were done only in the mammal and later extended to birds [3]. New evidence is showing that this kind of autophagy also can be functional in lower species such as fish [33], Drosophila [34], and C. elegans [35]. Thirdly, LAMP2A may not be the only one receptor for CMA with the following reasons. Knockout of LAMP2 does not alter the activity of CMA in the animal [36], and the LAMP2A decrease causes hippocampal dysfunction Danon disease, but lysosome still has a normal CMA function [37]. Fourthly, based on the findings on endosomal-microautophagy (eMI), this kind of autophagy looks like CMA but not MI with two reasons: (1) eMI is dependent of the chaperone Hsc70 to selectively target individual proteins with the KFERQ-like motif in the substrate proteins; and (2) eMI may be dependent of certain unidentified proteins as “receptors” to enclose its substrates to late endosomes [1]. Fifthly, besides more proteins identified as substrates for CMA, RNA and DNA may be suitable substrates for CMA, but this process seems to employ SIDT2 or LAMP2C as receptors [38, 39]. Production and degradation of RNA also are indispensable for cell survival [39]. Abnormal expression of RNA is related to several diseases, and SIDT2 or LAMP2C may be a target to develop new therapies [40]. Sixthly, the decline in CMA in aging is relative. In rat nucleus pulposus, LAMP2A is significantly higher in the 24-month group than in the 3-month group and CMA activity increases in the aging group [41]. Furthermore, LAMP2A increases in cardiac muscle but decreases in skeletal muscle during aging [2]. Finally, MA is the lysosome-mediated bulk degradation of cellular components for material recycling to maintain cellular homeostasis. MA was initially regarded as a nonselective process; however, recent evidence indicates that this process can be highly selective, especially for targeting and degrading organelles, invading pathogens and protein aggregates [42]. Just as mentioned above, part of MI such as eMI is also highly selective. Therefore, the selectivity for CMA is relative in lysosome-based autophagy. Taken together, the relativity stuffs into various aspects of CMA because the relativity is one of CMA characteristics, which may help us more deeply understanding of CMA; on the other hand, more researches need to address corresponding issues raised by the relativity of CMA to effectively regulate and employ CMA for both basic CMA mechanism research and clinic application.
Interplay of CMA with Other Proteolytic Pathways
Protein homeostasis, i.e., proteostasis is necessary for cell health and maintaining various physiological functions of cells, which is determined not only by the rate of protein synthesis but also the rate of degradation [43]. For many decades, scientists have been working to investigate the many reactions and process involved in protein biosynthesis, and until the last decade, more attention has been paid to the complementary processes of protein destruction, i.e., proteolytic pathways [44]. The cell runs a single proteolytic network consisting of the ubiquitin proteasome system (UPS) and the lysosome-based autophagy that shares notable similarity in many aspects and functionally cooperates with each other to maintain proteostasis [45].
Different proteolytic systems are wired through multi-levels of interactions to maintain proteostasis. Without exception, the contribution of CMA to cell health and disease is the fact that CMA does not work independent of other proteolytic systems [46]. As described above, CMA belongs to the lysosome-based autophagy system, which combines with MA and UPS to be responsible for degradation of most proteins in eukaryotic cells [47]. The CMA and MA pathways primarily degrades long-lived/aggregated proteins and cellular organelles, while the UPS is usually responsible for most of proteins, especially short-lived proteins such as cyclins which are quick degraded through a ubiquitin-mediated system for driving cell cycle process [48]. The crosstalk and interplay between CMA and MA/UPS are demonstrated as follows. Firstly, they share specific substrates and regulatory molecules [49]. CMA, MA, and UPS share some same substrates but depending on factors such as the functionality of each proteolytic pathway, cell type, and cellular conditions. For example, α-synuclein can be degraded by these three pathways [46]. Interestingly, one common denominator of MA and UPS is ubiquitination that assembles ubiquitin-based degrons on substrates [50]. As specifically for CMA, whether and how ubiquitination affects the CMA process are worth clarifying. Furthermore, CMA can degrade one MA component ATG5 to regulate the activity of MA [51]. Secondly, CMA can compensate the deficiency of either MA and UPS. The blockage of MA results in the constitutive activation of CMA, while the cells in culture respond to CMA blockage by upregulating MA [52]. But these two pathways are clearly not redundant, as CMA cannot degrade organelles, which can generally be turned over by MA [49, 53]. The inhibition of the UPS also results in the compensatory activation of CMA and MA. Proteasomal inhibition impairs ER-associated degradation (ERAD) but activated PERK and IRE1in the ER subsequently activate downstream pathways toward CMA and MA, respectively [24, 54]. Thirdly, CMA inhibition reduces the UPS activity. In contrast to the bi-directional relationship between CMA and MA, the crosstalk of CMA with the UPS seems to compensate only in one direction. During the acute stages of CMA blockage, there is an accumulation of polyubiquitinated proteins possibly by interfering with the turnover of specific proteasome subunits [52]. Interestingly, the maintenance of CMA efficiency in aging animals can positively preserve the UPS activity [49]. The relevant molecular mechanisms are currently under investigation. But the authors conjecture that CMA and MA may be at downstream of UPS. Finally, the interplay of CMA with CMA and UPS is altered in disease. The constitutive upregulation of CMA compensates for the dual defects of MA and UPS in a mouse model of HD. Activation of CMA in HD is achieved through increasing function of LAMP2A, but the ability of CMA to compensate for the severe proteolytic deficiency dramatically reduces with the progressive functional decline in CMA with age [49, 55]. Synergy between CMA and MA is also crucial in PD where the blockage of CMA is often compensated by activation of MA. This upregulation of MA is critical to remove α-synuclein [56].
Overall, the cross-talk between CMA and MA/UPS is still relatively unexplored, but it emerges as an essential mechanism that cells employs to its advantage during disease conditions. Therefore, understanding molecules and principles that regulate this cross-talk can provide an exciting novel therapeutic strategy to modulate intracellular protein degradation and to maintain protein homeostasis in disease settings. We cannot do worthwhile translational studies without a solid basic science. As proteolysis plays a vital role in various biological processes underlying human diseases [53], more detailed understanding of interplay of CMA with other proteolytic pathways will contribute to the development of therapeutic means to modulate proteostasis and the timely removal of pathogenic protein species with low side effects. In the subsequent sections, we mainly discuss some of the issues that we must deal with if CMA serves as a target for disease therapy. This review is not intended as comprehensive discussion of other proteolysis pathways. There are many such reviews available, and the interested readers are encouraged to read those on roles of MA and UPS in specific diseases as well as the more general ones [45, 47, 49, 53, 57].
Interaction Between CMA and Diseases
CMA can interfere with disease process by degrading disease-associated proteins [58]. Therefore, such substrates easily link the CMA process to disease conditions. The current research for CMA mainly focuses on identifying new substrates and elucidating the relationship between dysregulation of CMA and specific diseases. On the one hand, as more proteins are identified as CMA substrates, CMA functions have been associated with increasing physiological processes [59]. On the other hand, disease-associated factors, especially various proteins, can directly interact with key components LAMP2A and Hsc70 and alter their functions to the activity of CMA [60]. The altered activity of CMA has been implicated in increasing disease pathogeneses. The CMA activity decreases in various neurodegenerative diseases, especially in their late stages, and increases in many cancers and immune diseases.
Parkinson’s Disease
The hottest research on CMA is to investigate the relationship between PD and CMA. PD, the second most common form of neurodegenerative diseases after Alzheimer’s disease (AD), is pathologically characterized by the formation of Lewy bodies and loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNc). The accumulation of the protein α-synuclein in the DA neurons may be caused by impaired CMA, which is further involved in the loss of DA neurons [20]. CMA can interplay with PD: (1) six PD-associated proteins are identified as CMA substrates and (2) five mutant or modified PD-associated proteins may induce a compromise in the CMA activity.
CMA degrades PD-associated proteins as substrates to detoxify them or maintain their functions. α-Synuclein is the first CMA substrate identified among PD-associated proteins. Here, the steps to identify a CMA substrate are briefly described based on α-synuclein. Firstly, in the cell level, inhibition of lysosome activity can prevent degradation of α-synuclein, while induction of CMA can accelerate its degradation. Secondly, using in vitro binding and uptake assays, α-synuclein can bind to lysosomal membranes and transport into the lumen of lysosomes. Thirdly, both Hsc70 and LAMP2A can interact with α-synuclein. Finally, the sequence 95VKKDQ99 mutated into 95VKKAA99 can abolish the degradation of α-synuclein by CMA [20]. Mutations in leucine-rich repeat kinase 2 (LRRK2) are the most common cause of familial PD. Wild-type LRRK2 can be degraded in lysosomes via the CMA pathway, while mutations in LRRK2 can damage the CMA process (see below) [60]. Parkinson disease protein 7 (PARK7) is a ubiquitously expressed protein engaged in various cellular processes. Mutations in the PARK7 are the cause of autosomal recessive early-onset PD. PARK7 plays a key role in antioxidative response, and its dysfunction leads to mitochondrial defects. CMA degrades nonfunctional PARK7 proteins usually at oxidized format to maintain its normal functions mainly including mitochondrial homeostasis [61]. Raf kinase inhibitor protein (RKIP) is a major negative mediator of the ERK/MAPK pathway. In models of PD in vitro and in vivo, CDK5 can phosphorylate RKIP at T42, which facilitates the exposure and recognition of the CMA motif in the C-terminus of RKIP to Hsc70 and the subsequent degradation of RKIP by the CMA process, leading to the release of Raf-1, overactivation of the ERK/MAPK cascade, and neuronal loss [62]. The F-box protein Fbw7 can bind parkin in neurons and collaborate with parkin to regulate the Fbw7 target cyclin E1 for a neuroprotective role. The degradation of nonfunctional Fbw7β is important for neuronal survival, particularly under oxidative conditions. Like CMA substrates MEF2D and PARK7, Fbw7β can be more easily degraded in DA neuronal SN4741 cells treated with 6-hydroxydopamine. Further study shows that oxidized Fbw7β is a more suitable substrate for CMA and increases in postmortem PD brains where CMA is impaired [63].
Although wild-type α-synuclein is efficiently degraded by the CMA process, mutations in α-synuclein are poorly degraded by CMA [20]. Furthermore, mutant α-synucleins can block the lysosomal uptake and degradation of other CMA substrates such as MEF2D and PARK7 [21, 61]. Impaired CMA of pathogenic α-synuclein may promote toxic gains-of-functions by contributing to its modifications or aggregation. Mutant α-synucleins can also reduce the degradation of other long-lived cytosolic proteins, which may further contribute to cellular stress, adaptive response, and even apoptosis. Although pathogenic α-synuclein mutations are rare, wild-type α-synuclein undergoes can also underlie its accumulation in most forms of PD by its posttranslational modifications. Most of the α-synuclein posttranslational modifications impair degradation of this protein by CMA but do not affect degradation of other substrates, but dopamine-modified α-synuclein is not only poorly degraded but also inhibits degradation of other substrates by the CMA process [64]. Like mutations in α-synuclein, G2019S, the most common pathogenic mutant form of LRRK2, is poorly degraded by the CMA pathway. Different from the behavior of typical CMA substrates, lysosomal binding of both wild-type and several pathogenic mutant LRRK2 proteins increases in the presence of other CMA substrates, which interferes with the organization of the CMA translocation complex, leading to damage of CMA. Cells make an adaptive response to such LRRK2-mediated CMA compromise by increasing expression of LAMP2A. The impairment of CMA by mutant LRRK2 underlies the toxicity in PD by blocking the degradation of α-synuclein and other PD-associated proteins degraded by CMA [60]. The I93M mutation in ubiquitin C-terminal hydrolase L1 (UCH-L1) is associated with familial PD. UCH-L1 physically interacts with LAMP2A and Hsc70/Hsp90. These interactions are abnormally enhanced by the I93M mutation, leading to impaired activity of CMA. Therefore, I93M UCH-L1 can induce the CMA inhibition-associated increase in the amount of α-synuclein [65]. Vacuolar protein sorting-35 (VPS35) plays a key role in the endosome-to-Golgi retrieval of membrane proteins. Mutations in VPS35 have been identified in patients with PD. VPS35 is expressed in mouse DA neurons in SNc and striatum which are vulnerable to PD. VPS35-deficient mice show PD-like changes: accumulation of α-synuclein, loss of DA transmitter and neurons, and impairment of behaviors. Mechanical studies show that VPS35-deficient DA neurons or DA neurons expressing VPS35 mutant (D620N) impair endosome-to-Golgi retrieval of LAMP2A and accelerate its degradation, while expression of LAMP2A in VPS35-deficient DA neurons reduces α-synuclein [66]. Mutations in glucosidase beta acid 1 (GBA1) increase the risk of PD. GBA1 mutations confer a 20- to 30-fold increased risk for the PD development. The mechanism by which GBA1 mutations increase the risk for PD is still unknown, but one of the reasons may be that GBA1 deficit in sporadic PD damage the CMA process and cause the abnormal accumulation of α-synuclein [67].
Other Neurodegenerative Diseases
Dysregulation of the CMA activity is also implicated in other neurodegenerative diseases. AD, the most common neurodegenerative disease, is characterized by extracellular amyloid-containing senile and intracellular tau-based neurofibrillary tangle. A decrease in CMA activity has been reported in AD, and several AD-associated proteins have been shown to interplay with CMA [68]. Firstly, in AD patient brain, tau is a core component of neurofibrillary tangles, which start in the memory-associated areas and then march throughout the rest of the brain. Normal tau can be partially degraded by CMA, while mutant tau can bind to LAMP2A and disrupt its lysosomal membrane translocation. Consistently, neurofibrillary tangles increase when CMA is blocked. Together, impaired CMA may be one of the mechanisms underlying the form of neurofibrillary tangles [56, 69]. Secondly, amyloid precursor protein (APP) can be processed to produce β-amyloid (Aβ). The APP contains one KFERQ motif at its C-terminus, and deletion of this motif seems to keep it away from lysosomes, leading to the increase of C-terminal fragments (CTFs) and secreted N-terminal fragments of APP. Interestingly, KFERQ deletion does not reduce the interaction of APP or its cleaved products with Hsc70, but whether APP or its cleaved products are substrates or not for CMA needs to be identified because they contain at least eight KFERQ-like motifs [30]. On the other hand, in intra-hippocampal amyloid β-injected rats, amyloid β can reduce LAMP2A at 10 days after the injection with severe ER stress responses [70]. On the contrary, in human CSF, significantly higher levels of LAMP2 peptides are found in AD patients compared with non-AD controls [71]. Obviously, how the CMA activity changes under AD conditions need to be clarified. Thirdly, regulator of calcineurin 1 (RCAN1), identified in Down syndrome, has been implicated in the AD pathogenesis. RCAN1 expression increases in AD brains possibly by disruption of the CMA process. Mechanism study shows that RCAN1 protein is a typical substrate for CMA, and inhibition of RCAN1 degradation in cells reduces calcineurin-NFAT activity [72].
Huntington’s disease (HD) is caused by the accumulation of huntingtin (Htt) protein with polyQ tract. Aggregation of mutant Htt by aberrant degradation is the main HD pathology in striatal and cortical neurons. KFERQ-like motifs have been identified in the Htt exon 1 fragments that can be targeted by Hsc70 for CMA-mediated degradation. Therefore, wild-type Htt is a substrate for CMA, while mutant Htt protein impairs its uptake and is difficult to the degradation by lysosomes [26]. Encouragingly, increase of CMA activity by overexpression of LAMP2A and Hsc70 still can accelerate the degradation of mutant Htt, which provides therapeutic benefits [73].
Cancer
Altered activity of CMA is closely involved in the pathogenesis of various cancers [4, 74]. The role of CMA in cancer is under debate. Most scientists support the view that CMA promotes cancer development, but increasing findings show that CMA can inhibit cancer by specifically degrading tumor promotors [4, 74]. We collect evidence from both sides to discuss the relationship between cancer and CMA.
CMA positively coordinates with cancer development and promotes tumor growth. Firstly, consistent upregulation of CMA activity and main components has been found in diverse types of cancer cells regardless of the status of MA. CMA is required for cancer cell proliferation in vitro because it contributes to the maintenance of the metabolic alterations in malignant cells [74]. Secondly, CMA compensates for impaired MA in the cirrhotic liver to promote hepatocellular carcinoma [75]. Thirdly, overexpression of LAMP2A in breast cancer and hepatocellular carcinoma promotes cancer development [76]. Fourthly, tumor promoter MCL1 can be degraded by the ubiquitin-proteasome system, while the increase of CMA activity by enhancement of LAMP2A and Hsc70 inhibits the degradation of MCL1 in several lung cell lines, suggesting the existence of a CMA-mediated MCL1 stabilization system in cancer cells [77]. Finally, many tumor suppressors can serve as substrates of CMA, and CMA promotes cancer by degradation of these tumor suppressors, mainly including (1) ATG5 [51], (2) BBC3 [78], (3) cyclin D1 [79], (4) HMGB1 [80], (5) MST1 [81], (6) N-CoR [82], (7) p21Cip1/WAF1 [83], (8) p53 [84], (9) p300/CBP [85], (10) PED [86], and (11) RND3 [87]. The relevant findings are summarized in Table 1.
On the contrary, CMA plays a role of a tumor suppressor for cancer growth. CMA inhibition can cause the increase of total and nuclear MYC, promoting cancer cell proliferation and colony formation. Mechanism study shows that CMA regulates cellular MYC levels by inhibiting its proteasomal degradation [88]. With more lines of evidence, CMA may inhibit cancer growth by degradation of many tumor promoters, mainly include: (1) AF1Q [89], (2) Eps8 [90], (3) Galectin-3 [91], (4) HIF1α [92], (5) Histones H3-H4 [93], (6) HK2 [94], (7) HSD17B4 [95], (8) MDM2 [96], (9) mutant p53 [97], (10) p65 [98], (11) PKM2 [31], and (12) RhoH [99]. The relevant findings are summarized in Table 2.
Neurodegenerative disease-associated proteins can interfere with the key CMA proteins LAMP2A and Hsc70 and then dysregulate the activity of CMA [60]. However, whether and how cancer promoters and suppressors alter the activity of CMA remain enigmatic. The relevant research is still at the infant stage. The activity of CMA upregulates in many cancer cells in response to reactive oxygen species and ER stress possibly caused by tumor microenvironments, especially its metabolites and then contributes to the Warburg effect in cancer cells [51, 76]. Most recently, it has been reported that sorting nexin 10, a tumor suppressor, may promote the degradation of LAMP2A and then inhibit the activity of CMA, which reduce the degradation of p21 while both sorting nexin 10 and p21 are remarkably downregulated in human colorectal cancer [100]. Therefore, the effects of more popular tumor-associated proteins such as p53, PML, and Bcl2 on the CMA process urgently need to be determined to gain a better understanding of the relationship between cancer and CMA.
Immune Disease
Increasing evidence shows that CMA displays an essential role in the homeostasis of immune cells, antigen processing and presentation, and many other immune processes [101, 102]. Engagement of the T cell antigen receptor can induce LAMP2A and activate the CMA process to maintain the activity of T cells by degradation of Itch and RCAN1, two negative regulators of T cell activation. Therefore, defect of CMA activity can impair function of T cells. More importantly, the CMA pathway plays pivotal functions in major histocompatibility complex class II-mediated processing and presentation of several endogenous antigens for their transport from the cytosol to lysosomal compartments and contributes to herpes viral antigen presentation [102, 103]. Interestingly, perturbation of CMA is implicated in the autoimmune disorders such as lupus. LAMP2A expression and CMA activity upregulate in lupus B cells, providing a potential approach for the lupus therapy [103].
Targeting CMA for Disease Therapy
The connection between CMA and different human diseases has motivated a growing interest in understanding this fundamental cellular process and manipulating it for therapeutic purposes. Reduced CMA activity has been found in neurodegenerative diseases such as PD, and in metabolic disorders such as diabetes. Overexpression of LAMP2A in liver can improve the liver function through the reverse of aging-associated CMA decline. Therefore, interventions to improve the function of CMA in old organisms may have therapeutic value in age-related diseases.
Different neurodegenerative diseases share one characteristic: aggregation of deleterious proteins or inclusions in neurons, including α-synuclein in PD, amyloid-β and tau in the AD, and Htt protein in HD [104]. There is a strong link between a decline of CMA and the abnormal aggregation of such proteins in various models of neurodegenerative diseases, suggesting that CMA is a new and promising target for treating these neurodegenerative disorders [42]. In aging, the function of CMA is impaired, causing an inefficient stress response and the accumulation of damaged, oxidized or misfolded proteins, associated with aging-related diseases [105]. Restoration of CMA activity can be a promising therapeutic approach for such human disease conditions in which the upregulation of CMA activity may result in a dual effect: (1) promotion of the degradation of disease aggregation-prone proteins and (2) reverse of their detrimental effects on lysosomal function [106].
Strategy for PD Therapy
CMA impairment in PD has been demonstrated by the lower levels of the CMA markers LAMP2A and the chaperone Hsc70 in various regions of the PD brain compared to controls, while a vital role for the CMA process is proposed in the degradation of wild-type α-synuclein [107]. Indeed, in models of PD in vitro and in vivo, overexpression of LAMP2A in SH-SY5Y cells and rat primary cultured cortical neurons can increase the activity of CMA, decrease levels of α-synuclein, and protect cells against α-synuclein neurotoxicity [108]; and most importantly, LAMP2A overexpression in the rat substantia nigra effectively ameliorates dopaminergic neurodegeneration and increases the survival of nigral neurons [109]. Interestingly, the neuroprotective activity of CMA can be observed before the level of α-synuclein is unchanged, indicating that maintaining the function of CMA may be more important than the reduction of α-synuclein for PD therapy and even for other synucleinopathies such as dementia with Lewy bodies and multiple system atrophy [109]. Together, modulation of LAMP2A for CMA represents a therapeutic target for PD and other synucleinopathies in which α-synuclein accumulation and aggregation play a fundamental role.
Strategy for AD Therapy
The adaptor peptide-based CMA strategy has been used to knock down α-synuclein successfully [92]. According to this, the CMA strategy is proposed to be a powerful approach to regulate amyloid protein level and may become a promising therapy for the AD [110]. Resveratrol, a kind of polyphenol rich in red wine, can directly interfere with toxic β-amyloid protein (Aβ) aggregation. Using the AD model of transgenic C. elegans strain CL2006, expressing Aβ1–42 under control of a muscle-specific promoter and responding to Aβ-toxicity with paralysis, resveratrol can reduce paralysis by accelerating degradation of Aβ partially through a CMA-dependent manner, indicating the two things: (1) CMA may occur in the simple organisms besides mammals and (2) C. elegans can be used as a model to screen drug candidates for AD by targeting CMA [111].
Strategy for HD Therapy
Experimental upregulation of CMA has also been proven to be beneficial in HD models, although the activity of CMA compensatively increases in HD animals compared with control ones [26, 112, 113]. The phosphorylation of mutant Htt facilitates its clearance via CMA reduced Htt-mediated toxicity in rat brain slice cultures [114]. Also, the adaptor peptide, consisting of two parts: two HSC70-binding motifs and one interacting peptide for one targeted interesting protein, provides a general way to target a misfolded protein for degradation by CMA specifically. Mutant Htt contains an expanded polyglutamine (polyQ) tract. Interestingly, the polyglutamine binding peptide 1 (QBP1) can bind an expanded polyQ tract but not the polyQ motif in normal Htt proteins. Therefore, QBP1 can be used for selectively targeting mutant Htt for degradation by the CMA process. Notably, the delivery of a virus expressing the fusion peptide ameliorates the disease phenotype in the mouse model of HD [26].
Strategy for Cancer Therapy
In contract to neurodegenerative diseases, abnormal upregulation of CMA activity is common in many types of cancer [74] and also contributes to the pathogenesis of immune disorders such as lupus [115]. Inhibition of CMA activity may be of use as anti-cancer therapy. However, with extensive research for CMA, the alteration of CMA activity is not consistent in a kind of diseases, even in an individual disease [112, 113]. Indeed, it has been reported that the activation of CMA may be a benefit in therapy for some cancers [83, 98].
Although a dual role for CMA either protecting against or promoting cell death in cancer has been reported, targeting CMA for cancer therapy still seems to be promising [4]. The therapeutic role of CMA in diseases deserves to be investigated intensively and continuously regarding the field of oncology. Inhibition of CMA activity may be a potential therapeutic approach for cancers because CMA is consistently upregulated in many cancers and is required for optimal tumor growth and metastasis. Indeed, selective inhibition of CMA by knockdown of LAMP2A can reduce tumor growth, cause tumor cell death, and promote the regression of existing tumors [74]. Also, neutralization of lysosomal pH by chloroquine, currently used in anticancer clinical trials, exerts its effect in part by compromising CMA [112]. Drug resistance in cancer cells may be overcome through inhibition of CMA; therefore, targeting CMA provides a promising therapeutic strategy to enhance the effect of other anticancer therapies and to circumvent drug resistance [85]. However, the challenge remains in designing more selective small molecules to compromise CMA activity without affecting the other types of autophagy because the anti-MA may cause tumorigenesis and metastasis in several cancer conditions (51).
On the contrary, MA inhibition by spautin-1 can lead to the activation of CMA to mediate the degradation of mutant p53 and oncogenic HK2 which may display dominant oncogenic activities [116]. Furthermore, CMA activation induces the death of nonproliferating quiescent cancer cells, while normal cells are spared. For this, inducing CMA activation may be an approach for anticancer therapy [116]. Maximal CMA activation requires a combination of nutritional stress and a blockade of autophagy, which also may cause many off-target side effects. However, selective activation of CMA may be considered as a novel therapeutic strategy for the removal of pathological mutant proteins involved in specific human cancers [116, 117].
Strategy for Lupus Therapy
We give a special focus on CMA as a therapeutic target for the autoimmune disease. P140, one 21-amino acid peptide generated from the protein U1-70K (130-150aa), can target CMA as an inhibitor for lupus therapy [103]. In the mouse model of lupus, the lysosomal compartment expands, and the two key CMA components LAMP2A and Hsc70 dramatically increase in the splenic B cells, supporting the hyperactivation of CMA in lupus. The inhibitory effect of P140 on CMA is likely tied to its ability to alter the composition of Hsc70/LAMP2A heterocomplexes. P140 uses the clathrin-dependent endo-lysosomal pathway to enter B lymphocytes and accumulates in the lysosomal lumen where it may directly hamper lysosomal Hsc70 chaperoning functions and destabilize LAMP2A in lysosomes [102]. These effects may interfere with the endogenous autoantigen processing and present to primary histocompatibility complex class II molecules, inhibiting the activation of autoreactive T cells. Based on the unique mechanism of action on CMA, Lupuzor, the commercial name of P140, is in phase III clinical trials as a drug for lupus. According to the results from completing successful phases I and II, and initial phase III studies, Lupuzor displays protective activities in MRL/lpr lupus-prone mice and more importantly in lupus patients without adverse side effects [118].
Drug Candidates Targeting CMA
For the therapy purpose, dysregulated CMA substrates may be a more exciting target for the treatment of specific diseases. Therefore, how to regulate altered CMA for individual substrates is one of the hottest fields for CMA. For example, the CMA adaptor molecules increase the degradation of Htt protein or α-synuclein in animal models [26, 27]. But it is more popular that genetic and peptide/chemical-based approaches have been employed to rectify CMA failure for disease therapy. Yet, few drugs that modulate CMA exist and even these available are nonspecific and have pleiotropic effects. Encouragingly, Cuervo and her colleagues report RARα inhibitor and humanin can activate CMA and provide a kind of cellular protective activity against oxidative stress or proteotoxicity. Both enhance CMA activity by explicitly targeting the CMA machinery and may act as promising drug candidates for related diseases.
RARα Inhibitors
Signaling through retinoic acid receptor alpha (RARα) inhibits CMA activity. Synthetic derivatives of all-trans-retinoic acid can specifically neutralize this inhibitory effect. Such chemical enhancement of CMA can protect cells from oxidative stress and proteotoxicity, supporting a potential therapeutic opportunity when reduced CMA contributes to cellular dysfunction and disease. The protective effect against oxidation and proteotoxicity observed in response to the retinoid derivatives supports a possible therapeutic potential of these or related compounds in chronic age-related diseases. The efficient upregulation of CMA observed with the retinoid derivatives, and their lack of noticeable effects on MA makes them suitable for the selective modulation of CMA [119].
Humanin
Humanin/its analogs work as activators to increase the activity of CMA by antagonizing endogenous CMA inhibitors or agonizing interaction between CMA chaperone HSP90 and the CMA receptor LAMP2A. Such CMA activators have been demonstrated to have multiple protective effects against various stresses. Humanin and its analogs are potential drug candidates for diseases such as neurodegenerative diseases where CMA is at the low level [5].
Ketone
Ketone bodies consist of three compounds: β-hydroxybutyrate, acetoacetate, and acetone. During especially long-term starvation, oxidation of free fatty acids by liver generates ketone bodies, which release to the circulation system for energy usage by skeletal muscle and brain [120]. Interestingly, ketone bodies can induce CMA. Physiological concentrations of β-hydroxybutyrate and acetoacetate increase proteolysis in cells. Lysosomes from β-hydroxybutyrate-treated cells display an increased ability to degrade substrates for CMA. Such treatment does not affect the levels of LAMP2A or Hsc70. However, pretreatment of CMA substrates with β-hydroxybutyrate increase their rate of degradation by isolated lysosomes, suggesting that β-hydroxybutyrate acts on the substrates to increase their rates of proteolysis [121]. Consistently, lifestyle interventions by modulating ketone bodies, energy intake by caloric restriction, and energy expenditure by exercise can enhance brain health possibly by activation of the CMA process [122].
Conclusion and Remarks
CMA makes an adaptive response to address various disease-associated stresses; therefore, it is beneficial or vicious dependent of specific individual diseases. The significance of CMA in disease depends on cell types such as neurons versus cancer cells where CMA has protective activities but with different meanings and on disease conditions where CMA is downregulated versus upregulated. Based on the literature available on CMA, we draw a graph (Fig. 1) to interpret the status of CMA activity under basal and stress conditions in organisms from young (normal) to old: CMA positively (such as starvation where the capacity of CMA increases without increase of substrates) or passively (such as ER stress where the capacity of CMA increases following increase of substrates) addresses change of cellular microenvironments to restore an old cell homeostasis or establish a new cell homeostasis, but this capacity of CMA declines with high dietary, aging, or neurodegenerative disease while abnormally increases in many cancers.

The Yin-Yang theory for CMA status under different conditions. The Yin-Yang theory is a kind of philosophy to view the relationship between two opposite things in the system, profoundly rooting into the Traditional Chinese Medicine in which the healthy human body is a system with the balance between Yin and Yang, and the imbalance leads to disease while restoring balance can cure the disease. The CMA system at the philosophy level consists of CMA substrates (Yin) and CMA capacity (Yang). Under physiological condition, the CMA capacity matches the amount of substrate proteins, meaning a balance between Yin and Yang, i.e., homeostasis. Under pathological conditions, the CMA capacity declines with high dietary and aging, and the aging-associated decline of CMA capacity is further impaired in neurodegenerative disease where Yang is much less than Yin, while the CMA capacity increases under starvation and in many cancers where Yang is more than Yin, but in ER stress where Yang is still less than Yin. Under both pathological conditions, CMA substrates (Yin) and capacity (Yang) depend on and fight each other, leading to either balance or imbalance between Yin and Yang. For starvation, ER stress, and high dietary, the imbalance of CMA system is reversible, while for cancer and aging/neurodegenerative disease, the imbalance of CMA system is irreversible, which needs an intervention to establish a new balance. Note: the sizes of signs represent the amount of substrate proteins and the capacity of CMA, respectively
Deep understanding of CMA and regulation is a key to how to target CMA for disease therapy. Up to date, except Lupuzor, no drugs targeting CMA are available for specific diseases. Even for Lupuzor, its therapeutic effects on lupus are reported much earlier than its mechanism of action on CMA [102], which means that CMA is inadvertently manipulated for the therapeutic purpose. Nowadays, proposed targeting CMA therapy is only based on partially understanding of CMA and its interplay with a disease, which may mislead the development of drugs. The insufficiencies for CMA-associated research is evident as follows: (1) no research is employed to systematically identify CMA substrates in both basal and specific stress conditions; (2) the techniques and methods for CMA are relatively old and indirect, and no cutting-edge techniques such as crystal-EM-based method are used for CMA-associated research. CMA is one of the hottest research fields and urgently need more highly-expertise scientists to join this field. Furthermore, many open outstanding questions on CMA still are pending to be answered: (1) how does CMA identify its substrates for degradation? More specifically, how do the CMA chaperone proteins distinguish the malfunction substrates from functional ones? In the cancer-associated findings, CMA seems to degrade functional tumor promoters or suppressors but not their damaged forms, leading to reduction or loss of their functions. Based on the definition, the CMA only degrades damaged (misfolding or malfunction) proteins, but why CMA breaks down functional proteins in cancer instead? (2) what are differences of the molecular pathways regulating CMA among diverse types of cells and tissues under stress? The answer to this may help us to establish individual therapies; (3) why do modifications make substrates more readily degraded by CMA or non-substrates to substrates, or reversed? The answer to this may help us to manipulate substrates for therapy purposes; (4) what are crystal-EM structures of Hsc70 and LAMP2A with/without a substrate under basal and activated conditions? The relevant results will help us to manipulate the CMA process more efficiently; (5) what mechanisms and signal pathway underlie compensation and cross-talk between CMA and MA? and (6) what strategies can be employed to induce CMA without activating MA or other responses? Many stresses can activate MA and CMA simultaneously or sequentially. Furthermore, MA and CMA compensate each other when one of them is compromised [51]. Although targeting extracellular stress or intracellular upstream signal by chemicals can activate the CMA process, such broad effects may interfere with the MA process and cause severe responses to destroy the cells. Both types of autophagy may share a same upstream signal pathway, but they should have their own specific downstream signal pathways. For example, ER stress can activate both CMA and MA, but it employs PERK and IRE1 of three central UPR transducers to trigger CMA and MA, respectively [24, 54]. The manipulation of downstream CMA components, rather than broad CMA stimulation, may be an attractive strategy for the development of novel therapies in the CMA-associated diseases. In addition, targeting CMA substrates rather than its key components LAMP2A and Hsc70 may be another useful strategy to harness CMA for therapeutic purposes by selective degradation of targeted proteins [27]. More importantly, further characterizing the dysfunctional CMA in specific stages and molecular subtypes of diseases in combination with clinical drugs may pave an exciting new avenue for the CMA-based therapy because the altered CMA may be only one of many contributors to the diseases [123].
References
Tekirdag KA, Cuervo AM. Chaperone-mediated autophagy and endosomal microautophagy: joint by a chaperone. J Biol Chem. 2017:jbc.R117.818237.
Zhou J, Chong SY, Lim A, Singh BK, Sinha RA, Salmon AB, et al. Changes in macroautophagy, chaperone-mediated autophagy, and mitochondrial metabolism in murine skeletal and cardiac muscle during aging. Aging (Albany NY). 2017;9(2):583–99.
Patel B, Cuervo AM. Methods to study chaperone-mediated autophagy. Methods. 2015;75:133–40.
Tang Y, Wang XW, Liu ZH, Sun YM, Tang YX, Zhou DH. Chaperone-mediated autophagy substrate proteins in cancer. Oncotarget. 2017;8(31):51970–85.
Gong Z, Tasset I, Diaz A, Anguiano J, Tas E, Cui L, et al. Humanin is an endogenous activator of chaperone-mediated autophagy. J Cell Biol. 2017;
Bejarano E, Cuervo AM. Chaperone-mediated autophagy. Proc Am Thorac Soc. 2010;7(1):29–39.
Dice JF. Chaperone-mediated autophagy. Autophagy. 2007;3(4):295–9.
Arias E. Methods to study chaperone-mediated autophagy. Methods Enzymol. 2017;588:283–305.
Cuervo AM, Dice JF. Age-related decline in chaperone-mediated autophagy. J Biol Chem. 2000;275(40):31505–13.
Cuervo AM. Chaperone-mediated autophagy: Dice's 'wild' idea about lysosomal selectivity. Nat Rev Mol Cell Biol. 2011;12(8):535–41.
Dice JF. Altered degradation of proteins microinjected into senescent human fibroblasts. J Biol Chem. 1982;257(24):14624–7.
Backer JM, Bourret L, Dice JF. Regulation of catabolism of microinjected ribonuclease a requires the amino-terminal 20 amino acids. Proc Natl Acad Sci U S A. 1983;80(8):2166–70.
McElligott MA, Dice JF. Degradation of microinjected ribonuclease a and ribonuclease S-protein by lysosomal pathways. Prog Clin Biol Res. 1985;180:471–3.
Dice JF, Chiang HL, Spencer EP, Backer JM. Regulation of catabolism of microinjected ribonuclease a. Identification of residues 7-11 as the essential pentapeptide. J Biol Chem. 1986;261(15):6853–9.
Dice JF, Chiang HL. Peptide signals for protein degradation within lysosomes. Biochem Soc Symp. 1989;55:45–55.
Chiang HL, Terlecky SR, Plant CP, Dice JF. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science. 1989;246(4928):382–5.
Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273(5274):501–3.
Agarraberes FA, Terlecky SR, Dice JF. An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J Cell Biol. 1997;137(4):825–34.
Cuervo AM, Mann L, Bonten EJ, d'Azzo A, Dice JF. Cathepsin a regulates chaperone-mediated autophagy through cleavage of the lysosomal receptor. EMBO J. 2003;22(1):47–59.
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305(5688):1292–5.
Yang Q, She H, Gearing M, Colla E, Lee M, Shacka JJ, et al. Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science. 2009;323(5910):124–7.
Bandyopadhyay U, Sridhar S, Kaushik S, Kiffin R, Cuervo AM. Identification of regulators of chaperone-mediated autophagy. Mol Cell. 2010;39(4):535–47.
Arias E, Koga H, Diaz A, Mocholi E, Patel B, Cuervo AM. Lysosomal mTORC2/PHLPP1/Akt regulate chaperone-mediated autophagy. Mol Cell. 2015;59(2):270–84.
Li W, Zhu J, Dou J, She H, Tao K, Xu H, et al. Phosphorylation of LAMP2A by p38 MAPK couples ER stress to chaperone-mediated autophagy. Nat Commun. 2017;8(1):1763.
Zhang C, Cuervo AM. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med. 2008;14(9):959–65.
Bauer PO, Goswami A, Wong HK, Okuno M, Kurosawa M, Yamada M, et al. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat Biotechnol. 2010;28(3):256–63.
Fan X, Jin WY, Lu J, Wang J, Wang YT. Rapid and reversible knockdown of endogenous proteins by peptide-directed lysosomal degradation. Nat Neurosci. 2014;17(3):471–80.
Koga H, Martinez-Vicente M, Macian F, Verkhusha VV, Cuervo AM. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat Commun. 2011;2:386.
Li W, Yang Q, Mao Z. Chaperone-mediated autophagy: machinery, regulation and biological consequences. Cell Mol Life Sci. 2011;68(5):749–63.
Park JS, Kim DH, Yoon SY. Regulation of amyloid precursor protein processing by its KFERQ motif. BMB Rep. 2016;49(6):337–42.
Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell. 2011;42(6):719–30.
Bonhoure A, Vallentin A, Martin M, Senff-Ribeiro A, Amson R, Telerman A, et al. Acetylation of translationally controlled tumor protein promotes its degradation through chaperone-mediated autophagy. Eur J Cell Biol. 2017;96(2):83–98.
Yabu T, Imamura S, Mohammed MS, Touhata K, Minami T, Terayama M, et al. Differential gene expression of HSC70/HSP70 in yellowtail cells in response to chaperone-mediated autophagy. FEBS J. 2011;278(4):673–85.
Mukherjee A, Patel B, Koga H, Cuervo AM, Jenny A. Selective endosomal microautophagy is starvation-inducible in Drosophila. Autophagy. 2016;12(11):1984–99.
Eisermann DJ, Wenzel U, Fitzenberger E. Inhibition of chaperone-mediated autophagy prevents glucotoxicity in the Caenorhabditis elegans mev-1 mutant by activation of the proteasome. Biochem Biophys Res Commun. 2017;484(1):171–5.
Eskelinen EL, Schmidt CK, Neu S, Willenborg M, Fuertes G, Salvador N, et al. Disturbed cholesterol traffic but normal proteolytic function in LAMP-1/LAMP-2 double-deficient fibroblasts. Mol Biol Cell. 2004;15(7):3132–45.
Rothaug M, Stroobants S, Schweizer M, Peters J, Zunke F, Allerding M, et al. LAMP-2 deficiency leads to hippocampal dysfunction but normal clearance of neuronal substrates of chaperone-mediated autophagy in a mouse model for Danon disease. Acta Neuropathol Commun. 2015;3:6.
Aizawa S, Contu VR, Fujiwara Y, Hase K, Kikuchi H, Kabuta C, et al. Lysosomal membrane protein SIDT2 mediates the direct uptake of DNA by lysosomes. Autophagy. 2017;13(1):218–22.
Aizawa S, Fujiwara Y, Contu VR, Hase K, Takahashi M, Kikuchi H, et al. Lysosomal putative RNA transporter SIDT2 mediates direct uptake of RNA by lysosomes. Autophagy. 2016;12(3):565–78.
Fujiwara Y, Hase K, Wada K, Kabuta T. An RNautophagy/DNautophagy receptor, LAMP2C, possesses an arginine-rich motif that mediates RNA/DNA-binding. Biochem Biophys Res Commun. 2015;460(2):281–6.
Ye W, Xu K, Huang D, Liang A, Peng Y, Zhu W, et al. Age-related increases of macroautophagy and chaperone-mediated autophagy in rat nucleus pulposus. Connect Tissue Res. 2011;52(6):472–8.
Wu MY, Song JX, Wang SF, Cai CZ, Li M, Lu JH. Selective autophagy: the new player in the fight against neurodegenerative diseases? Brain Res Bull. 2017;137:79–90.
Sala AJ, Bott LC, Morimoto RI. Shaping proteostasis at the cellular, tissue, and organismal level. J Cell Biol. 2017;216(5):1231–41.
Chen Y, Klionsky DJ. The regulation of autophagy - unanswered questions. J Cell Sci. 2011;124(Pt 2):161–70.
Ji CH, Crosstalk KYT. Interplay between the ubiquitin-proteasome system and autophagy. Mol Cells. 2017;40(7):441–9.
Park C, Cuervo AM. Selective autophagy: talking with the UPS. Cell Biochem Biophys. 2013;67(1):3–13.
Wong E, Cuervo AM. Integration of clearance mechanisms: the proteasome and autophagy. Cold Spring Harb Perspect Biol. 2010;2(12):a006734.
Duronio RJ, Xiong Y. Signaling pathways that control cell proliferation. Cold Spring Harb Perspect Biol. 2013;5(3):a008904.
Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 2014;24(1):92–104.
Grumati P, Dikic I. Ubiquitin signaling and autophagy. J Biol Chem. 2017:jbc.TM117.000117.
Han Q, Deng Y, Chen S, Chen R, Yang M, Zhang Z, et al. Downregulation of ATG5-dependent macroautophagy by chaperone-mediated autophagy promotes breast cancer cell metastasis. Sci Rep. 2017;7(1):4759.
Massey AC, Follenzi A, Kiffin R, Zhang C, Cuervo AM. Early cellular changes after blockage of chaperone-mediated autophagy. Autophagy. 2008;4(4):442–56.
Kaushik S, Cuervo AM. Proteostasis and aging. Nat Med. 2015;21(12):1406–15.
Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26(24):9220–31.
Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci. 2010;13(5):567–76.
Wang Y, Martinez-Vicente M, Kruger U, Kaushik S, Wong E, Mandelkow EM, et al. Synergy and antagonism of macroautophagy and chaperone-mediated autophagy in a cell model of pathological tau aggregation. Autophagy. 2010;6(1):182–3.
Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010;584(7):1393–8.
Xilouri M, Brekk OR, Kirik D, Stefanis L. LAMP2A as a therapeutic target in Parkinson disease. Autophagy. 2013;9(12):2166–8.
Catarino S, Pereira P, Girao H. Molecular control of chaperone-mediated autophagy. Essays Biochem. 2017;61(6):663–74.
Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci. 2013;16(4):394–406.
Wang B, Cai Z, Tao K, Zeng W, Lu F, Yang R, et al. Essential control of mitochondrial morphology and function by chaperone-mediated autophagy through degradation of PARK7. Autophagy. 2016;12(8):1215–28.
Wen Z, Shu Y, Gao C, Wang X, Qi G, Zhang P, et al. CDK5-mediated phosphorylation and autophagy of RKIP regulate neuronal death in Parkinson's disease. Neurobiol Aging. 2014;35(12):2870–80.
Wang X, Zhai H, Wang F. 6-OHDA induces oxidation of F-box protein Fbw7beta by chaperone-mediated autophagy in Parkinson's model. Mol Neurobiol. 2017;
Martinez-Vicente M, Talloczy Z, Kaushik S, Massey AC, Mazzulli J, Mosharov EV, et al. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest. 2008;118(2):777–88.
Kabuta T, Furuta A, Aoki S, Furuta K, Wada K. Aberrant interaction between Parkinson disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J Biol Chem. 2008;283(35):23731–8.
Tang FL, Erion JR, Tian Y, Liu W, Yin DM, Ye J, et al. VPS35 in dopamine neurons is required for endosome-to-Golgi retrieval of Lamp2a, a receptor of chaperone-mediated autophagy that is critical for alpha-Synuclein degradation and prevention of pathogenesis of Parkinson's disease. J Neurosci. 2015;35(29):10613–28.
Migdalska-Richards A, Schapira AH. The relationship between glucocerebrosidase mutations and Parkinson disease. J Neurochem. 2016;139 Suppl 1:77–90.
Wang Y, Martinez-Vicente M, Kruger U, Kaushik S, Wong E, Mandelkow EM, et al. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet. 2009;18(21):4153–70.
Caballero B, Wang Y, Diaz A, Tasset I, Juste YR, Stiller B, et al. Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell. 2018;17(1):e12692.
Khodagholi F, Digaleh H, Motamedi F, Foolad F, Shaerzadeh F. Nitric oxide and protein disulfide isomerase explain the complexities of unfolded protein response following intra-hippocampal Abeta injection. Cell Mol Neurobiol. 2016;36(6):873–81.
Sjodin S, Ohrfelt A, Brinkmalm G, Zetterberg H, Blennow K, Brinkmalm A. Targeting LAMP2 in human cerebrospinal fluid with a combination of immunopurification and high resolution parallel reaction monitoring mass spectrometry. Clin Proteomics. 2016;13:4.
Liu H, Wang P, Song W, Sun X. Degradation of regulator of calcineurin 1 (RCAN1) is mediated by both chaperone-mediated autophagy and ubiquitin proteasome pathways. FASEB J. 2009;23(10):3383–92.
Qi L, Zhang XD, Wu JC, Lin F, Wang J, DiFiglia M, et al. The role of chaperone-mediated autophagy in huntingtin degradation. PLoS One. 2012;7(10):e46834.
Kon M, Kiffin R, Koga H, Chapochnick J, Macian F, Varticovski L, et al. Chaperone-mediated autophagy is required for tumor growth. Sci Transl Med. 2011;3(109):109ra17.
Chava S, Lee C, Aydin Y, Chandra PK, Dash A, Chedid M, et al. Chaperone-mediated autophagy compensates for impaired macroautophagy in the cirrhotic liver to promote hepatocellular carcinoma. Oncotarget. 2017;8(25):40019–36.
Saha T. LAMP2A overexpression in breast tumors promotes cancer cell survival via chaperone-mediated autophagy. Autophagy. 2012;8(11):1643–56.
Suzuki J, Nakajima W, Suzuki H, Asano Y, Tanaka N. Chaperone-mediated autophagy promotes lung cancer cell survival through selective stabilization of the pro-survival protein, MCL1. Biochem Biophys Res Commun. 2017;482(4):1334–40.
Xie W, Zhang L, Jiao H, Guan L, Zha J, Li X, et al. Chaperone-mediated autophagy prevents apoptosis by degrading BBC3/PUMA. Autophagy. 2015;11(9):1623–35.
Guo B, Li L, Guo J, Liu A, Wu J, Wang H, et al. M2 tumor-associated macrophages produce interleukin-17 to suppress oxaliplatin-induced apoptosis in hepatocellular carcinoma. Oncotarget. 2017;8(27):44465–76.
Wu JH, Guo JP, Shi J, Wang H, Li LL, Guo B, et al. CMA down-regulates p53 expression through degradation of HMGB1 protein to inhibit irradiation-triggered apoptosis in hepatocellular carcinoma. World J Gastroenterol. 2017;23(13):2308–17.
Li L, Fang R, Liu B, Shi H, Wang Y, Zhang W, et al. Deacetylation of tumor-suppressor MST1 in hippo pathway induces its degradation through HBXIP-elevated HDAC6 in promotion of breast cancer growth. Oncogene. 2016;35(31):4048–57.
Ali AB, Nin DS, Tam J, Khan M. Role of chaperone mediated autophagy (CMA) in the degradation of misfolded N-CoR protein in non-small cell lung cancer (NSCLC) cells. PLoS One. 2011;6(9):e25268.
Zhang S, Hu B, You Y, Yang Z, Liu L, Tang H, et al. Sorting nexin 10 acts as a tumor suppressor in tumorigenesis and progression of colorectal cancer through regulating chaperone mediated autophagy degradation of p21(Cip1/WAF1). Cancer Lett. 2018.
Aydin Y, Chatterjee A, Chandra PK, Chava S, Chen W, Tandon A, et al. Interferon-alpha-induced hepatitis C virus clearance restores p53 tumor suppressor more than direct-acting antivirals. Hepatol Commun. 2017;1(3):256–69.
Du C, Huang D, Peng Y, Yao Y, Zhao Y, Yang Y, et al. 5-fluorouracil targets histone acetyltransferases p300/CBP in the treatment of colorectal cancer. Cancer Lett. 2017;400:183–93.
Quintavalle C, Di Costanzo S, Zanca C, Tasset I, Fraldi A, Incoronato M, et al. Phosphorylation-regulated degradation of the tumor-suppressor form of PED by chaperone-mediated autophagy in lung cancer cells. J Cell Physiol. 2014;229(10):1359–68.
Zhou J, Yang J, Fan X, Hu S, Zhou F, Dong J, et al. Chaperone-mediated autophagy regulates proliferation by targeting RND3 in gastric cancer. Autophagy. 2016;12(3):515–28.
Gomes LR, Menck CFM, Cuervo AM. Chaperone-mediated autophagy prevents cellular transformation by regulating MYC proteasomal degradation. Autophagy. 2017;13(5):928–40.
Li P, Ji M, Lu F, Zhang J, Li H, Cui T, et al. Degradation of AF1Q by chaperone-mediated autophagy. Exp Cell Res. 2014;327(1):48–56.
Welsch T, Younsi A, Disanza A, Rodriguez JA, Cuervo AM, Scita G, et al. Eps8 is recruited to lysosomes and subjected to chaperone-mediated autophagy in cancer cells. Exp Cell Res. 2010;316(12):1914–24.
Li X, Ma Q, Wang J, Liu X, Yang Y, Zhao H, et al. C-Abl and Arg tyrosine kinases regulate lysosomal degradation of the oncoprotein Galectin-3. Cell Death Differ. 2010;17(8):1277–87.
Hubbi ME, Hu H, Kshitiz AI, Levchenko A, Semenza GL. Chaperone-mediated autophagy targets hypoxia-inducible factor-1alpha (HIF-1alpha) for lysosomal degradation. J Biol Chem. 2013;288(15):10703–14.
Cook AJ, Gurard-Levin ZA, Vassias I, Almouzni G. A specific function for the histone chaperone NASP to fine-tune a reservoir of soluble H3-H4 in the histone supply chain. Mol Cell. 2011;44(6):918–27.
Xia HG, Najafov A, Geng J, Galan-Acosta L, Han X, Guo Y, et al. Degradation of HK2 by chaperone-mediated autophagy promotes metabolic catastrophe and cell death. J Cell Biol. 2015;210(5):705–16.
Zhang Y, Xu YY, Yao CB, Li JT, Zhao XN, Yang HB, et al. Acetylation targets HSD17B4 for degradation via the CMA pathway in response to estrone. Autophagy. 2017;13(3):538–53.
Lu TL, Huang GJ, Wang HJ, Chen JL, Hsu HP, Lu TJ. Hispolon promotes MDM2 downregulation through chaperone-mediated autophagy. Biochem Biophys Res Commun. 2010;398(1):26–31.
Vakifahmetoglu-Norberg H, Kim M, Xia HG, Iwanicki MP, Ofengeim D, Coloff JL, et al. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013;27(15):1718–30.
Tang J, Zhan MN, Yin QQ, Zhou CX, Wang CL, Wo LL, et al. Impaired p65 degradation by decreased chaperone-mediated autophagy activity facilitates epithelial-to-mesenchymal transition. Oncogene. 2017;6(10):e387.
Troeger A, Chae HD, Senturk M, Wood J, Williams DA. A unique carboxyl-terminal insert domain in the hematopoietic-specific, GTPase-deficient rho GTPase RhoH regulates post-translational processing. J Biol Chem. 2013;288(51):36451–62.
Zhang S, Hu B, You Y, Yang Z, Liu L, Tang H, et al. Sorting nexin 10 acts as a tumor suppressor in tumorigenesis and progression of colorectal cancer through regulating chaperone mediated autophagy degradation of p21(Cip1/WAF1). Cancer Lett. 2018;419:116–27.
Lu W, Zhang Y, McDonald DO, Jing H, Carroll B, Robertson N, et al. Dual proteolytic pathways govern glycolysis and immune competence. Cell. 2014;159(7):1578–90.
Macri C, Wang F, Tasset I, Schall N, Page N, Briand JP, et al. Modulation of deregulated chaperone-mediated autophagy by a phosphopeptide. Autophagy. 2015;11(3):472–86.
Wang F, Muller S. Manipulating autophagic processes in autoimmune diseases: a special focus on modulating chaperone-mediated autophagy, an emerging therapeutic target. Front Immunol. 2015;6:252.
Ciechanover A, Kwon YT. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med. 2015;47:e147.
Kikis EA, Gidalevitz T, Morimoto RI. Protein homeostasis in models of aging and age-related conformational disease. Adv Exp Med Biol. 2010;694:138–59.
Xilouri M, Brekk OR, Polissidis A, Chrysanthou-Piterou M, Kloukina I, Stefanis L. Impairment of chaperone-mediated autophagy induces dopaminergic neurodegeneration in rats. Autophagy. 2016;12(11):2230–47.
Murphy KE, Gysbers AM, Abbott SK, Spiro AS, Furuta A, Cooper A, et al. Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson's disease. Mov Disord. 2015;30(12):1639–47.
Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem. 2008;283(35):23542–56.
Xilouri M, Brekk OR, Landeck N, Pitychoutis PM, Papasilekas T, Papadopoulou-Daifoti Z, et al. Boosting chaperone-mediated autophagy in vivo mitigates alpha-synuclein-induced neurodegeneration. Brain. 2013;136(Pt 7):2130–46.
Gao N, Chen YX, Zhao YF, Li YM. Chemical methods to knock down the amyloid proteins. Molecules. 2017;22(6)
Regitz C, Fitzenberger E, Mahn FL, Dussling LM, Wenzel U. Resveratrol reduces amyloid-beta (Abeta(1)(−)(4)(2))-induced paralysis through targeting proteostasis in an Alzheimer model of Caenorhabditis elegans. Eur J Nutr. 2016;55(2):741–7.
Wang G, Mao Z. Chaperone-mediated autophagy: roles in neurodegeneration. Transl Neurodegener. 2014;3:20.
Koga H, Martinez-Vicente M, Arias E, Kaushik S, Sulzer D, Cuervo AM. Constitutive upregulation of chaperone-mediated autophagy in Huntington's disease. J Neurosci. 2011;31(50):18492–505.
Wild EJ, Tabrizi SJ. Targets for future clinical trials in Huntington's disease: what's in the pipeline? Mov Disord. 2014;29(11):1434–45.
Ruiz-Cerda ML, Irurzun-Arana I, Gonzalez-Garcia I, Hu C, Zhou H, Vermeulen A, et al. Towards patient stratification and treatment in the autoimmune disease lupus erythematosus using a systems pharmacology approach. Eur J Pharm Sci. 2016;94:46–58.
Galan-Acosta L, Xia H, Yuan J, Vakifahmetoglu-Norberg H. Activation of chaperone-mediated autophagy as a potential anticancer therapy. Autophagy. 2015;11(12):2370–1.
Tracz-Gaszewska Z, Klimczak M, Biecek P, Herok M, Kosinski M, Olszewski MB, et al. Molecular chaperones in the acquisition of cancer cell chemoresistance with mutated TP53 and MDM2 up-regulation. Oncotarget. 2017;8(47):82123–43.
Maueroder C, Schall N, Meyer F, Mahajan A, Garnier B, Hahn J, et al. Capability of neutrophils to form NETs is not directly influenced by a CMA-targeting peptide. Front Immunol. 2017;8:16.
Anguiano J, Garner TP, Mahalingam M, Das BC, Gavathiotis E, Cuervo AM. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat Chem Biol. 2013;9(6):374–82.
Finn PF, Dice JF. Proteolytic and lipolytic responses to starvation. Nutrition. 2006;22(7–8):830–44.
Finn PF, Dice JF. Ketone bodies stimulate chaperone-mediated autophagy. J Biol Chem. 2005;280(27):25864–70.
Loos B, Klionsky DJ, Wong E. Augmenting brain metabolism to increase macro- and chaperone-mediated autophagy for decreasing neuronal proteotoxicity and aging. Prog Neurobiol. 2017;156:90–106.
Ouchida AT, Li Y, Geng J, Najafov A, Ofengeim D, Sun X, et al. Synergistic effect of a novel autophagy inhibitor and Quizartinib enhances cancer cell death. Cell Death Dis. 2018;9(2):138.
Acknowledgements
This work is supported by grants from National Institutes of Health: AG058866 and 1P50NS071669 (to W. L.)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
On behalf of all authors, the corresponding author states that there is no conflict interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Neuropharmacology
Rights and permissions
About this article

Cite this article
Li, W., Dou, J., Yang, J. et al. Targeting Chaperone-Mediated Autophagy for Disease Therapy. Curr Pharmacol Rep 4, 261–275 (2018). https://doi.org/10.1007/s40495-018-0138-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40495-018-0138-1