
Abstract
Chaperone-mediated autophagy (CMA) is the selective degradation process of intracellular components by lysosomes, which is required for the degradation of aggregate-prone proteins and contributes to proteostasis maintenance. Proteostasis is essential for normal cell function and survival, and it is determined by the balance of protein synthesis and degradation. Because postmitotic neurons are highly susceptible to proteostasis disruption, CMA is vital for the nervous system. Since Parkinson's disease (PD) was first linked to CMA dysfunction, an increasing number of studies have shown that CMA loss, as seen during aging, occurs in the pathogenetic process of neurodegenerative diseases. Here, we review the molecular mechanisms of CMA, as well as the physiological function and regulation of this autophagy pathway. Following, we highlight its potential role in neurodegenerative diseases, and the latest advances and challenges in targeting CMA in therapy of neurodegenerative diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
There are two main degradation pathways in eukaryotic cells: the ubiquitin–proteasome system (UPS) and the autophagy-lysosome pathway (ALP). Proteasome generally recognizes simply ubiquitinated substrates, whereas lysosome degradation is more complicated [1]. Autophagy commonly refers to ALP, which is a common concept for the pathways by which the cytoplasmic components are delivered to the lysosome in animal cells or the vacuole in plant and yeast cells [1]. Autophagy, as a dynamically cycling mechanism in the cell, maintains the homeostatic balance of protein synthesis, degradation, and organelle circulation [1]. Perhaps the significance of autophagy might be summed up as the removal of toxic substances and recycling of nutrients. In some cases, such as macroautophagy, the cytoplasmic components are isolated in vesicles that originate in the cytoplasm and subsequently combine with lysosomes to transport their contents for degradation. In other situations, such as microautophagy, the protein directly enter the vesicles formed by lysosomal membrane invagination [2]. Therefore, three types of autophagy have been identified in most mammalian systems based on how cargos are delivered to the lysosome: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) [3, 4]. Macroautophagy is a degradation pathway that occurs in bulk, recycling redundant or impaired organelles and proteins. Autophagosomes insulate cargo before fusing with lysosomes or late endosomal multivesicular bodies (MVB) to form autophago-lysosomes and amphisomes, severally [3]. Microautophagy, described in yeast, involves internalization of cytosolic cargo by invaginations of the lysosomal membrane, which has not been properly interpreted in eukaryotic cells [2]. Furthermore, Sahu et al. have found a microautophagy-like process that delivers soluble cytosolic proteins to the vesicles of late endosomes/MVB [5]. This pathway, referred to as endosomal microautophagy (e-MI), depends on cargo delivery by the 71 kDa heat shock cognate (HSC70) via electrostatic interactions with the limiting membrane of endosomes, but in contrast to CMA that occurs in lysosomes [5]. The third autophagic pathway, CMA, which is able to identify and degrade proteins with specific sequences by HSC70 and the lysosomal associated membrane protein 2A (LAMP2A), while being independent of vesicle formation, constitutes the focus of this review.
In postmitotic cells such as neurons, protein quality control provides for rigid regulation of protein homeostasis and its normal function, which subsequently modulates a range of intracellular processes [6]. Degradation of modified or damaged intracellular components prevents them from becoming cytotoxic, allowing cells to rapidly adapt to changing extracellular environments. CMA is a selective degradation process of intracellular components by lysosomes and is essential for the maintenance of neuronal homeostasis, as it is required for the degradation of aggregate-prone proteins. In other words, CMA controls a particular neuronal subproteome that is prone to aggregation and blocking CMA in neurons causes proteotoxicity and failure of the neurons [7]. Hence, CMA has been dubbed the "gatekeeper of neural proteostasis" [8]. Neurodegeneration is caused by the collapse of protein networks and loss of protein quality control. Toxic protein aggregation in neurons may be a sign of neurodegenerative diseases and brain aging [9]. Enhancing or maintaining CMA activity to promote neuronal proteostasis has a direct effect on neuronal health and brain function in neurodegenerative diseases.
In this review, we first outline the components and major steps of the CMA, as well as the physiological functions and regulation of this autophagy pathway. Following, we highlight current studies on the molecular mechanisms behind CMA dysfunction in the context of neurodegenerative diseases, along with the latest advances and challenges in targeting CMA under pathological conditions for neurodegenerative diseases therapy.
Mechanisms of CMA
CMA's unique features in terms of substrate recognition and intracytoplasmic transport have been validated by biochemical and genetic studies. There is no vesicle formation involved in the entire process, and CMA remains the sole pathway that allows monomer proteins to be degraded one by one through the lysosome. Because of this unparalleled selectivity, this precise process—eliminating misfolded or cytotoxic proteins while not interfering with the normal function of the same proteins—can be accomplished in the cell [4]. CMA differs from the remaining two lysosome-based autophagy processes in that substrate proteins must go through the following steps before being degraded by the lysosome.
Substrate selection
The prominent property of CMA is its selectivity, which is partially manifested in the substrate selection. Only proteins containing the KFERQ-like motif can be degraded by CMA, and the label (KFERQ-like motif) must be exposed rather than hidden by protein folding [10]. All targeting motifs contain positively charged residues, hydrophobic residues, negatively charged residues, and two glutamine [11]. Despite only about 40% of proteins in the mammalian proteome exhibiting this typical KFERQ-like motif, CMA's potential substrates could be expanded by a variety of post-translational modifications (PTMs). Even though some cytosolic proteins have multiple targeting motifs, adding tandem motifs in an experimental approach does not improve the CMA substrate characteristics of the proteins [11]. One motif may be sufficient for lysosomal targeting. The proteomic analysis of the lysosomal proteome is constantly evolving. Cuervo et al. have recently developed a free web-based resource (KFERQ finder) based on a proteome-wide analysis of KFERQ-like motifs across several species, which contributes to a better understanding of the physiological function of CMA [12].
Binding to HSC70
When a protein is chosen, it must first bind to HSC70 [13]. HSC70 is considered the front-line protein that protects peptides against misfolding and aggregation, together with the heat shock protein 90 (HSP90) chaperone and various accessory chaperones (co-chaperones) [14,15,16]. Although the specific subgroups of co-chaperones directly participating in CMA are uncertain, proteins including HSP90, HSP40, HSP70-HSP90 organizing protein (HOP), the HSP70-interacting protein (HIP), and the Bcl2-associated athanogene 1 protein (BAG-1) have been demonstrated to be involved in substrate recognition and unfolding [17]. The principal function of cytoplasmic HSC70 (CYT-HSC70) during CMA is to recognize substrate proteins expressing the KFERQ-like motif and facilitate their trafficking to the lysosome [13]. Furthermore, previous research has revealed that HSC70 is involved in the transport and circulation of synaptic vesicles, and HSC70 interacts with alpha-synuclein (α-Syn) to prevent oligomer formation in a novel manner that differs from its typical canonical substrate-binding site [18,19,20,21]. Strikingly, cytosolic protein delivery to late endosomes for degradation via microautophagy can also be mediated by HSC70 binding to the KFERQ-like motif [5]. Thus, CMA is only one of the responsibilities of HSC70.
Substrate targeting and translocation
HSC70 acts as a porter, delivering the substrate to LAMP2A [22]. The LAMP2 gene has three isoforms, each of which has a distinct function: LAMP2B participates in macroautophagy, LAMP2C is a kind of selective autophagy known as "RNautophagy," and only LAMP2A is essential for CMA [23, 24]. As a receptor for CMA substrates, LAMP2A is regarded as a landmark for CMA since the translocation step mediated by it is the rate-limiting step of the entire CMA process, and LAMP2A-deficient lysosomes in cells are not capable of CMA-driven degradation [22, 25]. Modulation of CMA activity is influenced by changes in LAMP2A synthesis rates, its controlled degradation at the lysosomal membrane, and its subcompartmentalization in this organelle [4]. It is not simply the presence of the KFERQ-like motif that identifies a protein as a CMA substrate; it is the dependency on LAMP2A that distinguishes CMA.
The binding of the substrate to LAMP2A in the translocation complex drives its dynamic assembly. LAMP2A exists as a monomer at the lysosomal membrane, but when HSC70 transports substrate proteins to the membrane, it can be activated to form a 700 kDa polymeric translocation complex [26]. Substrates can bind to LAMP2A in a folded state, but they must unfold to enter the lysosomal lumen [27], and HSC70, along with additional co-chaperones on the lysosomal membrane, is considered to mediate this process [27]. Notably, the substrate protein binds to the monomeric form of LAMP2A, which is then translocated by the polymeric form of LAMP2A. In addition, HSC70 and HSP90 are also present in the lysosomal lumen. Lysosomal-HSC70 (LYS-HSC70) is also involved in CMA by dragging substrates into the lysosome and preventing them from returning to the cytoplasm, facilitating substrate translocation [28]. Approximately half of lysosomal-HSP90 (LYS-HSP90) is located on the luminal side of the lysosomal membrane, and this chaperone may contribute to stabilizing the polymeric translocation complex by hiding the protease-sensitive areas of LAMP2A [26]. As the substrate is discharged into the lysosomal lumen, the complex instantly decomposes into monomers in a HSC70-dependent manner in order to bind to the next substrate.
Physiological function of CMA
The physiological function of CMA can be divided into three parts: respond to cellular stresses, proteostasis, and cell-specific functions. CMA responds to cellular stresses. In the state of starvation, macroautophagy and CMA are successively activated. Initially, essential amino acids for protein synthesis are obtained through macroautophagy. However, after 4–6 h of starvation, macroautophagy activity declines dramatically in many cells and tissues [25, 29]. As starvation persists, cells convert to CMA for great benefit: some necessary proteins are preserved, while other, fewer critical proteins are preferentially degraded via CMA [30]. CMA will remain active, as long as nutritional stress persists. This time-dependent transition from one autophagy to another implies that these two autophagy types interact to some extent. Certainly, as will be shown later, CMA can be compensated for suppressed macroautophagy and vice versa.
The second common function of CMA in most cells is to regulate protein quality, and for a long time, CMA's primary priority has been to preserve proteostasis [7]. CMA cannot remove aggregated proteins, but it may mediate the degradation of single misfolded proteins in order to enhance proteotoxicity resistance [31]. For example, CMA is activated during oxidative stress and contributes to the degradation of oxidative proteins [32]. In contrast, if CMA is not activated, there is a large deposition of oxidative damage proteins and a fall in cell viability [25]. Once proteins become irreversible oligomers or aggregates that cannot be completely unfolded, they can only be degraded by macroautophagy [33]. In addition, in the following sections, we will discuss in detail how CMA maintains neural proteostasis by degrading neurodegenerative proteins, such as tau protein, α-Syn, and others.
In addition to the general function of CMA, which is conserved by all cells, CMA activation contributes to the modulation of cell-specific functions. For example, CMA selectively degrades critical enzymes in glucose and lipid metabolism pathways, particularly in the starved state. The majority of glycolytic enzymes, lipogenesis enzymes, and lipid droplet coat proteins can be selective targets for CMA degradation [34]. Remarkably, lipid and carbohydrate metabolic changes driven by CMA have been linked to physiological aging of the brain [35] and neurodegeneration [36]. In addition, it has been reported that proteins involved in cell cycle regulation are targeted for degradation by CMA. Checkpoint kinase 1 (CHK1), a CMA substrate, is engaged in both regular and DNA damage-induced cell cycle arrest [37, 38]. Besides, CMA has been demonstrated to degrade hypoxia-inducible factor-1 subunit (HIF-1), which is in charge of cell cycle arrest as an adaptive response to organ damage [39]. The CMA is reported to be a key factor in determining the pluripotency of embryonic stem cells. The control of intracellular α-ketoglutarate, which affects histone and DNA methylation, by up-regulating CMA promotes differentiation [40]. And a recent study has demonstrated that CMA plays a crucial role in preserving hematopoietic stem-cell (HSC) function by regulating protein quality and providing energetic supply [41]. Through the selective elimination of antagonistic signaling regulators of T cell responses, CMA controls CD4 + T cell activation [42], which implies that CMA is also involved in the regulation of inflammation and immune responses. Sleep deprivation or alteration is linked to a variety of neurodegenerative disorders [43]. It should be noted that the latest research has found that CMA selectively degrades circadian proteins. CMA collaborates with different protein hydrolysis systems to remove clock proteins on time, allowing the internal timekeeping system to respond to the light/dark environment [44]. The LAMP2A knockout mice showed significant changes in circadian rhythm behavior, which were mirrored in older wild-type (WT) mice [45]. This lends credence to the idea that decreased CMA activity with aging may result in altered circadian rhythms in the elderly.
Regulation of CMA
The CMA has a wide range of physiological roles; hence, its physiological control is crucial. Cytoplasmic signaling pathways and lysosomal membrane signaling events have been found to regulate CMA in the presence of various stressors. The KFERQ-like motif serves as a passport to CMA for substances with various PTMs, which may allow additional proteins to become CMA substrates and provide a mechanism for CMA regulation. For example, only when the E3 ubiquitin-protein ligase STUB1 ubiquitinates hypoxia-inducible factor-1 on Lys63 can the CMA-dependent degradation appear [46]. On the other hand, acetylation of the Lys35 residue outside the MST1 KFERQ-like motif inhibits its lysosomal clearance, and CMA can only continue properly upon deacetylation [47]. The chaperone can also be employed as a modulation point to guarantee the overall efficiency of the CMA. Histone deacetylase 6 (HDAC6), as a tubulin deacetylase [48], mediates the deacetylation of HSP90 at the K489 site, which partly regulates the level of α-Syn oligomers and the survival of dopamine (DA) neurons [49]. This finding is in line with the previous findings [50]. HDAC6 is gaining popularity as a potential therapeutic target for neurodegenerative diseases [51]. Further research has discovered that the methyltransferase-like 21c methylates Lys-561 of HSC70 to enhance its stability [52]. These results reveal that epigenetic enzymes may be capable of regulating chaperones. Furthermore, Gong et al. discovered that partial humanin interacts with HSP90 on the cytoplasmic side of the lysosome, promoting substrate binding and uptake [53]. Interestingly, in addition to HSC70 and HSP90, the HSP40 family was discovered to prevent the development and propagation of toxic tau protein aggregates, demonstrating that chaperones from different families and classes serve distinct but complementary roles in preventing pathogenic protein aggregation [54]. Other co-chaperones of heat shock proteins are also involved in the degradation of certain pathogenic proteins [55,56,57,58,59], although their specific role in CMA remains unknown.
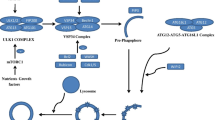
Since the binding of LAMP2A is the rate-limiting step in CMA, its expression, translocation, and stability are crucial for CMA regulation. The lysosomal form of glial fibrillary acidic protein (GFAP) and elongation factor 1-α (EF1α) can regulate CMA activity by binding to LAMP2A. GFAP stabilizes the CMA translocation complex against the catabolic activity of HSC70. Following substrate transit through the translocation complex, GFAP is self-bound to the phosphorylated GFAP usually covered by EF1α, with the consequent disassembly of the CMA translocation complex [60] (Fig. 1). Furthermore, nuclear factor, erythroid derived 2, like 2 (NFE2L2/NRF2) positively regulates CMA via binding to and activating transcription of LAMP2 gene regulatory regions [61] (Fig. 1). According to a recent study, cystinosin, RAB11, and RILP could rescue the LAMP2A trafficking defect [62]. Additionally, lysosomal Akt and the kinase target of rapamycin complex 2 could modulate the dynamics of CMA translocation complexes on lysosomal membranes [63]. Interestingly, LYS-HSP90 improves LAMP2A stability, whereas LYS-HSC70 promotes LAMP2A disassembly into a new cycle [26] (Fig. 1). And a recent study found that glucocorticoids negatively regulate CMA by reducing the level of LYS-HSC70 [64]. These findings shed light on the molecular components that regulate LAMP2A levels and assembly at the lysosomal membrane.

Regulatory targets of CMA translocation complex. Regulation of LAMP2A through major signaling pathways has been shown. TORC2 phosphorylates AKT1 and suppresses CMA activity, while PHLPP1 dephosphorylates AKT1, accelerating the LAMP2A dynamic cycle and activating CMA. NFAT and NFE2L2/NRF2 increase the transcription of LAMP2A, while RARα suppresses CMA by regulating LAMP2A-associated transcriptional programs. LYS-HSP90 improves LAMP2A stability, whereas LYS-HSC70 promotes LAMP2A disassembly into a new cycle. GFAP and EF1α can regulate CMA activity by binding to LAMP2A and contributing to the stabilization of LAMP2A against the catabolic activity of HSC70. PPCA promotes LAMP2A degradation, and with the suppression of PPCA, the rate of CMA degradation has increased
Role of CMA in neurodegenerative diseases
Proteostasis involves almost every tissue in the organism, but it has recently attracted special attention for its role in the central nervous system and neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson's disease (PD), Huntington's disease (HD), Amyotrophic lateral sclerosis (ALS), Frontotemporal dementia (FTD), Spinocerebellar ataxias (SCAs), and Prion disease. Protein degradation failure via the lysosome and proteasome pathways is thought to be at the root of neurodegenerative diseases [33]. In this review, we merely focus on the relationship between CMA and these neurodegenerative diseases.
Role of CMA in tauopathies and AD
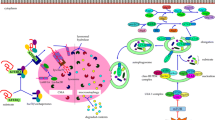
The tauopathies constitute a set of neurodegenerative diseases characterized by abnormal deposition of the microtubule binding protein tau (MAPT), which includes AD, FTD, Progressive Supranuclear Palsy (PSP), Corticobasal Degeneration (CBD), and so on. Under pathological conditions, tau loses its interaction with microtubules due to a variety of mutations or PTMs, resulting in the appearance of paired helical filaments (PHFs) and eventually forming neurofibrillary tangles (NFTs) [65, 66]. The repeat domain of tau with an FTDP-17 mutation (TauRD1K280) can be partially cracked into the F1 fragment [67]. The F1 fragment has two CMA-targeting motifs (336QVEVK340 and 347KDRVQ351), which can be targeted to LAMP2A by the HSC70. What’s more, in isolated lysosomes from mouse brains, Caballero et al. have discovered that the uptake rate for tau is extremely high with minimal binding [68]. The above results confirm that tau is a very well-characterized CMA substrate. Conversely, mutant (MT) tau cannot enter the lysosome for degradation due to various barriers (Fig. 3). The majority of MT tau significantly inhibit CMA-mediated degradation, but the step of the CMA process affected varies from the type of tau mutation [69] (Table 1). P301L, leading to autosomal-dominant FTD [70] and A152T, closely associated with FTD and AD [71], inhibits CMA and reroutes to poor e-MI [69]. The A152T mutation has a minor effect on its degradation via CMA by affecting translocation slightly [67], whereas the P301L mutation cannot bind to lysosomes and has a more marked suppressive effect [69] (Fig. 2). Furthermore, the modifications to the tau amino acid sequence influence the normal degradation of tau through CMA as well. The hTau40 ΔK280 (a deletion of lysine 280) is the first tau variant discovered to be limitedly removed by CMA and to have a detrimental effect on CMA (Table 1); this detrimental effect may be attributed in part to the accumulation of tau on the lysosomal membrane [67] (Fig. 2). Finally, PTMs of tau cannot be ignored in the context of CMA deficiency (Fig. 3). Caballero et al. investigated the effect of phosphorylated tau on CMA by mimicking phosphorylation at certain residues. The two pseudophosphorylated tau proteins (hTau40 AT8/AT100/PHF-1 and hTau40 4xKXGE) have a negative effect on the different steps of CMA [69] (Fig. 2).

Different pathogenic proteins damage various steps of CMA. (a) Targeting; (b) Binding: tau P301L, hTau40 ΔK280, and hTau40 AT8/AT100/PHF-1 cannot bind to lysosomes; (c) Internalization: tau A152T, hTau40 4xKXGE, acetylated tau, MT α-Syn, DA-mediated α-Syn, MT LRRK2, and MT GCase have an negative effect on internalization; and (d) Degradation: PTMs of GCase may impair lysosomal protein degradation?

CMA and neurodegenerative diseases. (1) Down-regulation of CMA in tauopathies (TP). WT tau and RCAN1 are CMA substrates. However, MT tau and PTMs of tau may suppress CMA. RCAN1 promotes the formation of NTFs and Aβ. CMA dysfunction results in RCAN1 up-regulation, tau aggregation, and Aβ production. (2) Impairment of the CMA in Parkinson’s disease (PD). CMA degrades α-Syn. MT α-Syn, PTMs of α-Syn, MT LRRK2, MT GCase, PTMs of GCase, MT UCH-L1, PTMs of UCH-L1, and MT VPS35 could damage CMA degradation and then lead to the α-Syn deposition. (3) Alterations of CMA in Huntington’s disease (HD). MT Htt binds with elevated affinity to HSC70 and LAMP2A, facilitating delayed delivery of misfolded proteins to the lysosome and development of Htt aggregation
Previous studies of pathogenic tau clearance have mainly focused on aberrantly aggregated tau. While acetylated tau is an early soluble variant in the AD brain, and reducing soluble acetylated tau can effectively improve cognitive function and neurodegeneration [72, 73]. It was recently suggested that acetylated tau hinders translocation complex disassembly and loses the PH sensitivity for HSC70 binding of LAMP2A, eventually reducing the efficiency of its lysosomal translocation and inhibiting CMA-mediated degradation [68] (Fig. 2). Interestingly, acetylated tau could reroute to e-MI, ultimately leading to its extracellular release and cell-to-cell propagation [68] (Table 1). Although the intact cell studies imply that e-MI has a minimal contribution to tau degradation, e-MI has a high efficiency for tau degradation [69]. This suggests that e-MI may become an effective way for tau degradation under certain cellular situations or upon specific tau modifications [69]. Tau cell-to-cell propagation has been identified as critical for the progression of tauopathies, and acetylation of tau, which inhibits CMA and reroutes to e-MI, may be the switch that allows tau to have prion-like seeding properties. The finding that TSC1 loss increases the risk for tauopathies by causing tau acetylation to spiral out of control and preventing its degradation via CMA further verifies acetylation of tau as a reasonable target for tauopathies therapy and diagnosis [74] (Table 1). While most evidence has revealed that acetylated tau is detrimental [68, 74,75,76], few studies have shown that acetylated tau represents a natural protective mechanism against aggregation [77, 78]. Future work on acetylated tau may better elucidate the effects of acetylation at various sites on autophagy. Overall, understanding the interplay between various types of human tau and CMA contributes to finding therapeutic targets for tauopathies.
AD, as a secondary tauopathy, is an age-related neurodegenerative disease. According to a recent work, CMA has been found in charge of maintaining the metastable proteome in a mouse model of AD-related proteotoxicity [7]. CMA deficiency and AD-driven proteotoxicity have a synergistic effect on the collapse of neuronal proteostasis. It is worth noting that the compensatory activation of macroautophagy reported in other cells following CMA blockade did not appear in neurons [7]. This demonstrates the potential role of pharmacological activation of CMA in age-related neurodegenerative diseases. Besides NFTs, amyloid plaque (composed of beta-amyloid (Aβ)) is also one of the characteristics of AD. According to Xu et al., amyloid-beta precursor protein (APP) is a CMA substrate that binds to HSC70 in an IKK/-dependent way [79]. A significant reduction in Aβ caused by the activation of CMA was unforeseen [7, 80], as CMA's substrates are soluble cytoplasmic proteins, whereas APP is a type I integral membrane protein that is not expected to be directly degraded by CMA. The C-terminus of APP has a KFERQ-like motif [81] (Table 1), and its split product, the APP intracellular domain, is readily accessible to the cytoplasm. These factors may contribute to the degradation of APP by CMA. The association between CMA and AD is not only through the degradation of APP and tau, but also through the degradation of AD-related regulatory proteins. The RCAN1 protein has been detected to be abundant in the AD brain, where it promotes the formation of NTFs and Aβ [82]. RCAN1 is degraded by CMA and failure of CMA with age increases RCAN1 in the brain [83], inevitably leading to tau hyperphosphorylation and Aβ production (Table 1, Fig. 3). The role of abnormal RCAN1 degradation by CMA in the pathogenesis of AD needs to be further investigated.
Role of CMA in PD
As the second-most common neurodegenerative disease after AD, PD is the first to be linked to CMA. At least six pathogenic genes that cause PD are associated with CMA: SNCA (PARK1), leucine-rich repeat kinase 2 (LRRK2/PRAK8), glucocerebrosidase gene (GBA), DJ-1 (PARK7), ubiquitin C-terminal hydrolase L1 (UCH-L1/PARK5), and vacuolar protein sorting 35 homolog gene (VPS35).
SNCA (encoding α-Syn) was the first gene tied to autosomal-dominant PD. α-Syn contains a KFERQ-like motif (VKKDQ), whose main catabolic pathway is CMA [84, 85] (Table 1). MT α-Syn (A30P and A53T) could still be recognized by chaperones and bound to LAMP2A, but it is poorly translocated into the lysosomal lumen for degradation (Fig. 3). MT α-Syn blocks the lysosomal uptake, compromising its own degradation as well as that of other substrates [84] (Table 1) (Fig. 2). However, the MT α-Syn emerged in a mere fraction of individuals, whereas α-Syn accumulates in most forms of PD. Hence, PTMs may be the basis of α-Syn aggregation in partial PD patients (Fig. 3). Nitration and oxidation of α-Syn marginally impair CMA, whereas phosphorylation of α-Syn and DA-mediated α-Syn completely prevent their own CMA degradation [33]. Strikingly, DA-mediated α-Syn inhibits CMA degradation of other substrates, acting similarly to MT α-Syn [86] (Table 1, Fig. 2). In the interaction between α-Syn aggregation and CMA, PTMs merit additional investigation. MT LRRK2 is prone to familial and sporadic PD. CMA degrades WT LRRK2, whereas MT LRRK2, including the most common G2019S MT, is poorly degraded by this pathway. Furthermore, MT LRRK2 and high levels of WT LRRK2 suppress CMA through restricting translocation complex activity in neurons (Fig. 2), which increases the susceptibility to α-Syn accumulation [87] (Fig. 3). And in the presence of α-Syn, the enhanced binding of LRRK2 to the LAMP2A serves to self-perpetuate the toxic effect of LRRK2 on CMA [87] (Table 1). It is a tragic duet for the CMA, to say the least [88]. Fortunately, in the MT LRRK2 knockin mouse model of PD, Ho et al. discovered that the activation of CMA could restore the inhibited lysosomal activity, induce LAMP2A transcription, and then reduce the aggregation of α-Syn [89] (Table 1). The MT β-glucocerebrosidase (GCase) induced by GBA mutations and PTMs of GCase impaires lysosomal protein degradation and causes a-Syn accumulation [90] (Table 1, Fig. 3). A recent study has demonstrated that the toxic effect of MT GCase on CMA is not attributable to a loss of GCase activity, but rather to the fact that MT GCase hinders the formation of CMA translocation complexes [91] (Fig. 2). Furthermore, Kuo et al. showed that unfolded GCase could be targeted to the lysosomal membrane for degradation by binding a CMA-targeting pentapeptide (QRDFI) to HSC70, despite the low efficiency of GCase translocation and degradation [91] (Table 1). It is noteworthy that the interaction of MT GCase and α-Syn is required for MT GCase-induced DA neuron injury [91]. Like LRRK2, the lysosomal activity of normal GCase could be blocked by α-Syn [92]. This also appears as part of a tragic duet, resulting in PD self-propagation. DJ-1 is a protective protein that has oxidation resistance and maintains mitochondrial homeostasis [93], while mutation and malfunction of it accelerate the accumulation of α-Syn in the early stage and cause early-onset familial PD [94]. CMA promotes DJ-1 degradation and preferential clearance of oxidatively damaged nonfunctional DJ-1, hence enhancing cell viability [95] (Table 1). Conversely, the level of nonfunctional DJ-1 increases in the context of deficient CMA [96] (Table 1). It is important to note that if DJ-1 were to mutate, not only would protection be lost, but also there could be harmful consequences. MT DJ-1 inhibits α-Syn degradation via CMA by down-regulating the level of LAMP2A and HSC70, boosting the accumulation of α-Syn [97] (Table 1) (Fig. 3). UCH-L1 could physically interact with CMA components, including LAMP2A, HSC70, and HSP90 [98]. The UCH-L1 I93M mutation and carbonyl modification of UCH-L1, which increased the level of α-Syn, abnormally strengthened these interactions [98, 99] (Table 1, Fig. 3). The UCH-L1 level in the cerebrospinal fluid (CSF) of PD patients considerably decreases [100], and it may become a biomarker for PD in future. Furthermore, in VPS35-deficient or MT VPS35-expressing DA neurons, LAMP2A retrieval from the endosomes to the Golgi apparatus is impaired, which promotes LAMP2A degradation and indirectly inhibits CMA [101] (Table 1). This process is accompanied by α-Syn deposition and locomotor function impairment (Fig. 3). The damaged CMA process could be one of the mechanisms through which VPS35 loss or MT VPS35 causes PD pathogenesis; however, this mechanism has to be further explored.
In addition to CMA, mitochondrial dysfunction plays a key role in the pathogenesis of PD as well. Excitingly, growing evidence has discovered a link between CMA and mitochondrial function. Membrane-associated ring-CH-type finger 5 (MARCHF5), an E3 ubiquitin ligase necessary for mitochondrial fission, has been identified as a CMA substrate. CMA activation enhances MARCHF5 turnover and decreases excessive mitochondria disintegration, which alleviates mitochondrial dysfunction under neuronal stress conditions such as oxidative stress and neurotoxin associated with PD [102] (Table 1). Overexpression of LAMP2A rescued DA neurons from mitochondrial damage and increased their viability in a model of PD [102], which provides compelling evidence that CMA is involved in maintaining mitochondrial homeostasis in PD. The myocyte enhancer binding factor-2 (MEF2) family has been identified as a nuclear factor required for neuronal survival, and a portion of MEF2D can also be found in rodent neurons' mitochondria [103]. α-Syn interferes with CMA-mediated degradation of MEF2D before substrate uptake (Table 1). Accumulation of nonfunctional MEF2D is found in CMA-inhibited neurons, resulting in neuronal death [104], which may underlie a portion of the pathogenic process in PD [105]. On the contrary, enhancing the chaperone HSC70 function in CMA would accelerate MEF2D degradation [106]. These findings indicate that CMA can influence mitochondrial activity via the regulation of MEF2D. Besides, as previously mentioned, CMA also regulates mitochondrial morphology and function by mediating the lysosome-dependent degradation of DJ-1 [95].
ER stress-induced unfolded protein response (UPR) and CMA, two major proteostasis-maintaining mechanisms, play crucial roles in PD [107]. In recent years, these two seemingly independent processes have been shown to be closely connected. As mentioned above, MEF2D, a well-known CMA substrate, is extremely sensitive to stress in neurons [104]. Interestingly, Li et al. found that ER stress inducers could reduce the level of MEF2D through the ER-P38 MAPK-CMA pathway [108] (Table 1). Concretely, ER stress activates MKK4 and recruits it to the lysosome, where it activates p38 MAPK. P38 MAPK phosphorylates LAMP2A directly, causing its membrane accumulation and conformational change, as well as increased CMA activity. Although p38 MAPK activation has an active effect on CMA through phosphorylation of LAMP2A, α-Syn could reduce the amount of available active MAPK [109]. It is unclear whether α-Syn affects phosphorylation and LAMP2A activity. In addition, LRRK2G2019S has been reported to interact with MKK4 [110, 111], which is unsure whether LRRK2G2019S is involved in the ER-P38 MAPK-CMA pathway. Remarkably, PARK7/DJ-1 can be overexpressed during ER stress, which may enhance CMA degradation of aggregated-prone proteins [112]. GCase is a lysosomal enzyme that reaches the lysosomal lumen after synthesis in the ER. A recent study showed that after ER retrotranslocation, MT GCase with the CMA-targeting motif could be targeted to lysosomes for degradation by CMA [113]. These findings support the idea that ER stress/UPR and CMA are related, and that a combined modulation of ER stress/UPR and CMA would be a more effective treatment for PD than either alone.
Role of CMA in HD
The aggregation of abnormal MT Huntington (Htt) protein with polyQ repeat expansion causes HD [114]. In contrast to tau and α-Syn, Htt has three potential KFERQ-like motifs: one at amino acids 99–103 (KDRVN) and one at amino acids 248–252 (NEIKV), with a third at the very amino-terminus (14-LKSFQ-18) deemed a KFERQ-like motif when serine 16 is phosphorylated [114]. Htt is undoubtedly one of the substrates of CMA, as evidenced by the findings that phosphorylated Htt may be eliminated by lysosomes and that purified lysosomes degrade Htt in a LAMP2A-dependent way [115,116,117,118] (Table 1). Interestingly, only the amino terminal fragment of Htt binds to the lysosome, not the full-length polyQ-Htt [116, 119] (Table 1). However, overexpression of LAMP2A or HSC70 to regulate CMA levels reduces endogenous full-length Htt [116]. Its specific mechanism is still under investigation. Although WT Htt clearance is driven by CMA, degradation of MT Htt is restricted by this route because MT Htt binds with high affinity to HSC70 and LAMP2A, promoting a delay in the delivery of misfolded protein to lysosomes and the development of intracellular aggregates [116] (Table 1, Fig. 3). Since the Htt truncated form is extremely cytotoxic and prevalent in the brains of HD patients and other HD models, CMA is an appealing therapeutic target. Accordingly, Bauer et al. reported that the polyglutamine binding peptide 1 (QBP1) specifically targeting the amino terminal of MT Htt fragments to CMA significantly reduced inclusion body formation and improved the HD phenotype in R6/2 mice [120]. This raises the possibility of manipulation of CMA as a promising treatment. Moreover, crosstalk between various autophages has been confirmed, which is not excluded in HD. CMA activity increased with macroautophagy failure in HD mouse models aged 2–7 months, but this up-regulation was no longer recorded in older mice [119].
Role of CMA in ALS-TDP and FTLD-TDP
The homeostasis of TAR DNA Binding Protein 43 kDa (TDP-43), a ribonuclear protein that regulates RNA metabolism, is crucial to ALS-TDP and FTLD-TDP [121,122,123,124]. The RRM1 domain of TDP-43 includes a KFERQ-like motif sequence (QVKKD) [125], and TDP-43 degradation could be competitively inhibited by CMA substrates such as GAPDH [126] (Table 1). Consistent with the above conclusions, endogenous TDP-43 is also found in the lysosomes isolated from the rat brain [126]. It follows that TDP-43 is one of the CMA substrates. Interestingly, despite the truncated TDP-43 fragments of 25 and 35 kDa do not contain QVKKD and do not bind to HSC70, in the context of CMA suppression, the blockage of TDP-43 degradation results in the accumulation of truncated TDP-43 fragments but not full-length TDP-43. This may be because full-length TDP-43 would be preferentially cleaved by active caspase-3 in the cytosol [125]. The TDP-25 fragment may serve as a seed for TDP-43 aggregation, so we should further explore the role of elevated full-length TDP-43 and TDP-25 fragments in the pathogenesis of TDP-43. Notably, QVKKD promotes the binding of ubiquitinated TDP-43 and HSC70, implying that certain ubiquitinated UPS substrates may shuttle into the lysosome for CMA clearance [125] (Table 1). This is probably due to compensatory up-regulation of CMA to compensate for UPS disability in TDP-43 proteinopathies, which needs additional research.
Prolonged periods of TDP-43 accumulation, like α-Syn, result in partial impairment of lysosomes connected with LAMP2A in cells expressing TDP-43 aggregates and a decrease in the lysosome perinuclear localization [126]. A recent study reported a considerable down-regulation of HSC70 in lymphomonocytes of sporadic ALS patients versus controls [127], further confirming that the altered expression of HSC70 is a universal pathological mechanism of ALS. Additionally, the TDP-43 nuclear export signal could be recognized by a single-chain variable fragment (scFv) generated from the 3B12A monoclonal antibody (MAb). In cultured cells and the mouse cerebral cortex, the addition of a CMA-related signal to 3B12A scFv stimulates HSP70 transcription against TDP-43 aggregation [128]. Specific elimination of TDP-43 by modulation of HSC70 may be a promising therapeutic strategy for the treatment of TDP-43 proteinopathies.
Role of CMA in SCAs
SCAS represent autosomal dominantly inherited neurodegenerative diseases, marked by loss of balance and coordination accompanied by vague speech [129]. Pathogenic proteins accumulate in the lysosomes of SCAs patients and model animals [130,131,132], suggesting that lysosomal impairment is involved in the pathogenesis of SCAs. Based on the locus of the causative gene, SCAs are grouped as SCA1 through SCA48. LAMP2A plays a critical role in the process of CMA. And a recent study discovered that selective knockdown LAMP2A in mouse cerebellar interneurons and granule cells triggers a progressive ataxic phenotype, gliosis, and subsequent cerebellar neurodegeneration [133]. This certainly implies that CMA is crucial for cerebellar neurons survival. Furthermore, CMA is decreased in the cellular model of SCA14 [134]and cellular and mouse models of SCA21 [132]. SCA14 and SCA21 are caused by MT kinase Cγ (PKCγ) [135] and MT transmembrane protein 240 (TMEM240) [136]. These findings provide significant evidence in favor of the hypothesis that CMA impairment is a common pathogenic factor of SCAs caused by various mutant proteins. Finally, initiation of macroautophagy in SCA7 might be related to a compensatory response to a CMA deficiency [137], in a mechanism similar to that reported in other neurodegenerative diseases. Taken together, CMA could be a potential therapeutic target for SCAs.
Role of CMA in prion disease
Prion disease is an unusual, incurable, and rapidly progressing neurodegenerative disease, with prion protein (PrP) misfolding being the most crucial step in the pathogenesis of prion protein disease [138]. Wang et al. initially found aberrant down-regulation of polo-like kinase 3 (PLK3) levels in the brains of scrapie (a prion-related disease) infected hamsters [139], and then revealed that the PLK3 kinase domain (KD) region is primarily responsible for the degradation of aberrant aggregated PrP MTs and scrapie PrPSc. Subsequent experiments further verified that the PLK3 KD region could bind PrP proteins expressed in mammalian cell lines, which have limited association with its kinase activity but are related to its co-chaperone activity in the CMA pathway [140]. Overexpression of the PLK3 KD region in the human embryonic kidney 293 T (HEK293T) cells expressing PG14-PrP effectively boosts the cellular levels of HSC70 and LAMP2A [141] (Table 1). Meanwhile, LAMP2A and HSC70 levels are significantly lower in HEK293T cells expressing PrP MT and scrapie-infected cell lines, and the PrP MT displays the ability to interact with LAMP2A and HSC70. Strikingly, a pentapeptide “RVVEQ”-motif is identified in the PrP protein's C-terminus [141]. More studies are needed to confirm if this motif is responsible for the putative interaction of PrP with HSC70. The mechanism of PLK3-induced increase in CMA does not seem to be well understood. However, the occurrence of enhanced CMA activity accompanied by anomalous PrP degradation by PLK3 provides compelling evidence that most of the PLK3 overexpression-mediated degradation of PrP MT and pathogenic PrPSc is via CMA. Furthermore, only lysosomal inhibitors, according to Wang et al., can prevent PLK3 overexpression-mediated degradation of PrP MTs and scrapie PrPSc, while ubiquitin–proteasome inhibitors (MG132) and macroautophagy inhibitors (3MA) cannot [141].
Potential therapeutic target
The fact that neuroprotection of CMA originates from its capacity to remove pathogenic forms of proteins and prevent additional lysosome damage has provided a motivation for interventions seeking to up-regulate CMA activity. CMA enhancement may be a therapeutic strategy for a variety of neurodegenerative diseases, and as a result, potential pathways and medicinally available targets for this goal have emerged.
Some MT proteins disrupt lysosomal acidification and proteolysis, which subsequently inhibits CMA. Approaches that only target the lysosomal lumen, which are suitable in contexts when the organelle malfunctions preferentially, are gaining momentum [142]. Acidic nanoparticles restoring lysosomal acidification [143] have been reported to be beneficial in PD-associated GCase mutation models, in which they alleviate neurodegeneration via rescue of compromised lysosomes after intracerebral injection in neurons [144]. Restoration of lysosomal pH attenuates CMA inhibition, demonstrating the utility of direct targeting of lysosomes in these conditions. However, CMA processes occurring at the lysosome surface are the most vulnerable to neurodegenerative protein toxicity. Up-regulation of LAMP2A levels and efficient assembly and disassembly of the CMA translocation complex are particularly vital in the context of CMA impairment.
The LAMP2A distribution between the lysosomal membrane and lumen is a dynamic process [145]. Protective protein/cathepsin A (PPCA) promotes LAMP2A degradation. With the suppression of PPCA, the level of LAMP2A and the rate of CMA degradation have increased [146]. Some LAMP2A proteins in the lysosomal membrane have been discovered to be clustered in cholesterol-rich, detergent-resistant areas [23, 147] (Fig. 1). The release of LAMP2A from these regions can be facilitated by cholesterol depletion, which enhances substrate degradation by CMA, whereas cholesterol overload has the reverse effect. Interestingly, PPCA readily catabolizes LAMP2A within lipid microdomains, and these regions represent LAMP2A degradation sites (Fig. 1). A part of LAMP2A is located in the lysosomal lumen or lipid microdomains. This LAMP2A is transported to other regions of the lysosomal membrane when required, where it can form translocation complexes when bound to substrates. Furthermore, three signaling pathways have been identified as being involved in the up-regulation of CMA. The first signaling mechanism found in the activation of LAMP2A, the calcineurin–NFAT pathway, has been shown to directly increase the transcription of LAMP2A during T cell activation [42] (Fig. 1). Production of reactive oxygen species (ROS) promotes nuclear translocation of NFAT. Furthermore, inhibiting the formation of ROS or using the calcineurin inhibitor cyclosporine A can block this up-regulation of LAMP2A, implying that ROS drives calcineurin-mediated NFAT activation of up-regulated LAMP2A expression, which further activates CMA [31]. In contrast, retinoic acid receptor alpha (RARα) may suppress CMA by regulating discrete LAMP2A-associated transcriptional programs [148] (Fig. 1). Not only LAMP2A, but also RAB11 or RILP, facilitating the translocation of LAMP2A to lysosomes, is also negatively regulated by this signaling pathway [62]. Based on this finding, the creation of all-trans retinoic acid derivatives can successfully prevent RAR-mediated inhibition of CMA without disrupting the RAR-dependent transcriptional program [148]. Given the prevalence of RAR expression, these atypical RAR inhibitors show significant potential for systemic recovery of aging-impaired CMA [31]. As mentioned previously, the state of the CMA translocation complex is dependent on the phosphorylation state of AKT1, which is present on the lysosomal membrane and whose activity is regulated by the phosphatase PH domain leucine-rich repeat-containing protein phosphatase 1 (PHLPP1) and the kinase TOR complex 2 (TORC2) on the lysosomal membrane [63]. TORC2 phosphorylates AKT1 and suppresses CMA activity, while PHLPP1 dephosphorylates AKT1, accelerating the LAMP2A dynamic cycle and activating CMA (Fig. 1). Although AKT1, mTORC2, and PHLPP1 are druggable CMA targets, their multiple biological activities make them inappropriate as selective CMA modulators [149]. Strikingly, a subset of retinoid derivatives has been identified to only affect a fraction of the RAR-dependent transcriptional activity [148]. The ability of these molecules to revert the functional decline of aged mouse and human stem cells [41], ameliorate retinal degeneration in retinitis pigmentosa mouse models [150], and to decrease neuronal and glial pathology in AD mouse models [7] has been demonstrated through optimization of these molecules to acquire drug-like attributes for in vivo use.
Some medications have also been found to improve neurodegenerative diseases, presumably via the CMA pathway. A recent study indicates that CMA enhancement by Metformin via activation of TAK1-IKKα/β signaling that leads to phosphorylation of HSC70 Ser85. Metformin significantly reduces the toxicity of APP and Aβ through CMA-mediated degradation and reverses the molecular and behavioral phenotypes of AD [79]. It is also worth noting that lactulose and trehalose ameliorate the toxicity of Aβ in AD animals by partially boosting HSC70 and LAMP2A levels, which enhance short-term memory and learning capacity in mice [151]. Furthermore, it was demonstrated in the PD models that chronic coffee [152], dihydromyricetin, and salvianolic acid B [153], 6-hydroxydopamine (6-OHDA) [154], and Sigma-2 receptor antagonists [155] may control LAMP2A expression to improve the PD phenotype and ameliorate α-Syn toxicity.
Another issue to consider is whether activating the CMA in certain situations may be harmful. According to a recent study, HSP70 eliminates tau amyloid at the cost of producing new seeds (monomers and oligomers), indicating that chaperone-mediated tau protein degradation is advantageous per se but may contribute to the propagation of prion-like tauopathies [59]. This validates the previously reported conclusion that HSC70 regulates the extracellular release of neurodegeneration-related proteins [15]. Therefore, identifying the pathological protein along with the WT version, the exact point at which CMA fails, and the optimal strategies for restoring CMA activity and maintaining neural proteostasis is critical for the development of therapies for neurodegenerative diseases [31, 142].
Summary and direction
In recent years, the discovery of additional CMA substrates and defects in CMA linked to a wide range of human diseases has expanded our view of the role of CMA in the maintenance of the metastable proteomes. Stirringly, progress has been particularly rapid in deconstructing the role of CMA in neurodegenerative disorders. However, research on CMA has a long way to go before it can approach the amount of knowledge accumulated on other forms of autophagy over the last two decades [31]. At the molecular level, modulation of LAMP2A multimerization is crucial for CMA regulation, while the signaling pathways governing CMA in the cellular environments remain imprecise, especially when considering the consequences of multiple CMA-activated stimuli. Evidence of a strong link between CMA and macroautophagy in neurodegenerative diseases highlights the need to understand the molecules involved in this interaction. To date, little is known about these crosstalk molecular modulators and their ubiquity [156].
Of course, this rapidly evolving area calls for innovative tools to not only better dissect the process, but also more effectively regulate it. The application of genetic manipulation of CMA in mouse, as well as the future extensions of tissue-specific conditioned models, will assist in understanding how alterations in tissue-specific pathways affect overall organism function. Most neurodegenerative diseases occur in the elderly, making genetic intervention clearly challenging. Recent studies, however, suggest that regulation of dietary lipid intake and the development of retinoic acid derivatives are more practical strategies [148, 157]. Pharmacological modulators aimed at improving neuroprotein homeostasis by manipulating CMA should be the next step in exploring therapeutic strategies.
Data availability
Not applicable.
References
Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147:728–741. https://doi.org/10.1016/j.cell.2011.10.026
Marzella L, Ahlberg J, Glaumann H (1981) Autophagy, heterophagy, microautophagy and crinophagy as the means for intracellular degradation. Virchows Arch B 36:219–234. https://doi.org/10.1007/bf02912068
Martinez-Lopez N, Athonvarangkul D, Singh R (2015) Autophagy and aging. Adv Exp Med Biol 847:73–87. https://doi.org/10.1007/978-1-4939-2404-2_3
Kaushik S, Cuervo AM (2012) Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol 22:407–417. https://doi.org/10.1016/j.tcb.2012.05.006
Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, Follenzi A, Potolicchio I, Nieves E, Cuervo AM, Santambrogio L (2011) Microautophagy of cytosolic proteins by late endosomes. Dev Cell 20:131–139. https://doi.org/10.1016/j.devcel.2010.12.003
Cuervo AM, Wong E (2014) Chaperone-mediated autophagy: roles in disease and aging. Cell Res 24:92–104. https://doi.org/10.1038/cr.2013.153
Bourdenx M, Martín-Segura A, Scrivo A, Rodriguez-Navarro JA, Kaushik S, Tasset I, Diaz A, Storm NJ, Xin Q, Juste YR, Stevenson E, Luengo E, Clement CC, Choi SJ, Krogan NJ, Mosharov EV, Santambrogio L, Grueninger F, Collin L, Swaney DL, Sulzer D, Gavathiotis E, Cuervo AM (2021) Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome. Cell 184:2696-2714.e25. https://doi.org/10.1016/j.cell.2021.03.048
Bourdenx M, Gavathiotis E, Cuervo AM (2021) Chaperone-mediated autophagy: a gatekeeper of neuronal proteostasis. Autophagy 17:2040–2042. https://doi.org/10.1080/15548627.2021.1935007
Loos B, Klionsky DJ, Wong E (2017) Augmenting brain metabolism to increase macro- and chaperone-mediated autophagy for decreasing neuronal proteotoxicity and aging. Prog Neurobiol 156:90–106. https://doi.org/10.1016/j.pneurobio.2017.05.001
Chiang HL, Dice JF (1988) Peptide sequences that target proteins for enhanced degradation during serum withdrawal. J Biol Chem 263:6797–6805
Dice JF (1990) Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci 15:305–309. https://doi.org/10.1016/0968-0004(90)90019-8
Kirchner P, Bourdenx M, Madrigal-Matute J, Tiano S, Diaz A, Bartholdy BA, Will B, Cuervo AM (2019) Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLoS Biol 17:e3000301. https://doi.org/10.1371/journal.pbio.3000301
Chiang HL, Terlecky SR, Plant CP, Dice JF (1989) A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 246:382–385. https://doi.org/10.1126/science.2799391
Chen BH, Chang YJ, Lin S, Yang WY (2020) Hsc70/Stub1 promotes the removal of individual oxidatively stressed peroxisomes. Nat Commun 11:5267. https://doi.org/10.1038/s41467-020-18942-3
Fontaine SN, Zheng D, Sabbagh JJ, Martin MD, Chaput D, Darling A, Trotter JH, Stothert AR, Nordhues BA, Lussier A, Baker J, Shelton L, Kahn M, Blair LJ, Stevens SM Jr, Dickey CA (2016) DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. Embo J 35:1537–1549. https://doi.org/10.15252/embj.201593489
Dokladny K, Myers OB, Moseley PL (2015) Heat shock response and autophagy–cooperation and control. Autophagy 11:200–213. https://doi.org/10.1080/15548627.2015.1009776
Agarraberes FA, Dice JF (2001) A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J Cell Sci 114:2491–2499. https://doi.org/10.1242/jcs.114.13.2491
Coyne AN, Lorenzini I, Chou CC, Torvund M, Rogers RS, Starr A, Zaepfel BL, Levy J, Johannesmeyer J, Schwartz JC, Nishimune H, Zinsmaier K, Rossoll W, Sattler R, Zarnescu DC (2017) Post-transcriptional inhibition of Hsc70-4/HSPA8 expression leads to synaptic vesicle cycling defects in multiple models of ALS. Cell Rep 21:110–125. https://doi.org/10.1016/j.celrep.2017.09.028
Ganguly A, Han X, Das U, Wang L, Loi J, Sun J, Gitler D, Caillol G, Leterrier C, Yates JR 3rd, Roy S (2017) Hsc70 chaperone activity is required for the cytosolic slow axonal transport of synapsin. J Cell Biol 216:2059–2074. https://doi.org/10.1083/jcb.201604028
Burmann BM, Gerez JA, Matečko-Burmann I, Campioni S, Kumari P, Ghosh D, Mazur A, Aspholm EE, Šulskis D, Wawrzyniuk M, Bock T, Schmidt A, Rüdiger SGD, Riek R, Hiller S (2020) Regulation of α-synuclein by chaperones in mammalian cells. Nature 577:127–132. https://doi.org/10.1038/s41586-019-1808-9
Banks SML, Medeiros AT, McQuillan M, Busch DJ, Ibarraran-Viniegra AS, Sousa R, Lafer EM, Morgan JR (2020) A Hsc70 ameliorates the vesicle recycling defects caused by excess α-synuclein at synapses. ENeuro. 7:1–10. https://doi.org/10.1523/eneuro.0448-19.2020
Cuervo AM, Dice JF (1996) A receptor for the selective uptake and degradation of proteins by lysosomes. Science 273:501–503. https://doi.org/10.1126/science.273.5274.501
Kaushik S, Massey AC, Cuervo AM (2006) Lysosome membrane lipid microdomains: novel regulators of chaperone-mediated autophagy. Embo J 25:3921–3933. https://doi.org/10.1038/sj.emboj.7601283
Gough NR, Hatem CL, Fambrough DM (1995) The family of LAMP-2 proteins arises by alternative splicing from a single gene: characterization of the avian LAMP-2 gene and identification of mammalian homologs of LAMP-2b and LAMP-2c. DNA Cell Biol 14:863–867. https://doi.org/10.1089/dna.1995.14.863
Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM (2006) Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci USA 103:5805–5810. https://doi.org/10.1073/pnas.0507436103
Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM (2008) The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol 28:5747–5763. https://doi.org/10.1128/mcb.02070-07
Salvador N, Aguado C, Horst M, Knecht E (2000) Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. J Biol Chem 275:27447–27456. https://doi.org/10.1074/jbc.M001394200
Agarraberes FA, Terlecky SR, Dice JF (1997) An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J Cell Biol 137:825–834. https://doi.org/10.1083/jcb.137.4.825
Finn PF, Dice JF (2005) Ketone bodies stimulate chaperone-mediated autophagy. J Biol Chem 280:25864–25870. https://doi.org/10.1074/jbc.M502456200
Orenstein SJ, Cuervo AM (2010) Chaperone-mediated autophagy: molecular mechanisms and physiological relevance. Semin Cell Dev Biol 21:719–726. https://doi.org/10.1016/j.semcdb.2010.02.005
Kaushik S, Cuervo AM (2018) The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol 19:365–381. https://doi.org/10.1038/s41580-018-0001-6
Kiffin R, Christian C, Knecht E, Cuervo AM (2004) Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell 15:4829–4840. https://doi.org/10.1091/mbc.e04-06-0477
Koga H, Cuervo AM (2011) Chaperone-mediated autophagy dysfunction in the pathogenesis of neurodegeneration. Neurobiol Dis 43:29–37. https://doi.org/10.1016/j.nbd.2010.07.006
Tasset I, Cuervo AM (2016) Role of chaperone-mediated autophagy in metabolism. FEBS J 283:2403–2413. https://doi.org/10.1111/febs.13677
Hallett PJ, Huebecker M, Brekk OR, Moloney EB, Rocha EM, Priestman DA, Platt FM, Isacson O (2018) Glycosphingolipid levels and glucocerebrosidase activity are altered in normal aging of the mouse brain. Neurobiol Aging 67:189–200. https://doi.org/10.1016/j.neurobiolaging.2018.02.028
Alfaro IE, Albornoz A, Molina A, Moreno J, Cordero K, Criollo A, Budini M (2018) Chaperone mediated autophagy in the crosstalk of neurodegenerative diseases and metabolic disorders. Front Endocrinol 9:778. https://doi.org/10.3389/fendo.2018.00778
Patil M, Pabla N, Dong Z (2013) Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell Mol Life Sci 70:4009–4021. https://doi.org/10.1007/s00018-013-1307-3
Andrade-Tomaz M, de Souza I, Rocha CRR, Gomes LR (2020) The role of chaperone-mediated autophagy in cell cycle control and its implications in cancer. Cells. https://doi.org/10.3390/cells9092140
Hubbi ME, Hu H, Kshitiz AI, Levchenko A, Semenza GL (2013) Chaperone-mediated autophagy targets hypoxia-inducible factor-1α (HIF-1α) for lysosomal degradation. J Biol Chem 288:10703–10714. https://doi.org/10.1074/jbc.M112.414771
Xu Y, Zhang Y, García-Cañaveras JC, Guo L, Kan M, Yu S, Blair IA, Rabinowitz JD, Yang X (2020) Chaperone-mediated autophagy regulates the pluripotency of embryonic stem cells. Science 369:397–403. https://doi.org/10.1126/science.abb4467
Dong S, Wang Q, Kao YR, Diaz A, Tasset I, Kaushik S, Thiruthuvanathan V, Zintiridou A, Nieves E, Dzieciatkowska M, Reisz JA, Gavathiotis E, D’Alessandro A, Will B, Cuervo AM (2021) Chaperone-mediated autophagy sustains haematopoietic stem-cell function. Nature 591:117–123. https://doi.org/10.1038/s41586-020-03129-z
Valdor R, Mocholi E, Botbol Y, Guerrero-Ros I, Chandra D, Koga H, Gravekamp C, Cuervo AM, Macian F (2014) Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat Immunol 15:1046–1054. https://doi.org/10.1038/ni.3003
Fleming A, Bourdenx M, Fujimaki M, Karabiyik C, Krause GJ, Lopez A, Martín-Segura A, Puri C, Scrivo A, Skidmore J, Son SM, Stamatakou E, Wrobel L, Zhu Y, Cuervo AM, Rubinsztein DC (2022) The different autophagy degradation pathways and neurodegeneration. Neuron 110:935–966. https://doi.org/10.1016/j.neuron.2022.01.017
Juste YR, Kaushik S, Bourdenx M, Aflakpui R, Bandyopadhyay S, Garcia F, Diaz A, Lindenau K, Tu V, Krause GJ, Jafari M, Singh R, Muñoz J, Macian F, Cuervo AM (2021) Reciprocal regulation of chaperone-mediated autophagy and the circadian clock. Nat Cell Biol 23:1255–1270. https://doi.org/10.1038/s41556-021-00800-z
Kaushik S, Juste YR and Cuervo AM (2022) Circadian remodeling of the proteome by chaperone-mediated autophagy. Autophagy 18: 1205. doi: https://doi.org/10.1080/15548627.2022.2038503
Ferreira JV, Soares AR, Ramalho JS, Pereira P, Girao H (2015) K63 linked ubiquitin chain formation is a signal for HIF1A degradation by chaperone-mediated autophagy. Sci Rep 5:10210. https://doi.org/10.1038/srep10210
Li L, Fang R, Liu B, Shi H, Wang Y, Zhang W, Zhang X, Ye L (2016) Deacetylation of tumor-suppressor MST1 in Hippo pathway induces its degradation through HBXIP-elevated HDAC6 in promotion of breast cancer growth. Oncogene 35:4048–4057. https://doi.org/10.1038/onc.2015.476
Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP (2002) HDAC6 is a microtubule-associated deacetylase. Nature 417:455–458. https://doi.org/10.1038/417455a
Du Y, Yang X, Li Z, Le W, Hao Y, Song Y, Wang F, Guan Y (2021) HDAC6-mediated Hsp90 deacetylation reduces aggregation and toxicity of the protein alpha-synuclein by regulating chaperone-mediated autophagy. Neurochem Int 149:105141. https://doi.org/10.1016/j.neuint.2021.105141
Su M, Guan H, Zhang F, Gao Y, Teng X, Yang W (2016) HDAC6 regulates the chaperone-mediated autophagy to prevent oxidative damage in injured neurons after experimental spinal cord injury. Oxid Med Cell Longev 2016:7263736. https://doi.org/10.1155/2016/7263736
Simões-Pires C, Zwick V, Nurisso A, Schenker E, Carrupt PA, Cuendet M (2013) HDAC6 as a target for neurodegenerative diseases: what makes it different from the other HDACs? Mol Neurodegener 8:7. https://doi.org/10.1186/1750-1326-8-7
Wang C, Arrington J, Ratliff AC, Chen J, Horton HE, Nie Y, Yue F, Hrycyna CA, Tao WA, Kuang S (2019) Methyltransferase-like 21c methylates and stabilizes the heat shock protein Hspa8 in type I myofibers in mice. J Biol Chem 294:13718–13728. https://doi.org/10.1074/jbc.RA119.008430
Gong Z, Tasset I, Diaz A, Anguiano J, Tas E, Cui L, Kuliawat R, Liu H, Kühn B, Cuervo AM, Muzumdar R (2018) Humanin is an endogenous activator of chaperone-mediated autophagy. J Cell Biol 217:635–647. https://doi.org/10.1083/jcb.201606095
Irwin R, Faust O, Petrovic I, Wolf SG, Hofmann H, Rosenzweig R (2021) Hsp40s play complementary roles in the prevention of tau amyloid formation. Elife. https://doi.org/10.7554/eLife.69601
Criado-Marrero M, Gebru NT, Blazier DM, Gould LA, Baker JD, Beaulieu-Abdelahad D, Blair LJ (2021) Hsp90 co-chaperones, FKBP52 and Aha1, promote tau pathogenesis in aged wild-type mice. Acta Neuropathol Commun 9:65. https://doi.org/10.1186/s40478-021-01159-w
Jinwal UK, Koren J 3rd, Borysov SI, Schmid AB, Abisambra JF, Blair LJ, Johnson AG, Jones JR, Shults CL, O’Leary JC 3rd, Jin Y, Buchner J, Cox MB, Dickey CA (2010) The Hsp90 cochaperone, FKBP51, increases Tau stability and polymerizes microtubules. J Neurosci 30:591–599. https://doi.org/10.1523/jneurosci.4815-09.2010
Shelton LB, Baker JD, Zheng D, Sullivan LE, Solanki PK, Webster JM, Sun Z, Sabbagh JJ, Nordhues BA, Koren J 3rd, Ghosh S, Blagg BSJ, Blair LJ, Dickey CA (2017) Hsp90 activator Aha1 drives production of pathological tau aggregates. Proc Natl Acad Sci USA 114:9707–9712. https://doi.org/10.1073/pnas.1707039114
Friesen EL, Zhang YT, Earnshaw R, De Snoo ML, O’Hara DM, Agapova V, Chau H, Ngana S, Chen KS, Kalia LV, Kalia SK (2020) BAG5 promotes alpha-synuclein oligomer formation and functionally interacts with the autophagy adaptor protein p62. Front Cell Dev Biol 8:716. https://doi.org/10.3389/fcell.2020.00716
Nachman E, Wentink AS, Madiona K, Bousset L, Katsinelos T, Allinson K, Kampinga H, McEwan WA, Jahn TR, Melki R, Mogk A, Bukau B, Nussbaum-Krammer C (2020) Disassembly of Tau fibrils by the human Hsp70 disaggregation machinery generates small seeding-competent species. J Biol Chem 295:9676–9690. https://doi.org/10.1074/jbc.RA120.013478
Bandyopadhyay U, Sridhar S, Kaushik S, Kiffin R, Cuervo AM (2010) Identification of regulators of chaperone-mediated autophagy. Mol Cell 39:535–547. https://doi.org/10.1016/j.molcel.2010.08.004
Pajares M, Rojo AI, Arias E, Díaz-Carretero A, Cuervo AM, Cuadrado A (2018) Transcription factor NFE2L2/NRF2 modulates chaperone-mediated autophagy through the regulation of LAMP2A. Autophagy 14:1310–1322. https://doi.org/10.1080/15548627.2018.1474992
Zhang J, Johnson JL, He J, Napolitano G, Ramadass M, Rocca C, Kiosses WB, Bucci C, Xin Q, Gavathiotis E, Cuervo AM, Cherqui S, Catz SD (2017) Cystinosin, the small GTPase Rab11, and the Rab7 effector RILP regulate intracellular trafficking of the chaperone-mediated autophagy receptor LAMP2A. J Biol Chem 292:10328–10346. https://doi.org/10.1074/jbc.M116.764076
Arias E, Koga H, Diaz A, Mocholi E, Patel B, Cuervo AM (2015) Lysosomal mTORC2/PHLPP1/Akt regulate chaperone-mediated autophagy. Mol Cell 59:270–284. https://doi.org/10.1016/j.molcel.2015.05.030
Sato M, Ueda E, Konno A, Hirai H, Kurauchi Y, Hisatsune A, Katsuki H, Seki T (2020) Glucocorticoids negatively regulates chaperone mediated autophagy and microautophagy. Biochem Biophys Res Commun 528:199–205. https://doi.org/10.1016/j.bbrc.2020.04.132
Jiang S, Bhaskar K (2020) Degradation and transmission of tau by autophagic-endolysosomal networks and potential therapeutic targets for tauopathy. Front Mol Neurosci 13:586731. https://doi.org/10.3389/fnmol.2020.586731
de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, Hyman BT (2010) Caspase activation precedes and leads to tangles. Nature 464:1201–1204. https://doi.org/10.1038/nature08890
Wang Y, Martinez-Vicente M, Krüger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, Mandelkow E (2009) Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet 18:4153–4170. https://doi.org/10.1093/hmg/ddp367
Caballero B, Bourdenx M, Luengo E, Diaz A, Sohn PD, Chen X, Wang C, Juste YR, Wegmann S, Patel B, Young ZT, Kuo SY, Rodriguez-Navarro JA, Shao H, Lopez MG, Karch CM, Goate AM, Gestwicki JE, Hyman BT, Gan L, Cuervo AM (2021) Acetylated tau inhibits chaperone-mediated autophagy and promotes tau pathology propagation in mice. Nat Commun 12:2238. https://doi.org/10.1038/s41467-021-22501-9
Caballero B, Wang Y, Diaz A, Tasset I, Juste YR, Stiller B, Mandelkow EM, Mandelkow E, Cuervo AM (2018) Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell. https://doi.org/10.1111/acel.12692
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, Heutink P (1998) Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393:702–705. https://doi.org/10.1038/31508
Coppola G, Chinnathambi S, Lee JJ, Dombroski BA, Baker MC, Soto-Ortolaza AI, Lee SE, Klein E, Huang AY, Sears R, Lane JR, Karydas AM, Kenet RO, Biernat J, Wang LS, Cotman CW, Decarli CS, Levey AI, Ringman JM, Mendez MF, Chui HC, Le Ber I, Brice A, Lupton MK, Preza E, Lovestone S, Powell J, Graff-Radford N, Petersen RC, Boeve BF, Lippa CF, Bigio EH, Mackenzie I, Finger E, Kertesz A, Caselli RJ, Gearing M, Juncos JL, Ghetti B, Spina S, Bordelon YM, Tourtellotte WW, Frosch MP, Vonsattel JP, Zarow C, Beach TG, Albin RL, Lieberman AP, Lee VM, Trojanowski JQ, Van Deerlin VM, Bird TD, Galasko DR, Masliah E, White CL, Troncoso JC, Hannequin D, Boxer AL, Geschwind MD, Kumar S, Mandelkow EM, Wszolek ZK, Uitti RJ, Dickson DW, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Ross OA, Rademakers R, Schellenberg GD, Miller BL, Mandelkow E, Geschwind DH (2012) Evidence for a role of the rare p. A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer’s diseases. Hum Mol Genet 21:3500–3512. https://doi.org/10.1093/hmg/dds161
Min SW, Chen X, Tracy TE, Li Y, Zhou Y, Wang C, Shirakawa K, Minami SS, Defensor E, Mok SA, Sohn PD, Schilling B, Cong X, Ellerby L, Gibson BW, Johnson J, Krogan N, Shamloo M, Gestwicki J, Masliah E, Verdin E, Gan L (2015) Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med 21:1154–1162. https://doi.org/10.1038/nm.3951
Irwin DJ, Cohen TJ, Grossman M, Arnold SE, Xie SX, Lee VM, Trojanowski JQ (2012) Acetylated tau, a novel pathological signature in Alzheimer’s disease and other tauopathies. Brain 135:807–818. https://doi.org/10.1093/brain/aws013
Alquezar C, Schoch KM, Geier EG, Ramos EM, Scrivo A, Li KH, Argouarch AR, Mlynarski EE, Dombroski B, DeTure M, Dickson DW, Yokoyama JS, Cuervo AM, Burlingame AL, Schellenberg GD, Miller TM, Miller BL, Kao AW (2021) TSC1 loss increases risk for tauopathy by inducing tau acetylation and preventing tau clearance via chaperone-mediated autophagy. Sci Adv 7:3897. https://doi.org/10.1126/sciadv.abg3897
Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, Meyers D, Cole PA, Ott M, Gan L (2010) Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67:953–966. https://doi.org/10.1016/j.neuron.2010.08.044
Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, Lee VM (2011) The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun 2:252. https://doi.org/10.1038/ncomms1255
Choi H, Kim HJ, Yang J, Chae S, Lee W, Chung S, Kim J, Choi H, Song H, Lee CK, Jun JH, Lee YJ, Lee K, Kim S, Sim HR, Choi YI, Ryu KH, Park JC, Lee D, Han SH, Hwang D, Kyung J, Mook-Jung I (2020) Acetylation changes tau interactome to degrade tau in Alzheimer’s disease animal and organoid models. Aging Cell 19:e13081. https://doi.org/10.1111/acel.13081
Xia Y, Bell BM, Giasson BI (2021) Tau K321/K353 pseudoacetylation within KXGS motifs regulates tau-microtubule interactions and inhibits aggregation. Sci Rep 11:17069. https://doi.org/10.1038/s41598-021-96627-7
Xu X, Sun Y, Cen X, Shan B, Zhao Q, Xie T, Wang Z, Hou T, Xue Y, Zhang M, Peng D, Sun Q, Yi C, Najafov A, Xia H (2021) Metformin activates chaperone-mediated autophagy and improves disease pathologies in an Alzheimer disease mouse model. Protein Cell 12:769–787. https://doi.org/10.1007/s13238-021-00858-3
Dou J, Su P, Xu C, Wen Z, Mao Z, Li W (2020) Targeting Hsc70-based autophagy to eliminate amyloid β oligomers. Biochem Biophys Res Commun 524:923–928. https://doi.org/10.1016/j.bbrc.2020.02.016
Park JS, Kim DH, Yoon SY (2016) Regulation of amyloid precursor protein processing by its KFERQ motif. BMB Rep 49:337–342. https://doi.org/10.5483/bmbrep.2016.49.6.212
Wu Y, Ly PT, Song W (2014) Aberrant expression of RCAN1 in Alzheimer’s pathogenesis: a new molecular mechanism and a novel drug target. Mol Neurobiol 50:1085–1097. https://doi.org/10.1007/s12035-014-8704-y
Liu H, Wang P, Song W, Sun X (2009) Degradation of regulator of calcineurin 1 (RCAN1) is mediated by both chaperone-mediated autophagy and ubiquitin proteasome pathways. FASEB J 23:3383–3392. https://doi.org/10.1096/fj.09-134296
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D (2004) Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305:1292–1295. https://doi.org/10.1126/science.1101738
Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L (2008) Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem 283:23542–23556. https://doi.org/10.1074/jbc.M801992200
Martinez-Vicente M, Talloczy Z, Kaushik S, Massey AC, Mazzulli J, Mosharov EV, Hodara R, Fredenburg R, Wu DC, Follenzi A, Dauer W, Przedborski S, Ischiropoulos H, Lansbury PT, Sulzer D, Cuervo AM (2008) Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest 118:777–788. https://doi.org/10.1172/jci32806
Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, Raya A, Sulzer D, Cuervo AM (2013) Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci 16:394–406. https://doi.org/10.1038/nn.3350
Yue Z, Yang XW (2013) Dangerous duet: LRRK2 and α-synuclein jam at CMA. Nat Neurosci 16:375–377. https://doi.org/10.1038/nn.3361
Ho PW, Leung CT, Liu H, Pang SY, Lam CS, Xian J, Li L, Kung MH, Ramsden DB, Ho SL (2020) Age-dependent accumulation of oligomeric SNCA/α-synuclein from impaired degradation in mutant LRRK2 knockin mouse model of Parkinson disease: role for therapeutic activation of chaperone-mediated autophagy (CMA). Autophagy 16:347–370. https://doi.org/10.1080/15548627.2019.1603545
Papadopoulos VE, Nikolopoulou G, Antoniadou I, Karachaliou A, Arianoglou G, Emmanouilidou E, Sardi SP, Stefanis L, Vekrellis K (2018) Modulation of β-glucocerebrosidase increases α-synuclein secretion and exosome release in mouse models of Parkinson’s disease. Hum Mol Genet 27:1696–1710. https://doi.org/10.1093/hmg/ddy075
Kuo SH, Tasset I, Cheng MM, Diaz A, Pan MK, Lieberman OJ, Hutten SJ, Alcalay RN, Kim S, Ximénez-Embún P, Fan L, Kim D, Ko HS, Yacoubian T, Kanter E, Liu L, Tang G, Muñoz J, Sardi SP, Li A, Gan L, Cuervo AM, Sulzer D (2022) Mutant glucocerebrosidase impairs α-synuclein degradation by blockade of chaperone-mediated autophagy. Sci Adv 8:6393. https://doi.org/10.1126/sciadv.abm6393
Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D (2011) Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146:37–52. https://doi.org/10.1016/j.cell.2011.06.001
De Miranda BR, Rocha EM, Bai Q, El Ayadi A, Hinkle D, Burton EA, Timothy Greenamyre J (2018) Astrocyte-specific DJ-1 overexpression protects against rotenone-induced neurotoxicity in a rat model of Parkinson’s disease. Neurobiol Dis 115:101–114. https://doi.org/10.1016/j.nbd.2018.04.008
Sala G, Marinig D, Arosio A, Ferrarese C (2016) Role of CHAPERONE-MEDIATED AUTOPHAGY DYSFUNCTIONS IN THE PATHOGENESIS OF PARKINSON’S DISEASE. Front Mol Neurosci 9:157. https://doi.org/10.3389/fnmol.2016.00157
Wang B, Cai Z, Tao K, Zeng W, Lu F, Yang R, Feng D, Gao G, Yang Q (2016) Essential control of mitochondrial morphology and function by chaperone-mediated autophagy through degradation of PARK7. Autophagy 12:1215–1228. https://doi.org/10.1080/15548627.2016.1179401
Brekk OR, Makridakis M, Mavroeidi P, Vlahou A, Xilouri M, Stefanis L (2019) Impairment of chaperone-mediated autophagy affects neuronal homeostasis through altered expression of DJ-1 and CRMP-2 proteins. Mol Cell Neurosci 95:1–12. https://doi.org/10.1016/j.mcn.2018.12.006
Xu CY, Kang WY, Chen YM, Jiang TF, Zhang J, Zhang LN, Ding JQ, Liu J, Chen SD (2017) DJ-1 inhibits α-synuclein aggregation by regulating chaperone-mediated autophagy. Front Aging Neurosci 9:308. https://doi.org/10.3389/fnagi.2017.00308
Kabuta T, Furuta A, Aoki S, Furuta K, Wada K (2008) Aberrant interaction between Parkinson disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J Biol Chem 283:23731–23738. https://doi.org/10.1074/jbc.M801918200
Kabuta T, Wada K (2008) Insights into links between familial and sporadic Parkinson’s disease: physical relationship between UCH-L1 variants and chaperone-mediated autophagy. Autophagy 4:827–829. https://doi.org/10.4161/auto.6560
Mondello S, Constantinescu R, Zetterberg H, Andreasson U, Holmberg B, Jeromin A (2014) CSF α-synuclein and UCH-L1 levels in Parkinson’s disease and atypical parkinsonian disorders. Parkinsonism Relat Disord 20:382–387. https://doi.org/10.1016/j.parkreldis.2014.01.011
Tang FL, Erion JR, Tian Y, Liu W, Yin DM, Ye J, Tang B, Mei L, Xiong WC (2015) VPS35 in dopamine neurons is required for endosome-to-golgi retrieval of Lamp2a, a receptor of chaperone-mediated autophagy that is critical for α-synuclein degradation and prevention of pathogenesis of parkinson’s disease. J Neurosci 35:10613–10628. https://doi.org/10.1523/jneurosci.0042-15.2015
Nie T, Tao K, Zhu L, Huang L, Hu S, Yang R, Xu P, Mao Z, Yang Q (2021) Chaperone-mediated autophagy controls the turnover of E3 ubiquitin ligase MARCHF5 and regulates mitochondrial dynamics. Autophagy 17:2923–2938. https://doi.org/10.1080/15548627.2020.1848128
She H, Yang Q, Shepherd K, Smith Y, Miller G, Testa C, Mao Z (2011) Direct regulation of complex I by mitochondrial MEF2D is disrupted in a mouse model of Parkinson disease and in human patients. J Clin Invest 121:930–940. https://doi.org/10.1172/jci43871
Yang Q, Mao Z (2009) The complexity in regulation of MEF2D by chaperone-mediated autophagy. Autophagy 5:1073–1074. https://doi.org/10.4161/auto.5.7.9824
Gao L, She H, Li W, Zeng J, Zhu J, Jones DP, Mao Z, Gao G, Yang Q (2014) Oxidation of survival factor MEF2D in neuronal death and Parkinson’s disease. Antioxid Redox Signal 20:2936–2948. https://doi.org/10.1089/ars.2013.5399
Huang L, Deng M, He Y, Lu S, Liu S, Fang Y (2016) β-asarone increases MEF2D and TH levels and reduces α-synuclein level in 6-OHDA-induced rats via regulating the HSP70/MAPK/MEF2D/Beclin-1 pathway: Chaperone-mediated autophagy activation, macroautophagy inhibition and HSP70 up-expression. Behav Brain Res 313:370–379. https://doi.org/10.1016/j.bbr.2016.07.028
Ren H, Zhai W, Lu X, Wang G (2021) The cross-links of endoplasmic reticulum stress, autophagy, and neurodegeneration in parkinson’s disease. Front Aging Neurosci 13:691881. https://doi.org/10.3389/fnagi.2021.691881
Li W, Zhu J, Dou J, She H, Tao K, Xu H, Yang Q, Mao Z (2017) Phosphorylation of LAMP2A by p38 MAPK couples ER stress to chaperone-mediated autophagy. Nat Commun 8:1763. https://doi.org/10.1038/s41467-017-01609-x
Iwata A, Maruyama M, Kanazawa I, Nukina N (2001) Alpha-Synuclein affects the MAPK pathway and accelerates cell death. J Biol Chem 276:45320–45329. https://doi.org/10.1074/jbc.M103736200
Chen CY, Weng YH, Chien KY, Lin KJ, Yeh TH, Cheng YP, Lu CS, Wang HL (2012) (G2019S) LRRK2 activates MKK4-JNK pathway and causes degeneration of SN dopaminergic neurons in a transgenic mouse model of PD. Cell Death Differ 19:1623–1633. https://doi.org/10.1038/cdd.2012.42
Zhu Y, Wang C, Yu M, Cui J, Liu L, Xu Z (2013) ULK1 and JNK are involved in mitophagy incurred by LRRK2 G2019S expression. Protein Cell 4:711–721. https://doi.org/10.1007/s13238-013-3910-3
Yang J, Kim KS, Iyirhiaro GO, Marcogliese PC, Callaghan SM, Qu D, Kim WJ, Slack RS, Park DS (2019) DJ-1 modulates the unfolded protein response and cell death via upregulation of ATF4 following ER stress. Cell Death Dis 10:135. https://doi.org/10.1038/s41419-019-1354-2
Kuo SH, Tasset I, Cuervo AM, Sulzer D (2022) Misfolded GBA/β-glucocerebrosidase impairs ER-quality control by chaperone-mediated autophagy in Parkinson disease. Autophagy 1:1–3. https://doi.org/10.1080/15548627.2022.2071383
Cortes CJ, La Spada AR (2014) The many faces of autophagy dysfunction in Huntington’s disease: from mechanism to therapy. Drug Discov Today 19:963–971. https://doi.org/10.1016/j.drudis.2014.02.014
Thompson LM, Aiken CT, Kaltenbach LS, Agrawal N, Illes K, Khoshnan A, Martinez-Vincente M, Arrasate M, O’Rourke JG, Khashwji H, Lukacsovich T, Zhu YZ, Lau AL, Massey A, Hayden MR, Zeitlin SO, Finkbeiner S, Green KN, LaFerla FM, Bates G, Huang L, Patterson PH, Lo DC, Cuervo AM, Marsh JL, Steffan JS (2009) IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J Cell Biol 187:1083–1099. https://doi.org/10.1083/jcb.200909067
Qi L, Zhang XD, Wu JC, Lin F, Wang J, DiFiglia M, Qin ZH (2012) The role of chaperone-mediated autophagy in huntingtin degradation. PLoS ONE 7:e46834. https://doi.org/10.1371/journal.pone.0046834
Choi SH, Cho K (2021) LAMP2A-mediated autophagy involved in Huntington’s disease progression. Biochem Biophys Res Commun 534:561–567. https://doi.org/10.1016/j.bbrc.2020.11.042
Qi L, Zhang XD (2014) Role of chaperone-mediated autophagy in degrading Huntington’s disease-associated huntingtin protein. Acta Biochim Biophys Sin 46:83–91. https://doi.org/10.1093/abbs/gmt133
Koga H, Martinez-Vicente M, Arias E, Kaushik S, Sulzer D, Cuervo AM (2011) Constitutive upregulation of chaperone-mediated autophagy in Huntington’s disease. J Neurosci 31:18492–18505. https://doi.org/10.1523/jneurosci.3219-11.2011
Bauer PO, Goswami A, Wong HK, Okuno M, Kurosawa M, Yamada M, Miyazaki H, Matsumoto G, Kino Y, Nagai Y, Nukina N (2010) Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat Biotechnol 28:256–263. https://doi.org/10.1038/nbt.1608
Suk TR, Rousseaux MWC (2020) The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol Neurodegener 15:45. https://doi.org/10.1186/s13024-020-00397-1
Walker AK, Spiller KJ, Ge G, Zheng A, Xu Y, Zhou M, Tripathy K, Kwong LK, Trojanowski JQ, Lee VM (2015) Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol 130:643–660. https://doi.org/10.1007/s00401-015-1460-x
Montalbano M, McAllen S, Cascio FL, Sengupta U, Garcia S, Bhatt N, Ellsworth A, Heidelman EA, Johnson OD, Doskocil S, Kayed R (2020) TDP-43 and tau oligomers in alzheimer’s disease, amyotrophic lateral sclerosis, and frontotemporal dementia. Neurobiol Dis 146:105130. https://doi.org/10.1016/j.nbd.2020.105130
de Boer EMJ, Orie VK, Williams T, Baker MR, De Oliveira HM, Polvikoski T, Silsby M, Menon P, van den Bos M, Halliday GM, van den Berg LH, Van Den Bosch L, van Damme P, Kiernan MC, van Es MA, Vucic S (2020) TDP-43 proteinopathies: a new wave of neurodegenerative diseases. J Neurol Neurosurg Psychiatry 92:86–95. https://doi.org/10.1136/jnnp-2020-322983
Huang CC, Bose JK, Majumder P, Lee KH, Huang JT, Huang JK, Shen CK (2014) Metabolism and mis-metabolism of the neuropathological signature protein TDP-43. J Cell Sci 127:3024–3038. https://doi.org/10.1242/jcs.136150
Ormeño F, Hormazabal J, Moreno J, Riquelme F, Rios J, Criollo A, Albornoz A, Alfaro IE, Budini M (2020) Chaperone mediated autophagy degrades TDP-43 protein and is affected by TDP-43 aggregation. Front Mol Neurosci 13:19. https://doi.org/10.3389/fnmol.2020.00019
Arosio A, Cristofani R, Pansarasa O, Crippa V, Riva C, Sirtori R, Rodriguez-Menendez V, Riva N, Gerardi F, Lunetta C, Cereda C, Poletti A, Ferrarese C, Tremolizzo L, Sala G (2020) HSC70 expression is reduced in lymphomonocytes of sporadic ALS patients and contributes to TDP-43 accumulation. Amyotroph Lateral Scler Frontotemporal Degener 21:51–62. https://doi.org/10.1080/21678421.2019.1672749
Tamaki Y, Shodai A, Morimura T, Hikiami R, Minamiyama S, Ayaki T, Tooyama I, Furukawa Y, Takahashi R, Urushitani M (2018) Elimination of TDP-43 inclusions linked to amyotrophic lateral sclerosis by a misfolding-specific intrabody with dual proteolytic signals. Sci Rep 8:6030. https://doi.org/10.1038/s41598-018-24463-3
Klockgether T, Mariotti C, Paulson HL (2019) Spinocerebellar ataxia. Nat Rev Dis Primers 5:24. https://doi.org/10.1038/s41572-019-0074-3
Alves S, Cormier-Dequaire F, Marinello M, Marais T, Muriel MP, Beaumatin F, Charbonnier-Beaupel F, Tahiri K, Seilhean D, El Hachimi K, Ruberg M, Stevanin G, Barkats M, den Dunnen W, Priault M, Brice A, Durr A, Corvol JC, Sittler A (2014) The autophagy/lysosome pathway is impaired in SCA7 patients and SCA7 knock-in mice. Acta Neuropathol 128:705–722. https://doi.org/10.1007/s00401-014-1289-8
Unno T, Wakamori M, Koike M, Uchiyama Y, Ishikawa K, Kubota H, Yoshida T, Sasakawa H, Peters C, Mizusawa H, Watase K (2012) Development of Purkinje cell degeneration in a knockin mouse model reveals lysosomal involvement in the pathogenesis of SCA6. Proc Natl Acad Sci USA 109:17693–17698. https://doi.org/10.1073/pnas.1212786109
Seki T, Sato M, Kibe Y, Ohta T, Oshima M, Konno A, Hirai H, Kurauchi Y, Hisatsune A, Katsuki H (2018) Lysosomal dysfunction and early glial activation are involved in the pathogenesis of spinocerebellar ataxia type 21 caused by mutant transmembrane protein 240. Neurobiol Dis 120:34–50. https://doi.org/10.1016/j.nbd.2018.08.022
Sato M, Ohta T, Morikawa Y, Konno A, Hirai H, Kurauchi Y, Hisatsune A, Katsuki H, Seki T (2021) Ataxic phenotype and neurodegeneration are triggered by the impairment of chaperone-mediated autophagy in cerebellar neurons. Neuropathol Appl Neurobiol 47:198–209. https://doi.org/10.1111/nan.12649
Seki T, Yoshino KI, Tanaka S, Dohi E, Onji T, Yamamoto K, Hide I, Paulson HL, Saito N, Sakai N (2012) Establishment of a novel fluorescence-based method to evaluate chaperone-mediated autophagy in a single neuron. PLoS ONE 7:e31232. https://doi.org/10.1371/journal.pone.0031232
Seki T, Adachi N, Abe-Seki N, Shimahara T, Takahashi H, Yamamoto K, Saito N, Sakai N (2011) Elucidation of the molecular mechanism and exploration of novel therapeutics for spinocerebellar ataxia caused by mutant protein kinase Cγ. J Pharmacol Sci 116:239–247. https://doi.org/10.1254/jphs.11r04cp
Delplanque J, Devos D, Huin V, Genet A, Sand O, Moreau C, Goizet C, Charles P, Anheim M, Monin ML, Buée L, Destée A, Grolez G, Delmaire C, Dujardin K, Dellacherie D, Brice A, Stevanin G, Strubi-Vuillaume I, Dürr A, Sablonnière B (2014) TMEM240 mutations cause spinocerebellar ataxia 21 with mental retardation and severe cognitive impairment. Brain 137:2657–2663. https://doi.org/10.1093/brain/awu202
Duncan C, Papanikolaou T, Ellerby LM (2010) Autophagy: polyQ toxic fragment turnover. Autophagy 6:312–314. https://doi.org/10.4161/auto.6.2.11139
Vallabh SM, Minikel EV, Schreiber SL, Lander ES (2020) Towards a treatment for genetic prion disease: trials and biomarkers. Lancet Neurol 19:361–368. https://doi.org/10.1016/s1474-4422(19)30403-x
Wang H, Tian C, Xu Y, Xie WL, Zhang J, Zhang BY, Ren K, Wang K, Chen C, Wang SB, Shi Q, Shao QX, Dong XP (2013) Abortive cell cycle events in the brains of scrapie-infected hamsters with remarkable decreases of PLK3/Cdc25C and increases of PLK1/cyclin B1. Mol Neurobiol 48:655–668. https://doi.org/10.1007/s12035-013-8455-1
Wang H, Tian C, Fan XY, Chen LN, Lv Y, Sun J, Zhao YJ, Zhang LB, Wang J, Shi Q, Gao C, Chen C, Shao QX, Dong XP (2015) Polo-like kinase 3 (PLK3) mediates the clearance of the accumulated PrP mutants transiently expressed in cultured cells and pathogenic PrP(Sc) in prion infected cell line via protein interaction. Int J Biochem Cell Biol 62:24–35. https://doi.org/10.1016/j.biocel.2015.02.011
Wang H, Tian C, Sun J, Chen LN, Lv Y, Yang XD, Xiao K, Wang J, Chen C, Shi Q, Shao QX, Dong XP (2017) Overexpression of PLK3 mediates the degradation of abnormal prion proteins dependent on chaperone-mediated autophagy. Mol Neurobiol 54:4401–4413. https://doi.org/10.1007/s12035-016-9985-0
Scrivo A, Bourdenx M, Pampliega O, Cuervo AM (2018) Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol 17:802–815. https://doi.org/10.1016/s1474-4422(18)30238-2
Lee JH, McBrayer MK, Wolfe DM, Haslett LJ, Kumar A, Sato Y, Lie PP, Mohan P, Coffey EE, Kompella U, Mitchell CH, Lloyd-Evans E, Nixon RA (2015) Presenilin 1 maintains lysosomal Ca(2+) homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep 12:1430–1444. https://doi.org/10.1016/j.celrep.2015.07.050
Bourdenx M, Daniel J, Genin E, Soria FN, Blanchard-Desce M, Bezard E, Dehay B (2016) Nanoparticles restore lysosomal acidification defects: implications for Parkinson and other lysosomal-related diseases. Autophagy 12:472–483. https://doi.org/10.1080/15548627.2015.1136769
Cuervo AM, Dice JF (2000) Regulation of lamp2a levels in the lysosomal membrane. Traffic 1:570–583. https://doi.org/10.1034/j.1600-0854.2000.010707.x
Cuervo AM, Mann L, Bonten EJ, d’Azzo A, Dice JF (2003) Cathepsin A regulates chaperone-mediated autophagy through cleavage of the lysosomal receptor. EMBO J 22:47–59. https://doi.org/10.1093/emboj/cdg002
Catarino S, Pereira P, Girão H (2017) Molecular control of chaperone-mediated autophagy. Essays Biochem 61:663–674. https://doi.org/10.1042/ebc20170057
Anguiano J, Garner TP, Mahalingam M, Das BC, Gavathiotis E, Cuervo AM (2013) Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat Chem Biol 9:374–382. https://doi.org/10.1038/nchembio.1230
Kaushik S, Tasset I, Arias E, Pampliega O, Wong E, Martinez-Vicente M, Cuervo AM (2021) Autophagy and the hallmarks of aging. Ageing Res Rev 72:101468. https://doi.org/10.1016/j.arr.2021.101468
Gomez-Sintes R, Xin Q, Jimenez-Loygorri JI, McCabe M, Diaz A, Garner TP, Cotto-Rios XM, Wu Y, Dong S, Reynolds CA, Patel B, de la Villa P, Macian F, Boya P, Gavathiotis E, Cuervo AM (2022) Targeting retinoic acid receptor alpha-corepressor interaction activates chaperone-mediated autophagy and protects against retinal degeneration. Nat Commun 13:4220. https://doi.org/10.1038/s41467-022-31869-1
Lee YS, Lai DM, Huang HJ, Lee-Chen GJ, Chang CH, Hsieh-Li HM, Lee GC (2021) Prebiotic lactulose ameliorates the cognitive deficit in alzheimer’s disease mouse model through macroautophagy and chaperone-mediated autophagy pathways. J Agric Food Chem 69:2422–2437. https://doi.org/10.1021/acs.jafc.0c07327
Luan Y, Ren X, Zheng W, Zeng Z, Guo Y, Hou Z, Guo W, Chen X, Li F, Chen JF (2018) Chronic caffeine treatment protects against α-synucleinopathy by reestablishing autophagy activity in the mouse striatum. Front Neurosci 12:301. https://doi.org/10.3389/fnins.2018.00301
Wu JZ, Ardah M, Haikal C, Svanbergsson A, Diepenbroek M, Vaikath NN, Li W, Wang ZY, Outeiro TF, El-Agnaf OM, Li JY (2019) Dihydromyricetin and Salvianolic acid B inhibit alpha-synuclein aggregation and enhance chaperone-mediated autophagy. Transl Neurodegener 8:18. https://doi.org/10.1186/s40035-019-0159-7
Wang X, Zhai H, Wang F (2018) 6-OHDA induces oxidation of F-box protein Fbw7β by chaperone-mediated autophagy in Parkinson’s model. Mol Neurobiol 55:4825–4833. https://doi.org/10.1007/s12035-017-0686-0
Limegrover CS, Yurko R, Izzo NJ, LaBarbera KM, Rehak C, Look G, Rishton G, Safferstein H, Catalano SM (2021) Sigma-2 receptor antagonists rescue neuronal dysfunction induced by Parkinson’s patient brain-derived α-synuclein. J Neurosci Res 99:1161–1176. https://doi.org/10.1002/jnr.24782
Cui H, Norrbacka S, Myöhänen TT (2022) Prolyl oligopeptidase acts as a link between chaperone-mediated autophagy and macroautophagy. Biochem Pharmacol 197:114899. https://doi.org/10.1016/j.bcp.2021.114899
Rodriguez-Navarro JA, Kaushik S, Koga H, Dall’Armi C, Shui G, Wenk MR, Di Paolo G, Cuervo AM (2012) Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc Natl Acad Sci USA 109:E705–E714. https://doi.org/10.1073/pnas.1113036109
Funding
This work was supported by grants from the National Natural Science Foundation of China (81971032), Taishan Scholars Program of Shandong Province (tsqn20161078), and Medical Science Research Guidance Plan of Qingdao (2021-WJZD001).
Author information
Authors and Affiliations
Contributions
YL, MST, and LT did the manuscript preparation and drafting. MST and LT are responsible for the study conception and design. YL and MST wrote the first draft of the manuscript. All authors have contributed to the manuscript revising and editing critically for important intellectual content and given final approval of the version and agreed to be accountable for all aspects of the work presented here.
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethical approval
Not applicable.
Research involving human participants and/or animals
Not applicable.
Informed consent
Not applicable.
Consent to participate
Not applicable.
Consent for publication
All authors have given final approval of the version and agreed with the publication of this study here.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liu, Y., Tan, L. & Tan, MS. Chaperone-mediated autophagy in neurodegenerative diseases: mechanisms and therapy. Mol Cell Biochem 478, 2173–2190 (2023). https://doi.org/10.1007/s11010-022-04640-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-022-04640-9