
Abstract
Purpose of Review
It is well established that T helper type 2 (TH2) immune responses are necessary not only to provide protection against helminth parasites but also to promote the detrimental inflammation associated with allergies and asthma. Given the importance of type 2 immunity and inflammation, many studies have focused on better understanding the factors that regulate TH2 cell development and activation. As a result, significant progress has been made in understanding the signaling pathways and molecular events necessary to promote TH2 cell polarization. In addition to the adaptive compartment, emerging studies are better defining the innate immune pathways needed to promote TH2 cell responses. Given the recent and substantial growth of this field, the purpose of this review is to highlight recent studies defining the innate immune events that promote immunity to helminth parasites and allergic inflammation.
Recent Findings
Emerging studies have begun to elucidate the importance of cytokine alarmins such as thymic stromal lymphopoietin (TSLP), IL-25 (IL-17E), and IL-33 in promoting type 2 immunity and inflammation following helminth challenge or exposure to allergens. Specifically, recent reports have begun to define the complex cellular networks these alarmins activate and their contribution to type 2 immunity and inflammation.
Summary
Our increased understanding of the pathways that regulate type 2 cytokine-mediated immunity and inflammation have revealed novel therapeutic targets to treat both helminth infections and allergic disease states.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
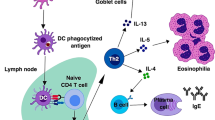
Helminth parasites have infected humans for millennia and continue to be a problem in developing countries where they affect billions of people [1]. Helminth infections cause malnutrition, anemia, and impaired cognitive functions as well as greatly impact impoverished areas of the world, which generally lack access to proper sanitation [2]. Mammals mount a potent T helper type 2 (TH2) immune response to protect against helminth infections, which usually occur at mucosal or barrier surfaces [3]. The induction of type 2 immunity involves the activation and coordinated interplay between hematopoietic and non-hematopoietic cells that release a complex array of cytokines and other inflammatory mediators necessary to promote immunity [3, 4]. The proper initiation of TH2 responses not only results in reduced parasite burdens but also promotes wound-healing events necessary to maintain the integrity of affected tissues. Type 2 immune responses involve the cooperative actions of epithelial cells, dendritic cells (DCs), basophils, mast cells, eosinophils, type 2 innate lymphoid cells (ILC2s), alternatively activated M2 macrophages, TH2 cells, and IgE-producing B cells. In addition to these terminally differentiated cell types, there is also a growing appreciation for the role that hematopoietic stem/progenitor cells (HSPCs) play in promoting and maintaining type 2 inflammation [5].
A variety of cytokines are produced in the context of type 2 responses and they promote well-defined events that regulate TH2 cell development and type 2 inflammation. The classical type 2 cytokines, which include interleukin (IL)-4, IL-5, IL-9, and IL-13, are known to assist in polarizing naïve CD4+ T cells into TH2 cells as well as to promote other critical events [6]. For example, IL-4 promotes isotype class switching and IgE production by B cells. IL-5 is crucial in the expansion and activation of eosinophils. IL-9 has been demonstrated to activate mast cells and ILC2s. IL-13 is implicated to play a critical role in promoting goblet cell hyperplasia, mucus hypersecretion, and smooth muscle hyperactivity. Furthermore, IL-13 and IL-4 induce the polarization of macrophages to a M2 or alternatively activated phenotype [7]. In addition to type 2 cytokines, recent reports have also defined the importance of cytokine alarmins (TSLP, IL-25, and IL-33) in promoting many of the events described above [8]. The below sections will highlight the cellular regulators of the above effector molecules and pathways, describe their unique and distinct contribution to type 2 inflammatory responses, and outline how these pathways promote host-protective responses in the context of helminth infections.
In addition to immunity to helminth parasites, type 2 cytokine responses also promote the inflammation associated with allergic disease states, which are reaching epidemic proportions in developed areas of the world. For example, in the United States, 28% of children are sensitized to food allergens and among them, 5 to 7% suffer from food allergies [9]. Further, the prevalence of asthma has dramatically increased over the past several decades in a number of developed nations [10]. Allergic responses occur when certain innocuous allergens trigger dysregulated acute and/or chronic TH2 inflammation leading to conditions such as asthma, atopic dermatitis, eosinophilic esophagitis, and allergic rhinitis [11]. Therefore, this review will also briefly highlight how many of the cellular and molecular events initiated following a helminth challenge are known to contribute to these pathologic conditions. First, we will briefly mention the well-defined role of the adaptive immune compartment in TH2 responses. Then, we will highlight the recent studies defining the contribution of innate immune cells to type 2 immunity and inflammation. Finally, we will describe how better understanding these pathways may inform the development of new therapeutic strategies to treat helminth infections and allergic inflammation.
Adaptive Immunity
TH2 Lymphocytes
The contributions of T lymphocytes to anti-helminth immunity and allergic disorders have been highly studied, and the importance of T cells in regulating these inflammatory responses is well defined. For example, seminal murine studies illustrated that depletion of CD4+ T cells resulted in impaired worm expulsion following infection with Nippostrongylus brasiliensis [12], Heligmosomoides polygyrus [13], Trichuris muris [14], and Trichinella spiralis [15] as well as prevented the development of protective immunity after vaccination with Schistosoma mansoni [16]. Similarly, CD4+ T cell-depletion prevented the development of allergic immunopathology after exposure to allergens such as house dust mite (HDM) [17] and in models of antigen-induced allergic inflammation [18]. Collectively, these studies have left little doubt of the importance of T cells in promoting type 2 inflammatory responses as reviewed here [6, 19]. In the context of both helminth infections and allergic inflammation, CD4+ T cells are exposed to IL-4 and IL-13, which bind to the IL-4 receptor alpha (IL-4Rα) on T cells and activate the signal transducer and activation of transcription 6 (STAT6) signaling pathway inducing a very specific transcriptional program, chracterized by the expression of the transcription factor GATA3 [6, 19]. Activated TH2 cells secrete cytokines such as IL-4, IL-5, IL-9, and IL-13, which promote many aspects of type 2 immunity that are described above and are comprehensively reviewed in several articles [6, 19]. Thus, the production of these effector molecules by TH2 cells is capable of directly regulating the important events necessary to promote immunity to helminths and allergic inflammation.
In addition to the classic induction of TH2 cells during helminth infection and allergic inflammation, novel studies suggest that other CD4+ T cell subsets such as TH9, TH17, TH22, and TFH might regulate type 2 inflammation [19]. However, the precise interactions between these CD4+ T cell subsets during helminth infections and allergic diseases remain to be fully defined and require further investigation.
Antigen Presentation
The antigen presenting cells (APCs) required to promote TH2 cell development have been a much studied and debated topic and therefore will only be briefly addressed in this section. The balance of the data strongly suggests that the antigen-specific activation of TH2 cells is mediated mainly by dendritic cells [20], although other immune cells such as eosinophils, basophils, and ILC2s have been shown to contribute to the establishment of optimal type 2 responses as will be described below. The priming of TH2 cells by DCs seems to be partially dependent on cell-cell surface interactions especially by the signaling molecule OX40L. Interestingly, cytokine alarmins and helminth-derived products have been shown to induce the expression of OX40L on DCs [21, 22], supporting the notion that a complex interplay of early events regulates TH2 cell development. Furthermore, the establishment of allergic inflammation can be prevented by blockade of OX40 or OX40L [23, 24], demonstrating the important role this particular signaling pathway can play in the development of TH2 responses. A more comprehensive review of the role of DCs and other APCs in promoting type 2 immunity and inflammation can be found in the following articles [25, 26].
B Lymphocytes
B cell activation is a common feature of both helminth infections and allergic inflammation. B cells activated in the context of strong type 2 immune responses are known to undergo class switching and produce robust levels of antigen-specific IgE [27]. Antigen-specific IgE is capable of binding to the surface of FcεRIα-expressing cell populations such as basophils and mast cells. Upon secondary exposure to helminth-derived products or allergens, surface-bound IgE can interact with relevant antigens and trigger the degranulation and release of effector molecules from mast cell and basophil populations. This process is well defined and has been shown to play a role in promoting immunity to several helminth parasites in the context of a secondary challenge [28]. Further, IgE-mediated inflammatory reactions to allergens are the prevailing causes of most allergic diseases and are responsible for life-threatening anaphylaxis reactions caused by exposure to food antigens or insect stings. However, IgE antibodies seem to be dispensable for protective immunity to H. polygyrus, N. brasiliensis, or S. mansoni [29]. By contrast, IgG and IgM antibodies have been reported to provide protection against H. polygyrus, Schistosoma species, N. brasiliensis, T. muris, T. spiralis, and B. malayi [29]. Further, IgG1+ B cells were required for the generation of memory IgE responses to N. brasiliensis infection [30]. Interestingly, B cell-deficient mice exhibit increased worm burdens after secondary infection with H. polygyrus but not after infection with N. brasiliensis or T. spiralis [29], suggesting that B cells may play distinct roles in a parasite-specific manner. In addition to antibody production, B cells have been proposed to contribute to TH2 polarization via expression of co-stimulatory molecules such as OX40L and B7 after infection with N. brasiliensis and H. polygyrus [31, 32]. In return, T cell-derived IL-4 and IL-13 signal through STAT6 on B cells, promoting the generation of germinal center responses [33]. This pathway and its clinical importance are compressively described in several review articles [29, 34, 35].
Innate Immunity
Type 2 Innate Lymphoid Cells
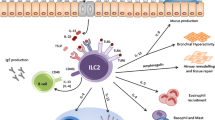
Despite the necessary contributions of adaptive responses to anti-helminth immunity and allergic inflammation as described above, the development of substantial type 2 inflammation in lymphocyte-deficient mice demonstrates that complex and potent innate immune pathways are initiated upstream of T cell responses [36]. Recently, independent groups reported the presence of a small population of tissue-resident cells that contributed to protective immunity to N. brasiliensis via their potent production of IL-5 and IL-13 [37,38,39]. Remarkably, this population lacked the expression of lineage markers associated with common lymphoid and myeloid lineages and has since been defined as type 2 innate lymphoid cells (ILC2s) as a result of their expression of the TH2 cell-associated transcription factor GATA-3 and ability to promote type 2 inflammation [40]. ILC2 populations have been found in lymphoid and non-lymphoid peripheral sites such as the lungs, intestines, skin, liver, nasal polyps, and adipose tissue of humans and mice [41,42,43]. Despite the absence of common lineage markers, ILC2s can be identified by the expression of surface molecules such as CD25, CD90, CD127, CRTH2, MHCII, ICOS, and KLRG1. Although the expression of pattern recognition receptors (PRRs) has not been observed in ILC2s, the activation of ILC2 responses appears to be mediated by soluble mediators including cytokines (IL-25, IL-33, TSLP, IL-1β, IL-2, IL-7, IL-4, IL-9, and TL1A) and lipid mediators (prostaglandins and leukotrienes) [41,42,43]. In response to these signals, ILC2s promote host-protective responses via the secretion of soluble effector molecules such as IL-4, IL-5, IL-9, IL-13, and amphiregulin (Areg) [41,42,43]. Defining the developmental pathways of ILC2s has also been an area of active research. Briefly, mature ILC2s arise from bone marrow lymphoid progenitors, which express transcription factors such as Id2, PLZF, RORα, and GATA-3 [41,42,43]. ILC2 precursors are reported to traffic into specific mucosal sites via the actions of specific chemokine signals early during development [41,42,43]. In addition, ILC2s have a remarkable proliferative potential, which allows them to expand dramatically at peripheral sites in response to appropriate stimuli and contribute to inflammatory responses.
The essential role of ILC2s to host-protective responses against the nematode N. brasiliensis has been an active area of investigation. Infection with N. brasiliensis results in substantial tissue damage at mucosal sites such as the lungs and the gut. The damage to the epithelium induces the activation of ILC2 populations via the secretion of cytokine alarmins such as IL-25 and IL-33 [37,38,39] or the expression of lipid mediators such as prostaglandin D2 [44]. More specifically, recent studies have demonstrated that tuft cells are a specialized intestinal epithelial cell population capable of producing robust amounts of IL-25 following N. brasiliensis challenge, which in turn induces activation of ILC2s and promotes their secretion of IL-13 stimulating tuft-cell hyperplasia following infection [45, 46]. IL-25 is also reported to induce the expansion of a specific population of ILC2s in vivo, which has been termed “inflammatory ILC2s” [47]. Inflammatory ILC2s lack expression of the IL-33 receptor ST2, but express high levels of KLRG1 and show high levels of cell proliferation in response to IL-25 in vivo [47]. Remarkably, inflammatory ILC2s arise following infection with N. brasiliensis and acquire the expression of ST2 after activation [47], suggesting that “inflammatory ILC2s” may be an ILC2 progenitor or represent a distinct state of activation. Activated ILC2s produce robust amounts of TH2 cytokines including IL-5, IL-9, and IL-13 as well as other tissue remodeling factors such as arginase 1 (Arg1) [48] and Areg. ILC2-derived IL-5 induces the accumulation of eosinophils into affected tissues, while ILC2-derived IL-13 promotes goblet cell hyperplasia and contraction of smooth muscle cells to promote worm expulsion. In addition to alarmins and lipid mediators, the mast cell-activating cytokine IL-9 has also been shown to promote ILC2 activation [49], suggesting that ILC2-derived IL-9 may act in an autocrine manner and amplify cellular activation. Finally, ILC2-derived Areg has been shown to promote wound healing of the tissues affected by the passage of helminth parasites [50], suggesting that ILC2s can perform host-protective responses following parasite challenge.
In addition to their own effector functions, ILC2s have recently been shown to cooperate with CD4+ T cells to promote the alternative activation of macrophages and establish protective immunity against reinfection with N. brasiliensis [51]. Likewise, ILC2s have been shown to promote TH2 differentiation via their expression of MHCII [52], reinforcing the concept that cooperative interactions between innate and adaptive responses are required to promote optimal immunity and inflammation. To date, the contributions of ILC2s to host-protective responses in the context of other helminths are less defined. A recent study by Shimokawa et al. has shown that mast cells cooperate with ILC2s to promote immunity to H. polygyrus [53]. Specifically, they reported that ATP-activated mast cells promote ILC2 responses via their secretion of IL-33 and that these events contribute to reduced parasite burdens [53]. Additional studies report that ILC2s promote the differentiation of TH2 cells following H. polygyrus infection via their secretion of IL-4 [54], and preliminary reports suggest that ILC2s may contribute to host-protective responses against Strongyloides venezuelensis, T. muris, and Schistosoma species [55,56,57]. However, further studies are required to define the precise contribution of ILC2s in these infectious models.
In contrast to their less defined role in promoting anti-helminthic immunity, ILC2s are reported to play a major role in promoting the immunopathology associated with various mouse models of allergic disease. Prolonged exposure to protease allergens, such as papain and HDM, induces allergic-like pathology characterized by eosinophil accumulation, expression of TH2 cytokines, tissue remodeling, and airway hyper-responsiveness, even in the absence of adaptive immune responses [58, 59]. Intranasal treatment with papain results in elevated expression of IL-33 in the airway epithelium, which induces the activation of ILC2s and the development of airway inflammation [58, 60]. Specifically, type 2 alveolar cells have been shown to be a major producer of IL-33 in the lung and are thought to regulate ILC2s via production of this alarmin [56]. Further, a recent study by de Kleer et al. [61] demonstrated that ILC2s accumulate in the pulmonary tissue of newborn mice in an IL-33-dependent manner. They further went on to demonstrate that early exposure to HDM allergen resulted in exacerbated airway inflammation as compared to allergen exposure at later developmental stages, which could be prevented by blockade with decoy sST2 [61]. Similarly, studies have also demonstrated that ILC2s contribute to OVA-induced airway inflammation and pathology [60]. These elegant murine studies are supported by multiple reports demonstrating increased ILC2 numbers in patients suffering from asthma [62, 63]. Similar to IL-33 in the lung, the alarmin TSLP appears to be especially important for the activation of skin-resident ILC2s. For instance, TSLP-dependent ILC2s were shown to promote pathology independently of IL-33 and IL-25 in a murine model of AD-like disease [64]. These studies are consistent with reports demonstrating that patients suffering from atopic dermatitis have elevated levels of TSLP and ILC2 populations in lesional skin [64, 65]. However, IL-25 and IL-33 have also been shown to contribute to the development of AD-like disease in murine models mediated by the delivery of IL-2 complexes or IL-33 overexpression [66, 67]. Moreover, it is thought that ILC2s might have a specific metabolic programming that further allows them to promote allergic responses. Specifically, it has been demonstrated that ILC2s constitutively express Arg1, and inhibition of this pathway is reported to ameliorate papain-induced allergic inflammation [48].
In addition to alarmins, other mechanisms have been proven to mediate ILC2 activation during allergic responses including basophil-derived IL-4 [59, 68] (Fig. 1), leukotrienes (LTC4, LTD4, and LTE4) [69, 70], the TNF-family cytokine TL1A [71], ICOS-ICOSL interactions [72], and the previously mentioned autocrine IL-9 pathways [49]. Remarkably, three independent groups have recently highlighted a novel mechanism for ILC2 activation via the action of the neuropeptide neuromedin U (NMU) [73,74,75]. ILC2s were shown to express high levels of the NMU receptor 1 as compared to other immune cells and to co-localize with cholinergic neurons in the respiratory and gastrointestinal tracts. Further, NMU treatment induced ILC2 activation in vitro and in vivo, which correlated with accelerated expulsion of N. brasiliensis larvae and pulmonary inflammation [73,74,75]. Collectively, these studies illustrate the complex cellular networks participating in the context of helminth infections and allergic diseases.

Following exposure to helminth parasites or allergens, epithelial cells and some hematopoietic cells produce cytokine alarmins that directly influence ILC2 activation. Activated ILC2s produce robust levels of effector cytokines such as IL-4, IL-5, IL-9, and IL-13 that support the development of immunity and inflammation at barrier surfaces. Further, activated ILC2s also produce Areg that serves to promote the integrity of affected tissues. In addition to cytokine alarmins, recent studies have demonstrated that innate immune cells such as basophils are also capable of directly regulating ILC2 activation. Collectively, these studies demonstrate that distinct mechanisms are capable of influencing ILC2 responses
ILC2s appear to not only mediate the early activation of immune responses during acute exposure to allergens but are also thought to contribute to the development of persistent asthmatic responses as well. For example, it has been reported that allergen-exposed ILC2s showed enhanced cytokine production and proliferative capacity [76]. Further, ILC2s have been shown to contribute to the development of adaptive immune responses by inducing dendritic cell activation and promoting TH2 memory responses [77, 78]. The many functions of ILC2s in the context of models of allergic disease suggest that ILC2 populations might represent a fruitful therapeutic target. It is also possible that negative regulators of ILC2 responses such as type 1 [79] and type 2 interferons [80], IL-27 [81, 82], KLRG1 signaling by E-cadherin [66], lipoxin A4 [83], and T regulatory cells [84, 85] could also be manipulated to reduce ILC2 activation in the context of allergic inflammation.
Eosinophils
Eosinophils are granulocytes that were first described by Paul Ehrlich in 1879 for their remarkable capacity to be stained by acidophilic dyes [86, 87]. Eosinophils develop in the bone marrow from myeloid progenitors and circulate as mature cells in the periphery under homeostasis. However, recent studies have also reported the presence of eosinophil progenitors in the blood and bronchial biopsies of asthmatic patients [88, 89], suggesting they can also develop in the tissue microenvironment. These findings were later confirmed by the presence of committed eosinophil progenitors in the lungs of mice subjected to OVA-induced allergic inflammation [90]. Eosinophil development is dependent on the expression of transcription factors such as GATA-1, PU.1, and C/EBPα [86, 87]. The exact molecular interactions that drive eosinophil maturation remain to be fully defined; however, IL-5 is reported to play a major role in this process. For example, IL-5 is known to promote eosinophil differentiation in the bone marrow and to regulate eosinophil trafficking to the blood circulation [91]. In fact, IL-5Rα is a dominant surface marker used to identify committed eosinophil progenitors [92]. In addition to IL-5, other cytokines contribute to eosinophil differentiation during homeostasis such as IL-3 and GM-CSF [86, 87]. Once in the periphery, eosinophils are reported to migrate to distinct portions of the gastrointestinal tract under homeostatic conditions. Eosinophil-tissue homing is known to be regulated by different mechanisms including the secretion of CCL11 (eotaxin-1) by Ly6Chi monocytes, intestinal epithelial cells [93, 94] and IL-5 release by ILC2s following activation of the GI neuropeptide vasoactive intestinal peptide (VIP) in response to metabolic and circadian signals [95]. In summary, the presence of tissue-resident eosinophils appears to be regulated by complex cellular networks that include the interaction of the gastrointestinal, nervous, and immune system. Eosinophils contribute to type 2 immunity via multiple mechanisms including the secretion of cytotoxic proteins, including major basic protein (MBP-1 and MBP-2), eosinophil peroxidase (EPO), eosinophil-derived neurotoxin (EDN), and eosinophil cationic protein (ECP), TH2 cytokines (IL-4 and IL-13) and pro-fibrotic factors (TGF-β, VEGF, and bFGF) as well as expression of MHCII [86, 87].
Peripheral eosinophilia is a shared characteristic of helminth infections [96], suggesting that eosinophils play a pivotal role in host-protective immunity. Classic in vitro studies described the anti-helminthic actions of eosinophils co-cultured with antigen-specific antibodies and complement [97, 98]. These results are supported by in vivo studies showing the essential contribution of eosinophils to mediate protection against reinfection with N. brasiliensis and T. spiralis [99, 100]. Furthermore, expression of MHCII and co-stimulatory molecules on activated eosinophils following infection with T. muris and Brugia malayi [101, 102] suggests that eosinophils might contribute to establish long-term protection against helminths by promoting adaptive immune responses. However, it has also been shown that host-protective responses remain largely intact in eosinophil-deficient mice following primary infection with S. mansoni, T. muris, S. stercolaris, and N. brasiliensis [103]. By contrast, mice lacking eosinophils exhibit increased worm burdens after primary infection with H. polygyrus and B. malayi [103], suggesting that the role of eosinophils in promoting immunity may be parasite-specific. The site of infection might also have an impact on the anti-parasitic properties of eosinophils as suggested by observations in the context of T. spiralis infection. While eosinophil deficiency did not impact intestinal adult worm counts or fecundity [104], absence of eosinophils resulted in diminished newborn larvae burdens, reduced TH2 cell infiltrate and elevated iNOS expression by neutrophils and macrophages in the skeletal muscle [104, 105]. Further, it was shown that eosinophil-derived IL-10 promoted the secretion of IL-10 by dendritic cells and CD4+CD25− T cells, which was reported to inhibit the expression of iNOS in the local microenvironment thereby promoting larval growth [106]. In summary, the role of eosinophils in promoting protective immunity appears to be parasite- and tissue-specific; however, further studies are required to better define the precise contribution of eosinophils to anti-helminth immunity.
Similar to helminth infections, eosinophil accumulation is a common feature of allergic inflammation in both animal models of allergic disease and human patient populations [107]. Migration of eosinophils to affected tissues is mediated by the synergistic action of chemokines (eotaxins), TH2 cytokines (IL-5 and IL-13), and lipid mediators (PGD2 and 5-oxo-ETE). Once in the tissue, eosinophils contribute to the pathology of allergic diseases by multiple mechanisms such as directly inducing damage to the mucosal epithelium via secretion of cytotoxic mediators [108] as well as promoting airway hyperreactivity, mucus production, and bronchoconstriction via their production of IL-13 [109]. Further, eosinophil-derived TGF-β induces tissue remodeling by activating pro-fibrotic responses [110]. The intrinsic functions of eosinophils are enhanced by their interaction with other immune cells, including mast cells, DCs, and CD4+ T lymphocytes. Eosinophil-derived MBP is reported to activate mast cells and induce their secretion of lipid inflammatory mediators as well as pro-fibrotic factors that promote inflammatory responses [111]. Moreover, eosinophils license adaptive immune responses by modulating dendritic cell activation and promoting TH2 polarization [112, 113]. Collectively, these studies suggest that targeting eosinophil responses may prove beneficial for the treatment of allergic disease and asthma.
Despite their contributions to the pathology of allergic inflammation, recent studies have also unveiled the essential role of eosinophils during homeostatic conditions, especially in the regulation of the beiging of white adipose tissue (WAT). Under homeostasis, ILC2s are enriched in WAT and induce eosinophil recruitment and activation via secretion of IL-5 [114, 115]. In return, eosinophil-derived IL-4 and IL-13 induce the polarization of M2 macrophages, which promote the expression of uncoupling protein 1 (UCP-1) and regulate the caloric expenditure via the production of epinephrine and catecholamines [114, 115]. Additionally, eosinophil-derived IL-4 can directly activate PDGFRa+ adipose progenitors and promote the growth of beige fat [116]. Collectively, these studies show that eosinophils and type 2 responses contribute to adipose tissue homeostasis and might contribute to diseases such as obesity and metabolic syndrome. Interestingly, epidemiological studies have established a correlation between the prevalence of obesity and allergic disease [117,118,119]. Further, a high-fat diet has been shown to increase eosinophil trafficking from the bone marrow to the lungs [120], and eosinophils are known to be reduced in WAT of obese mice [114, 115], suggesting that tissue redistribution of eosinophils may represent an indirect mechanism through which obesity promotes allergic inflammation. A more detailed discussion about this association has been extensively reviewed previously [121, 122].
Mast Cells
Mast cells are granular tissue-resident myeloid cells that originate from bone marrow hematopoietic precursors. Commitment of hematopoietic stem cells (HSC) to the mast cell lineage requires exposure of the stem cell to the appropriate cytokines and subsequent expression of associated transcription factors [123]. According to a commonly used model of evaluating malignant and normal hematopoiesis, a long-term (LT)-HSC develops into a common myeloid progenitor which subsequently differentiates into a granulocyte-monocyte progenitor (GMP) [124]. Mast cells are derived from the bipotent basophil-mast cell progenitor (BMCP), which has been shown to originate from GMPs in vitro [125]. Specifically, BMCPs give rise to the mast cell progenitor (MCP, Lin−CD34+β7hiFcεR1α loFcγRII/III+c-kitloThy-1−), which can then differentiate into mast cells depending on the tissue microenvironment [125]. Mast cells are reported to fully mature in peripheral tissues and are present as progenitors in general circulation [125]. Their development into either connective tissue or mucosal mast cells is dependent on signals received from the local tissue environment [126]. Our recent work suggests that the lineage commitment of peripheral progenitor cell populations to the mast cell lineage is mediated, in part, by expression of the enzyme carbonic anhydrase I (Car1) [127]. For example, it was shown that inhibition of Car1 was sufficient to prevent mast cell responses in murine models of helminth infection and allergic inflammation. Consistent with murine models, Car1 inhibition also prevented the development of human mast cells from blood-resident progenitor cells [127].
Mast cells are reported to play an important role in tissue homeostasis and repair, and host defense against numerous pathogens such as helminth parasites [128, 129]. They are found in many tissues particularly throughout the skin and mucosa where they are in contact with the external environment. Mast cells express the high affinity IgE receptor (FcεRIα), which induces the release of early and late phase inflammatory mediators following crosslinking of IgE-antigen complexes. Leukotrienes, prostaglandins, chymases, and tryptases are among the early phase mediators known to be released by activated mast cells. Hours after crosslinking, mast cells are also capable of releasing cytokines and chemokines such as IL-4, IL-5, IL-10, and macrophage inflammatory protein 1 alpha (MIP-1α). In summary, the vast array of inflammatory mediators mast cells express and their proximity to barrier surfaces enable them to function efficiently as immune sentinels capable of modulating immune responses at mucosal sites. Consistent with this notion, mast cell responses have been shown to be critically important in promoting immunity to T. spiralis. In these studies, it was demonstrated that mast cell-deficient mice exhibit delayed clearance of Trichinella larvae [130]. It was subsequently shown that interrupting mast cell development by targeting Car1 also results in reduced protective immunity to T. spiralis [127]. In addition to T. spiralis, Hepworth and colleagues demonstrated that mast cell degranulation was essential for establishing immunity to H. polygyrus and T. muris [131]. Here, treatment with cromolyn sodium, a mast cell stabilizing agent, inhibited worm clearance, TH2 priming, and type 2 cytokine production. Interestingly, mast cell degranulation was reported to induce the intestinal expression of cytokine alarmins, thereby initiating anti-helminth immunity [131]. Recent work has also shown that Spi-B-deficient mice, which have increased numbers of mast cells, exhibit significantly improved immunity to H. polygyrus. Specifically, mast cell-derived IL-33 was shown to promote IL-13-producing ILC2s leading to goblet cell hyperplasia and rapid worm expulsion in Spi-B-deficient mice [53]. In addition to promoting anti-helminth immunity, mast cells are well recognized for their contributions to allergic disease states as compressively highlighted in the following review articles [132, 133].
Given the importance of mast cells in initiating TH2 inflammation in the context of both helminth infection and allergic disease, they represent an ideal target for developing novel therapies. Therefore, recent discoveries such as the importance of mast cell-derived IL-33 and Car1 in promoting mast cell-mediated inflammation may offer new and exciting therapeutic targets.
Basophils
Basophils are the least common cells among peripheral blood leukocytes at the steady state. Similar to mast cells, basophils originate from the bone marrow LT-HSC. However, the BMCP develops into the basophil progenitor which subsequently differentiates into basophils [125]. On a molecular level, it is suggested that differentiation into basophils or mast cells by GMPs or BMCPs is dependent on differential expression of C/EBPα, GATA-2, and MITF [125, 134]. However, the specific contributions of these transcription factors in basophil and mast cell commitment remain to be fully defined. Basophil development is also regulated by the cytokines IL-3 and TSLP. Several groups including Lantz et al. and Ohmori et al. demonstrated that basophilia following N. brasiliensis or S. venezuelensis infection is critically dependent on IL-3–IL-3 receptor signaling [135, 136]. In addition, TSLP-dependent basophils have been identified in the context of T. spiralis and T. muris infection, and in a murine model of atopic dermatitis (AD)-like disease [137, 138].
Basophils are very similar to mast cells in that they are granular, possess FcεRIα, and release similar pro-inflammatory mediators, such as histamine upon IgE-antigen stimulation [132]. They are also known to produce IL-4 in response to antigen-IgE or cytokine stimulation and expand during certain helminth infections or in response to parasite antigens [132]. Consequently, they have been implicated in type 2 responses and are thought to provide an early source of IL-4 that contributes to the polarization of naïve CD4+ T cells into TH2 cells [132]. Although similar to mast cells, basophils have now been shown to uniquely contribute to immunity to several helminth parasites. They are thought to be important in the primary responses against T. spiralis and T. muris and during the secondary responses against N. brasiliensis and H. polygyrus bakeri [138,139,140,141]. For example, work by Giacomin and colleagues showed that depleting basophils resulted in impaired production of IL-4, IL-5, and IL-13 by mesenteric lymph node CD4+ T cells following T. spiralis infection [138]. Although, basophils were reported to be dispensable during primary N. brasiliensis infection, they play an important role in preventing invasion of parasitic larvae during secondary infection. Specifically, basophil-derived IL-4 induced M2 macrophage responses that were capable of trapping and killing parasitic larvae in the skin following a secondary challenge [141].
In addition to helminth infections, basophil populations are known to expand in the context of several models of allergic disease and are reported to contribute to allergen-induced inflammation. For example, basophils have been shown to play a role in the induction of allergic inflammation in murine models of AD-like disease and have also been shown to promote optimal type 2 cytokine responses in a model of airway inflammation [64, 68]. Consistent with these studies, basophils are increased in the lesional skin of patients suffering from AD and are elevated in the airways of patients suffering from asthma [68, 142]. Moreover, murine studies have also demonstrated a non-redundant role for basophils in prompting the pathology associated with eosinophilic esophagitis (EoE)-like inflammation. EoE is a chronic allergic inflammatory disorder characterized by the presence of eosinophils in the esophagus due to inhaled or ingested antigen and is often associated with disease states such as food allergy and atopic dermatitis [143]. Recently, Noti et al. developed a novel murine model of EoE-like disease that facilitated the investigation and manipulation of the pathways necessary to promote inflammation of the esophagus. Interestingly, their findings demonstrated that the alarmin TSLP was essential for the development of EoE-like disease. Further, they showed that TSLP elicited a potent basophil population that was expanded in peripheral tissues and was found to be present in the inflamed esophagus. Critically, lineage-specific depletion of basophils resulted in reduced inflammation and characteristics of EoE-like disease [144]. In addition to the above studies, Venturelli and colleagues employed additional murine models of EoE-like disease and have also found an important role for basophils in promoting esophageal inflammation. However, these pathways were dependent on the alarmin IL-33 rather than TSLP [145]. These important murine studies are supported by human data demonstrating that increased expression of TSLP and IL1RL1/ST2 are found in the esophageal biopsies of patients suffering from EoE [145, 146]. Further, basophil populations have also been found to be significantly increased in esophageal biopsies taken from EoE patients [144]. In conclusion, there is a growing body of evidence suggesting that basophils contribute to the development of several allergic disease states.
Macrophages
Macrophages are key immune cells that are known for their phagocytic activity and ability to present antigens. Similar to CD4+ T cells, macrophages are known for their phenotypic plasticity and are capable of initiating distinct transcriptional programs in the context of either a TH1 or TH2 immune response. Specifically, macrophage populations are capable of adopting a classical M1 or alternative M2 macrophage phenotype [147, 148]. M1 macrophages, generated upon exposure to TH1 cytokines such as IFN-γ, have enhanced anti-microbial activity against intracellular pathogens and thus will not be discussed in this review article. By contrast, alternatively activated M2 macrophages are induced in the context of helminth infection and allergic inflammation following activation by the TH2 cytokines IL-4 and IL-13 [147]. Similar to T cells, IL-4 and IL-13 signaling occurs via the activation of IL-4Rα and is dependent on transcription factors such as STAT6, KLF4, and IRF4. IL-4 and IL-13 signaling induce the acquisition of a distinct gene signature in M2 macrophages including the upregulation of arg1, relma, chi3l3, igf1, and mmp13 as well as downregulation of nos2 [147, 148]. M2 macrophages are also characterized by the expression of cell surface proteins such as IL-4Rα, IL-13 receptor alpha 1 (IL-13Rα1), IL-13Rα2, C-type mannose receptor 1 (Mrc1, CD206), dectin-1 (Clec7a), CD163, programmed death ligand 1 (PD-L1) and PD-L2, as well as the secretion of anti-inflammatory cytokines (IL-10 and IL1ra), chemokines (CCL17), lipids (omega 3 fatty acids, lipoxins, and palmitoleic acid), and tissue remodeling factors (TGF-β and arginase 1). In addition, M2 macrophages shape their cellular metabolism away from the production of pro-inflammatory cytokines and nitric oxide and towards the synthesis of anti-inflammatory and tissue repair factors by activation of the transcription factors PPARγ and PPARδ. These factors promote an aerobic metabolism in M2 macrophages, characterized by β-oxidation of fatty acids and enable the long-term maintenance of M2 macrophages during the chronic stages of helminth infections [147,148,149].
Although the roles of M2 macrophages remain to be fully defined, they have been implicated in playing critical host-protective roles in the context of helminth infections and allergic disease and have been extensively reviewed elsewhere [147, 148]. Briefly, depletion of macrophages impaired host-protective responses following infection with N. brasiliensis, H. polygyrus, and Schistosoma species [147, 148]. Remarkably, the polarization of M2 macrophages following helminth infection was partially mediated by IL-13-producing neutrophils, which acquired a distinct transcriptional program as compared to LPS-activated neutrophils [150]. Further, epithelial cell-derived chitinase-like proteins were shown to drive neutrophil recruitment by inducing the secretion of IL-17 by γδ T cells, suggesting that TH17 and neutrophil responses might be required for the initiation of TH2 inflammation and M2 polarization [151]. Supporting this notion, neutrophils and macrophages were shown to collaborate to immobilize S. stercolaris larvae via the release of neutrophil extracellular traps [152]. In addition to their roles in anti-helminthic immunity, M2 macrophages also contribute to the tissue remodeling following helminth infection [153, 154], suggesting that M2 macrophages have essential functions in different aspects of TH2 immunity. Altogether, these studies highlight the complex cascade of events that lead to the establishment of TH2 responses.
In contrast to their role in helminth infections, the contributions of M2 macrophages to allergic diseases are less defined. Pulmonary expression of M2-associated markers has been reported in allergic models of mice challenged with OVA and Aspergillus fumigatus [147, 148]. However, deletion of IL-4Rα in myeoloid cells did not result in significant changes in the lung pathology following OVA challenge [155]. Conversely, transfer of M2 macrophages resulted in an exacerbated eosinophil accumulation in OVA-challenged mice [156]. Furthermore, TSLP was shown to contribute to the polarization of M2 macrophages during allergic airway inflammation [157]. Despite these advances, further studies are required to define the precise contributions of M2 macrophages to allergic disease.
Hematopoietic Stem/Progenitor Cells
Hematopoietic stem cells (HSCs) are undifferentiated pluripotent cells that can self-renew and differentiate into all blood cells. Conversely, hematopoietic progenitor cells are progeny of HSCs, but unlike stem cells, have limited self-renewal capacity and restricted lineage potential. CD34, a surface marker often used to identify hematopoietic stem/progenitor cells (HSPCs), is downregulated as HSPCs mature with the exception of some mast cells [5]. HSPCs are mainly present in a specialized bone marrow niche that regulates their survival, proliferation, self-renewal, and differentiation [5]. Hematopoiesis typically occurs in the bone marrow in response to signals received from stromal cells and growth factors. However, differentiation does occur in peripheral tissues by a process called in situ hematopoiesis that is an important innate effector mechanism in TH2 inflammation [5]. Under homeostatic conditions, HSPCs leave the bone marrow and circulate in the bloodstream but their egress is increased in response to inflammatory signals. Many factors control this process including C-X-C Chemokine receptor type 4 (CXCR4), stromal cell-derived factor 1α (SDF-1α), and epithelial cytokine alarmins [158, 159].
In the context of TH2 responses, the epithelial cell-derived cytokines IL-25, IL-33, and TSLP are reported to promote the recruitment of TH2-promoting HSPCs to mucosal tissues following helminth infection or the induction of allergic inflammation [160, 161]. For example, Saenz et al. demonstrated that type 2 multipotent progenitors (MPPtype2) are recruited to gut-associated lymphoid tissues following infection with T. muris and N. brasiliensis. Genetic ablation of the IL-25 receptor (IL-17RB) or treatment with IL-25 neutralizing antibodies impaired the helminth-induced expansion of MPPtype2 cells highlighting their dependency on IL-25-IL-25R signaling. Further, MPPtype2 cells were capable of developing into mast cells, basophils, and macrophages, all of which contribute to anti-helminth immunity [160]. In addition to IL-25, TSLP was also found to promote TH2-promoting HSPC responses in the context of T. spiralis infection or a murine model of atopic dermatitis (AD)-like disease [64, 138]. Similar to MPPtype2 cells, the TSLP-elicited progenitors were able to develop into mast cells, basophils, and macrophages [161]. Further, it is well established that the IL-33 receptor (ST2) is expressed on a variety of type 2 cytokine-related HSPCs including eosinophil, basophil, and mast cell progenitors suggesting IL-33 may also be capable of promoting progenitor cell responses. This is supported by recent work demonstrating that IL-33 plays a critical role in regulating murine eosinophil hematopoiesis [162]. Supporting these murine studies, elevated CD34+ progenitor cell populations have been identified in the inflamed tissues of patients suffering from nasal polyposis and asthma [162, 163]. Collectively, these studies suggest that cytokine alarmins not only recruit HSPCs to peripheral tissues but also alter their functional properties, enabling them to selectively promote type 2 responses (Fig. 2). Future studies are needed to better define the cellular and molecular events regulating these progenitor cell responses in order to better understand their exciting therapeutic potential.

A common feature of helminth infections and allergic inflammation is the production of cytokine alarmins by both non-hematopoietic and hematopoietic cells. Recent studies have demonstrated that cytokine alarmins are capable of influencing the activation state of several terminally differentiated innate immune cells including mast cells, macrophages, eosinophils, dendritic cells, basophils, and ILC2s. In addition to terminally differentiated cells, emerging studies have shown that cytokine alarmins also influence the accumulation and activation of hematopoietic stem/progenitor cells at the barrier surface. Activated HSPCs are capable of undergoing in situ hematopoiesis and differentiating into innate immune cells that further support the development of immunity and inflammation
Summary
Type 2 cytokine-mediated inflammation is a multifaceted process that not only promotes anti-helminth immunity but also regulates the development of allergic inflammation. Many recent studies have highlighted the important roles a variety of innate immune cells play such as ILC2s, eosinophils, mast cells, basophils, macrophages, and HSPCs in regulating this inflammatory pathway (Fig. 1). Recent data also demonstrate that non-hematopoietic cells contribute to type 2 responses via their production of cytokine alarmins known to activate the above immune cell populations. Developing a better understanding of these pathways and how they counter regulate each other will inform the development of novel therapeutic strategies to address two major public health concerns (helminth infection and allergic disease).
References
Savioli L, Albonico M. Soil-transmitted helminthiasis. Nat Rev Microbiol. 2004;2(8):618–9.
King CH. Health metrics for helminthic infections. Adv Parasitol. 2010;73:51–69.
Patel N, Kreider T, Urban JF Jr, Gause WC. Characterisation of effector mechanisms at the host:parasite interface during the immune response to tissue-dwelling intestinal nematode parasites. Int J Parasitol. 2009;39(1):13–21.
Maizels RM, Hewitson JP, Smith KA. Susceptibility and immunity to helminth parasites. Curr Opin Immunol. 2012;24(4):459–66.
Hui CCK, McNagny KM, Denburg JA, Siracusa MC. In situ hematopoiesis: a regulator of TH2 cytokine-mediated immunity and inflammation at mucosal surfaces. Mucosal Immunol [Review]. 2015;8(4):701–11.
Lloyd CM, Hessel EM. Functions of T cells in asthma: more than just T(H)2 cells. Nat Rev Immunol. 2010;10(12):838–48.
Wynn TA. Type 2 cytokines: mechanisms and therapeutic strategies. Nat Rev Immunol. 2015;15(5):271–82.
Hammad H, Lambrecht BN. Barrier epithelial cells and the control of type 2 immunity. Immunity. 2015;43(1):29–40.
McGowan EC, Keet CA. Prevalence of self-reported food allergy in the National Health and Nutrition Examination Survey (NHANES) 2007–2010. J Allergy Clin Immunol. 2013;132(5):1216–9.e5.
Eder W, Ege MJ, von Mutius E. The asthma epidemic. N Engl J Med. 2006;355(21):2226–35.
Galli SJ, Tsai M, Piliponsky AM. The development of allergic inflammation. Nature. 2008;454(7203):445–54. https://doi.org/10.1038/nature07204.
Katona IM, Urban JF Jr, Finkelman FD. The role of L3T4+ and Lyt-2+ T cells in the IgE response and immunity to Nippostrongylus brasiliensis. J Immunol. 1988;140(9):3206–11.
Urban JF Jr, Katona IM, Finkelman FD. Heligmosomoides polygyrus: CD4+ but not CD8+ T cells regulate the IgE response and protective immunity in mice. Exp Parasitol. 1991;73(4):500–11.
Koyama K, Tamauchi H, Ito Y. The role of CD4+ and CD8+ T cells in protective immunity to the murine nematode parasite Trichuris muris. Parasite Immunol. 1995;17(3):161–5.
Walls RS, Carter RL, Leuchars E, Davies AJ. The immunopathology of trichiniasis in T-cell deficient mice. Clin Exp Immunol. 1973;13(2):231–42.
Vignali DA, Crocker P, Bickle QD, Cobbold S, Waldmann H, Taylor MG. A role for CD4+ but not CD8+ T cells in immunity to Schistosoma mansoni induced by 20 krad-irradiated and Ro 11-3128-terminated infections. Immunology. 1989;67(4):466–72.
Raemdonck K, Baker K, Dale N, Dubuis E, Shala F, Belvisi MG, et al. CD4(+) and CD8(+) T cells play a central role in a HDM driven model of allergic asthma. Respir Res. 2016;17:45.
Gavett SH, Chen X, Finkelman F, Wills-Karp M. Depletion of murine CD4+ T lymphocytes prevents antigen-induced airway hyperreactivity and pulmonary eosinophilia. Am J Respir Cell Mol Biol. 1994;10(6):587–93.
Bouchery T, Kyle R, Ronchese F, Le Gros G. The differentiation of CD4(+) T-helper cell subsets in the context of helminth parasite infection. Front Immunol. 2014;5:487.
Na H, Cho M, Chung Y. Regulation of Th2 cell immunity by dendritic cells. Immune Network. 2016 02/25, 12/17/received, 01/22/revised, 01/26/accepted;16(1):1–12.
Ito T, Wang YH, Duramad O, Hori T, Delespesse GJ, Watanabe N, et al. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J Exp Med. 2005;202(9):1213–23.
Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23(5):479–90.
Hoshino A, Tanaka Y, Akiba H, Asakura Y, Mita Y, Sakurai T, et al. Critical role for OX40 ligand in the development of pathogenic Th2 cells in a murine model of asthma. Eur J Immunol. 2003;33(4):861–9.
Salek-Ardakani S, Song J, Halteman BS, Jember AG, Akiba H, Yagita H, et al. OX40 (CD134) controls memory T helper 2 cells that drive lung inflammation. J Exp Med. 2003;198(2):315–24.
Hussaarts L, Yazdanbakhsh M, Guigas B. Priming dendritic cells for Th2 polarization: lessons learned from helminths and implications for metabolic disorders. Frontiers in Immunology. [Mini Review]. 2014 2014-October-20;5(499).
León B, Ballesteros-Tato A, Lund FE. Dendritic cells and B cells: unexpected partners in Th2 development. Journal of immunology (Baltimore, Md : 1950). 2014;193(4):1531–7.
Nawa Y, Miller HR, Hall E, Jarrett EE. Adoptive transfer of total and parasite-specific IgE responses in rats infected with Nippostrongylus brasiliensis. Immunology. 1981;44(1):119–23.
Appleton JA, McGregor DD. Characterization of the immune mediator of rapid expulsion of Trichinella spiralis in suckling rats. Immunology. 1987;62(3):477–84.
Harris N, Gause WC. To B or not to B: B cells and the Th2-type immune response to helminths. Trends Immunol. 2011;32(2):80–8.
Turqueti-Neves A, Otte M, Schwartz C, Schmitt ME, Lindner C, Pabst O, et al. The extracellular domains of IgG1 and T cell-derived IL-4/IL-13 are critical for the polyclonal memory IgE response in vivo. PLoS Biol. 2015;13(11):e1002290.
Linton PJ, Bautista B, Biederman E, Bradley ES, Harbertson J, Kondrack RM, et al. Costimulation via OX40L expressed by B cells is sufficient to determine the extent of primary CD4 cell expansion and Th2 cytokine secretion in vivo. J Exp Med. 2003;197(7):875–83.
Liu Q, Liu Z, Rozo CT, Hamed HA, Alem F, Urban JF Jr, et al. The role of B cells in the development of CD4 effector T cells during a polarized Th2 immune response. J Immunol. 2007;179(6):3821–30.
Turqueti-Neves A, Otte M, Prazeres da Costa O, Hopken UE, Lipp M, Buch T, et al. B-cell-intrinsic STAT6 signaling controls germinal center formation. Eur J Immunol. 2014;44(7):2130–8.
Gould HJ, Sutton BJ, Beavil AJ, Beavil RL, McCloskey N, Coker HA, et al. The biology of IGE and the basis of allergic disease. Annu Rev Immunol. 2003;21:579–628.
Gould HJ, Sutton BJ. IgE in allergy and asthma today. Nat Rev Immunol. 2008;8(3):205–17.
Fort MM, Cheung J, Yen D, Li J, Zurawski SM, Lo S, et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. 2001;15(6):985–95.
Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464(7293):1367–70.
Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature. 2010;463(7280):540–4.
Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci U S A. 2010;107(25):11489–94.
Hoyler T, Klose CS, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity. 2012;37(4):634–48.
Artis D, Spits H. The biology of innate lymphoid cells. Nature. 2015;517(7534):293–301.
Klose CSN, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nature immunology [Review]. 2016;17(7):765–74.
Scanlon ST, McKenzie AN. Type 2 innate lymphoid cells: new players in asthma and allergy. Curr Opin Immunol. 2012;24(6):707–12.
Wojno ED, Monticelli LA, Tran SV, Alenghat T, Osborne LC, Thome JJ, et al. The prostaglandin D(2) receptor CRTH2 regulates accumulation of group 2 innate lymphoid cells in the inflamed lung. Mucosal Immunol. 2015;8(6):1313–23.
von Moltke J, Ji M, Liang HE, Locksley RM. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature. 2016;529(7585):221–5.
Howitt MR, Lavoie S, Michaud M, Blum AM, Tran SV, Weinstock JV, et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science. 2016;351(6279):1329–33.
Huang Y, Guo L, Qiu J, Chen X, Hu-Li J, Siebenlist U, et al. IL-25-responsive, lineage-negative KLRG1(hi) cells are multipotential 'inflammatory' type 2 innate lymphoid cells. Nat Immunol. 2015;16(2):161–9.
Monticelli LA, Buck MD, Flamar AL, Saenz SA, Tait Wojno ED, Yudanin NA, et al. Arginase 1 is an innate lymphoid-cell-intrinsic metabolic checkpoint controlling type 2 inflammation. Nat Immunol. 2016;17(6):656–65.
Wilhelm C, Hirota K, Stieglitz B, Van Snick J, Tolaini M, Lahl K, et al. An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nat Immunol. 2011;12(11):1071–7.
Turner JE, Morrison PJ, Wilhelm C, Wilson M, Ahlfors H, Renauld JC, et al. IL-9-mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J Exp Med. 2013;210(13):2951–65.
Bouchery T, Kyle R, Camberis M, Shepherd A, Filbey K, Smith A, et al. ILC2s and T cells cooperate to ensure maintenance of M2 macrophages for lung immunity against hookworms. Nat Commun [Research Support, Non-US Gov't]. 2015;6:6970.
Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity. 2014;41(2):283–95.
Shimokawa C, Kanaya T, Hachisuka M, Ishiwata K, Hisaeda H, Kurashima Y, et al. Mast cells are crucial for induction of group 2 innate lymphoid cells and clearance of helminth infections. Immunity. 2017;46(5):863–74.e4.
Pelly VS, Kannan Y, Coomes SM, Entwistle LJ, Ruckerl D, Seddon B, et al. IL-4-producing ILC2s are required for the differentiation of TH2 cells following Heligmosomoides polygyrus infection. Mucosal Immunol. 2016;9(6):1407–17.
Zaiss DM, Yang L, Shah PR, Kobie JJ, Urban JF, Mosmann TR. Amphiregulin, a TH2 cytokine enhancing resistance to nematodes. Science. 2006;314(5806):1746.
Yasuda K, Muto T, Kawagoe T, Matsumoto M, Sasaki Y, Matsushita K, et al. Contribution of IL-33-activated type II innate lymphoid cells to pulmonary eosinophilia in intestinal nematode-infected mice. Proc Natl Acad Sci U S A. 2012;109(9):3451–6.
Nausch N, Appleby LJ, Sparks AM, Midzi N, Mduluza T, Mutapi F. Group 2 innate lymphoid cell proportions are diminished in young helminth infected children and restored by curative anti-helminthic treatment. PLoS Negl Trop Dis. 2015;9(3):e0003627.
Halim TY, Krauss RH, Sun AC, Takei F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity. 2012;36(3):451–63.
Motomura Y, Morita H, Moro K, Nakae S, Artis D, Endo TA, et al. Basophil-derived interleukin-4 controls the function of natural helper cells, a member of ILC2s, in lung inflammation. Immunity. 2014;40(5):758–71.
Oboki K, Ohno T, Kajiwara N, Arae K, Morita H, Ishii A, et al. IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc Natl Acad Sci U S A. 2010;107(43):18581–6.
de Kleer IM, Kool M, de Bruijn MJ, Willart M, van Moorleghem J, Schuijs MJ, et al. Perinatal activation of the Interleukin-33 pathway promotes type 2 immunity in the developing lung. Immunity. 2016;45(6):1285–98.
Smith SG, Chen R, Kjarsgaard M, Huang C, Oliveria JP, O'Byrne PM, et al. Increased numbers of activated group 2 innate lymphoid cells in the airways of patients with severe asthma and persistent airway eosinophilia. J Allergy Clin Immunol. 2016;137(1):75–86.e8.
Liu T, Wu J, Zhao J, Wang J, Zhang Y, Liu L, et al. Type 2 innate lymphoid cells: a novel biomarker of eosinophilic airway inflammation in patients with mild to moderate asthma. Respir Med. 2015;109(11):1391–6.
Kim BS, Siracusa MC, Saenz SA, Noti M, Monticelli LA, Sonnenberg GF, et al. TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci Transl Med. 2013;5(170):170ra16.
Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol. 2002;3(7):673–80.
Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med. 2013;210(13):2939–50.
Imai Y, Yasuda K, Sakaguchi Y, Haneda T, Mizutani H, Yoshimoto T, et al. Skin-specific expression of IL-33 activates group 2 innate lymphoid cells and elicits atopic dermatitis-like inflammation in mice. Proc Natl Acad Sci U S A. 2013;110(34):13921–6.
Kim BS, Wang K, Siracusa MC, Saenz SA, Brestoff JR, Monticelli LA, et al. Basophils promote innate lymphoid cell responses in inflamed skin. J Immunol. 2014;193(7):3717–25.
Salimi M, Stoger L, Liu W, Go S, Pavord I, Klenerman P, et al. Cysteinyl leukotriene E4 activates human group 2 innate lymphoid cells and enhances the effect of prostaglandin D2 and epithelial cytokines. J Allergy Clin Immunol. 2017 20.
Lund SJ, Portillo A, Cavagnero K, Baum RE, Naji LH, Badrani JH, et al. Leukotriene C4 Potentiates IL-33-induced group 2 innate lymphoid cell activation and lung inflammation. Journal of immunology. 2017 30.
Meylan F, Hawley ET, Barron L, Barlow JL, Penumetcha P, Pelletier M, et al. The TNF-family cytokine TL1A promotes allergic immunopathology through group 2 innate lymphoid cells. Mucosal Immunol. 2014;7(4):958–68.
Maazi H, Patel N, Sankaranarayanan I, Suzuki Y, Rigas D, Soroosh P, et al. ICOS:ICOS-ligand interaction is required for type 2 innate lymphoid cell function, homeostasis, and induction of airway hyperreactivity. Immunity. 2015;42(3):538–51.
Wallrapp A, Riesenfeld SJ, Burkett PR, Abdulnour RE, Nyman J, Dionne D, et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature. 2017;549(7672):351–6.
Cardoso V, Chesne J, Ribeiro H, Garcia-Cassani B, Carvalho T, Bouchery T, et al. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature. 2017;549(7671):277–81.
Klose CSN, Mahlakoiv T, Moeller JB, Rankin LC, Flamar AL, Kabata H, et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature. 2017;549(7671):282–6.
Martinez-Gonzalez I, Matha L, Steer CA, Ghaedi M, Poon GF, Takei F. Allergen-experienced group 2 innate lymphoid cells acquire memory-like properties and enhance allergic lung inflammation. Immunity. 2016;45(1):198–208.
Halim TY, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, McNagny KM, et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity. 2014;40(3):425–35.
Halim TY, Hwang YY, Scanlon ST, Zaghouani H, Garbi N, Fallon PG, et al. Group 2 innate lymphoid cells license dendritic cells to potentiate memory TH2 cell responses. Nat Immunol. 2016;17(1):57–64.
Duerr CU, McCarthy CD, Mindt BC, Rubio M, Meli AP, Pothlichet J, et al. Type I interferon restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat Immunol. 2016;17(1):65–75.
Molofsky AB, Van Gool F, Liang HE, Van Dyken SJ, Nussbaum JC, Lee J, et al. Interleukin-33 and interferon-gamma counter-regulate group 2 innate lymphoid cell activation during immune perturbation. Immunity. 2015;43(1):161–74.
McHedlidze T, Kindermann M, Neves AT, Voehringer D, Neurath MF, Wirtz S. IL-27 suppresses type 2 immune responses in vivo via direct effects on group 2 innate lymphoid cells. Mucosal Immunol. 2016;9(6):1384–94.
Moro K, Kabata H, Tanabe M, Koga S, Takeno N, Mochizuki M, et al. Interferon and IL-27 antagonize the function of group 2 innate lymphoid cells and type 2 innate immune responses. Nat Immunol. 2016;17(1):76–86.
Barnig C, Cernadas M, Dutile S, Liu X, Perrella MA, Kazani S, et al. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci Transl Med. 2013;5(174):174ra26.
Krishnamoorthy N, Burkett PR, Dalli J, Abdulnour RE, Colas R, Ramon S, et al. Cutting edge: maresin-1 engages regulatory T cells to limit type 2 innate lymphoid cell activation and promote resolution of lung inflammation. J Immunol. 2015;194(3):863–7.
Morita H, Arae K, Unno H, Miyauchi K, Toyama S, Nambu A, et al. An Interleukin-33-mast cell-Interleukin-2 axis suppresses papain-induced allergic inflammation by promoting regulatory T cell numbers. Immunity. 2015;43(1):175–86.
Rosenberg HF, Dyer KD, Foster PS. Eosinophils: changing perspectives in health and disease. Nat Rev Immunol. 2013;13(1):9–22. https://doi.org/10.1038/nri3341.
McBrien CN, Menzies-Gow A. The biology of eosinophils and their role in asthma. Front Med (Lausanne). 2017;4:93.
Sehmi R, Howie K, Sutherland DR, Schragge W, O'Byrne PM, Denburg JA. Increased levels of CD34+ hemopoietic progenitor cells in atopic subjects. Am J Respir Cell Mol Biol. 1996;15(5):645–55.
Robinson DS, Damia R, Zeibecoglou K, Molet S, North J, Yamada T, et al. CD34(+)/interleukin-5Ralpha messenger RNA+ cells in the bronchial mucosa in asthma: potential airway eosinophil progenitors. Am J Respir Cell Mol Biol. 1999;20(1):9–13.
Southam DS, Widmer N, Ellis R, Hirota JA, Inman MD, Sehmi R. Increased eosinophil-lineage committed progenitors in the lung of allergen-challenged mice. J Allergy Clin Immunol. 2005;115(1):95–102.
Faccioli LH, Mokwa VF, Silva CL, Rocha GM, Araujo JI, Nahori MA, et al. IL-5 drives eosinophils from bone marrow to blood and tissues in a guinea-pig model of visceral larva migrans syndrome. Mediat Inflamm. 1996;5(1):24–31.
Mori Y, Iwasaki H, Kohno K, Yoshimoto G, Kikushige Y, Okeda A, et al. Identification of the human eosinophil lineage-committed progenitor: revision of phenotypic definition of the human common myeloid progenitor. J Exp Med. 2009;206(1):183–93.
Waddell A, Ahrens R, Steinbrecher K, Donovan B, Rothenberg ME, Munitz A, et al. Colonic eosinophilic inflammation in experimental colitis is mediated by Ly6C(high) CCR2(+) inflammatory monocyte/macrophage-derived CCL11. J Immunol. 2011;186(10):5993–6003.
Ahrens R, Waddell A, Seidu L, Blanchard C, Carey R, Forbes E, et al. Intestinal macrophage/epithelial cell-derived CCL11/eotaxin-1 mediates eosinophil recruitment and function in pediatric ulcerative colitis. J Immunol. 2008;181(10):7390–9.
Nussbaum JC, Van Dyken SJ, von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature. 2013;502(7470):245–8.
Basten A, Boyer MH, Beeson PB. Mechanism of eosinophilia. I. Factors affecting the eosinophil response of rats to Trichinella spiralis. J Exp Med. 1970;131(6):1271–87.
Buys J, Wever R, van Stigt R, Ruitenberg EJ. The killing of newborn larvae of Trichinella spiralis by eosinophil peroxidase in vitro. Eur J Immunol. 1981;11(10):843–5.
Capron M, Torpier G, Capron A. In vitro killing of S. mansoni schistosomula by eosinophils from infected rats: role of cytophilic antibodies. J Immunol. 1979;123(5):2220–30.
Huang L, Gebreselassie NG, Gagliardo LF, Ruyechan MC, Luber KL, Lee NA, et al. Eosinophils mediate protective immunity against secondary nematode infection. J Immunol. 2015;194(1):283–90.
Knott ML, Matthaei KI, Giacomin PR, Wang H, Foster PS, Dent LA. Impaired resistance in early secondary Nippostrongylus brasiliensis infections in mice with defective eosinophilopoeisis. Int J Parasitol. 2007;37(12):1367–78.
Mawhorter SD, Pearlman E, Kazura JW, Boom WH. Class II major histocompatibility complex molecule expression on murine eosinophils activated in vivo by Brugia malayi. Infect Immun. 1993;61(12):5410–2.
Svensson M, Bell L, Little MC, DeSchoolmeester M, Locksley RM, Else KJ. Accumulation of eosinophils in intestine-draining mesenteric lymph nodes occurs after Trichuris muris infection. Parasite Immunol. 2011;33(1):1–11.
Huang L, Appleton JA. Eosinophils in helminth infection: defenders and dupes. Trends Parasitol. 2016;32(10):798–807.
Fabre V, Beiting DP, Bliss SK, Gebreselassie NG, Gagliardo LF, Lee NA, et al. Eosinophil deficiency compromises parasite survival in chronic nematode infection. J Immunol. 2009;182(3):1577–83.
Gebreselassie NG, Moorhead AR, Fabre V, Gagliardo LF, Lee NA, Lee JJ, et al. Eosinophils preserve parasitic nematode larvae by regulating local immunity. J Immunol. 2012;188(1):417–25.
Huang L, Gebreselassie NG, Gagliardo LF, Ruyechan MC, Lee NA, Lee JJ, et al. Eosinophil-derived IL-10 supports chronic nematode infection. J Immunol. 2014;193(8):4178–87.
Bousquet J, Chanez P, Lacoste JY, Barneon G, Ghavanian N, Enander I, et al. Eosinophilic inflammation in asthma. N Engl J Med. 1990;323(15):1033–9.
Gundel RH, Letts LG, Gleich GJ. Human eosinophil major basic protein induces airway constriction and airway hyperresponsiveness in primates. J Clin Invest. 1991;87(4):1470–3.
Walsh ER, Thakar J, Stokes K, Huang F, Albert R, August A. Computational and experimental analysis reveals a requirement for eosinophil-derived IL-13 for the development of allergic airway responses in C57BL/6 mice. J Immunol. 2011;186(5):2936–49.
Ohno I, Nitta Y, Yamauchi K, Hoshi H, Honma M, Woolley K, et al. Transforming growth factor beta 1 (TGF beta 1) gene expression by eosinophils in asthmatic airway inflammation. Am J Respir Cell Mol Biol. 1996;15(3):404–9.
Piliponsky AM, Gleich GJ, Bar I, Levi-Schaffer F. Effects of eosinophils on mast cells: a new pathway for the perpetuation of allergic inflammation. Mol Immunol. 2002;38(16–18):1369.
Yang D, Chen Q, Rosenberg HF, Rybak SM, Newton DL, Wang ZY, et al. Human ribonuclease A superfamily members, eosinophil-derived neurotoxin and pancreatic ribonuclease, induce dendritic cell maturation and activation. J Immunol. 2004;173(10):6134–42.
Yang D, Rosenberg HF, Chen Q, Dyer KD, Kurosaka K, Oppenheim JJ. Eosinophil-derived neurotoxin (EDN), an antimicrobial protein with chemotactic activities for dendritic cells. Blood. 2003;102(9):3396–403.
Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature. 2015;519(7542):242–6.
Molofsky AB, Nussbaum JC, Liang HE, Van Dyken SJ, Cheng LE, Mohapatra A, et al. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med. 2013;210(3):535–49.
Lee MW, Odegaard JI, Mukundan L, Qiu Y, Molofsky AB, Nussbaum JC, et al. Activated type 2 innate lymphoid cells regulate beige fat biogenesis. Cell. 2015;160(1–2):74–87.
Beuther DA, Sutherland ER. Overweight, obesity, and incident asthma: a meta-analysis of prospective epidemiologic studies. Am J Respir Crit Care Med. 2007;175(7):661–6.
Beckett WS, Jacobs DR Jr, Yu X, Iribarren C, Williams OD. Asthma is associated with weight gain in females but not males, independent of physical activity. Am J Respir Crit Care Med. 2001;164(11):2045–50.
Dixon AE, Pratley RE, Forgione PM, Kaminsky DA, Whittaker-Leclair LA, Griffes LA, et al. Effects of obesity and bariatric surgery on airway hyperresponsiveness, asthma control, and inflammation. J Allergy Clin Immunol. 2011;128(3):508–15. e1-2
Calixto MC, Lintomen L, Schenka A, Saad MJ, Zanesco A, Antunes E. Obesity enhances eosinophilic inflammation in a murine model of allergic asthma. Br J Pharmacol. 2010;159(3):617–25.
Julia V, Macia L, Dombrowicz D. The impact of diet on asthma and allergic diseases. Nat Rev Immunol. 2015;15(5):308–22.
Hersoug LG, Linneberg A. The link between the epidemics of obesity and allergic diseases: does obesity induce decreased immune tolerance? Allergy. 2007;62(10):1205–13.
Kondo M, Wagers AJ, Manz MG, Prohaska SS, Scherer DC, Beilhack GF, et al. Biology of hematopoietic stem cells and progenitors: implications for clinical application. Annu Rev Immunol. 2003;21(1):759–806.
Iwasaki H, Akashi K. Myeloid lineage commitment from the hematopoietic stem cell. Immunity. 2007;26(6):726–40.
Arinobu Y, Iwasaki H, Gurish MF, Mizuno S-I, Shigematsu H, Ozawa H, et al. Developmental checkpoints of the basophil/mast cell lineages in adult murine hematopoiesis. Proc Natl Acad Sci U S A. 2005;102(50):18105–10.
Gurish MF, Austen KF. Developmental origin and functional specialization of mast cell subsets. Immunity. 2012;37(1):25–33.
Henry EK, Sy CB, Inclan-Rico JM, Espinosa V, Ghanny SS, Dwyer DF, et al. Carbonic anhydrase enzymes regulate mast cell-mediated inflammation. J Exp Med. 2016;213(9):1663–73.
Ng MF. The role of mast cells in wound healing. Int Wound J. 2010;7(1):55–61.
Kennelly R, Conneely JB, Bouchier-Hayes D, Winter DC. Mast cells in tissue healing: from skin to the gastrointestinal tract. Curr Pharm Des. 2011;17(34):3772–5.
Urban JF Jr, Schopf L, Morris SC, Orekhova T, Madden KB, Betts CJ, et al. Stat6 signaling promotes protective immunity against Trichinella spiralis through a mast cell- and T cell-dependent mechanism. J Immunol. 2000;164(4):2046–52.
Hepworth MR, Danilowicz-Luebert E, Rausch S, Metz M, Klotz C, Maurer M, et al. Mast cells orchestrate type 2 immunity to helminths through regulation of tissue-derived cytokines. Proc Natl Acad Sci U S A. 2012;109(17):6644–9.
Voehringer D. Protective and pathological roles of mast cells and basophils. Nat Rev Immunol. 2013;13(5):362–75.
Bischoff SC. Role of mast cells in allergic and non-allergic immune responses: comparison of human and murine data. Nat Rev Immunol. 2007;7(2):93–104.
Iwasaki H, Mizuno S-I, Arinobu Y, Ozawa H, Mori Y, Shigematsu H, et al. The order of expression of transcription factors directs hierarchical specification of hematopoietic lineages. Genes Dev. 2006;20(21):3010–21.
Lantz CS, Boesiger J, Song CH, Mach N, Kobayashi T, Mulligan RC, et al. Role for interleukin-3 in mast-cell and basophil development and in immunity to parasites. Nature. 1998;392(6671):90–3.
Ohmori K, Luo Y, Jia Y, Nishida J, Wang Z, Bunting KD, et al. IL-3 induces basophil expansion in vivo by directing granulocyte-monocyte progenitors to differentiate into basophil lineage-restricted progenitors in the bone marrow and by increasing the number of basophil/mast cell progenitors in the spleen. Journal of immunology (Baltimore, Md : 1950). 2009;182(5):2835–41.
Siracusa MC, Saenz SA, Hill DA, Kim BS, Headley MB, Doering TA, et al. TSLP promotes interleukin-3-independent basophil haematopoiesis and type 2 inflammation. Nature [Research Support, NIH, Extramural Research Support, Non-US Gov't]. 2011;477(7363):229–33.
Giacomin PR, Siracusa MC, Walsh KP, Grencis RK, Kubo M, Comeau MR, et al. Thymic stromal lymphopoietin-dependent basophils promote Th2 cytokine responses following intestinal helminth infection. J Immunol. 2012;189(9):4371–8.
Sullivan BM, Liang HE, Bando JK, Wu D, Cheng LE, McKerrow JK, et al. Genetic analysis of basophil function in vivo. Nat Immunol. 2011;12(6):527–35.
Ohnmacht C, Schwartz C, Panzer M, Schiedewitz I, Naumann R, Voehringer D. Basophils orchestrate chronic allergic dermatitis and protective immunity against helminths. Immunity. 2010;33(3):364–74.
Obata-Ninomiya K, Ishiwata K, Tsutsui H, Nei Y, Yoshikawa S, Kawano Y, et al. The skin is an important bulwark of acquired immunity against intestinal helminths. J Exp Med. 2013;210(12):2583–95.
Mashiko S, Mehta H, Bissonnette R, Sarfati M. Increased frequencies of basophils, type 2 innate lymphoid cells and Th2 cells in skin of patients with atopic dermatitis but not psoriasis. Journal of dermatological science. 2017 15.
Rocha R, Vitor AB, Trindade E, Lima R, Tavares M, Lopes J, et al. Omalizumab in the treatment of eosinophilic esophagitis and food allergy. Eur J Pediatr. 2011;170(11):1471–4.
Noti M, Wojno ED, Kim BS, Siracusa MC, Giacomin PR, Nair MG, et al. Thymic stromal lymphopoietin-elicited basophil responses promote eosinophilic esophagitis. Nat Med. 2013;19(8):1005–13.
Venturelli N, Lexmond WS, Ohsaki A, Nurko S, Karasuyama H, Fiebiger E, et al. Allergic skin sensitization promotes eosinophilic esophagitis through the IL-33-basophil axis in mice. J Allergy Clin Immunol. 2016;138(5):1367–80.e5.
Sherrill JD, Gao PS, Stucke EM, Blanchard C, Collins MH, Putnam PE, et al. Variants of thymic stromal lymphopoietin and its receptor associate with eosinophilic esophagitis. J Allergy Clin Immunol. 2010;126(1):160–5.e3.
Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–83.
Van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Annu Rev Immunol. 2013;31:317–43.
Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, Non-P.H.S.]. 2011;332(6035):1284–8.
Chen F, Wu W, Millman A, Craft JF, Chen E, Patel N, et al. Neutrophils prime a long-lived effector macrophage phenotype that mediates accelerated helminth expulsion. Nat Immunol. 2014;15(10):938–46.
Sutherland TE, Logan N, Ruckerl D, Humbles AA, Allan SM, Papayannopoulos V, et al. Chitinase-like proteins promote IL-17-mediated neutrophilia in a tradeoff between nematode killing and host damage. Nat Immunol. 2014;15(12):1116–25.
Bonne-Annee S, Kerepesi LA, Hess JA, O'Connell AE, Lok JB, Nolan TJ, et al. Human and mouse macrophages collaborate with neutrophils to kill larval Strongyloides stercoralis. Infect Immun. 2013;81(9):3346–55.
Marsland BJ, Kurrer M, Reissmann R, Harris NL, Kopf M. Nippostrongylus brasiliensis infection leads to the development of emphysema associated with the induction of alternatively activated macrophages. Eur J Immunol. 2008;38(2):479–88.
Chen F, Liu Z, Wu W, Rozo C, Bowdridge S, Millman A, et al. An essential role for TH2-type responses in limiting acute tissue damage during experimental helminth infection. Nat Med. 2012;18(2):260–6.
Nieuwenhuizen NE, Kirstein F, Jayakumar J, Emedi B, Hurdayal R, Horsnell WG, et al. Allergic airway disease is unaffected by the absence of IL-4Ralpha-dependent alternatively activated macrophages. J Allergy Clin Immunol. 2012;130(3):743–50.e8.
Moon KA, Kim SY, Kim TB, Yun ES, Park CS, Cho YS, et al. Allergen-induced CD11b+ CD11c(int) CCR3+ macrophages in the lung promote eosinophilic airway inflammation in a mouse asthma model. Int Immunol. 2007;19(12):1371–81.
Han H, Headley MB, Xu W, Comeau MR, Zhou B, Ziegler SF. Thymic stromal lymphopoietin amplifies the differentiation of alternatively activated macrophages. Journal of immunology (Baltimore, Md : 1950). 2013;190(3):904–12.
Kopp HG, Avecilla ST, Hooper AT, Rafii S. The bone marrow vascular niche: home of HSC differentiation and mobilization. Physiology (Bethesda, Md). 2005;20:349–56.
Mazo IB, Massberg S, von Andrian UH. Hematopoietic stem and progenitor cell trafficking. Trends Immunol. 2011;32(10):493–503.
Saenz SA, Siracusa MC, Perrigoue JG, Spencer SP, Urban JF Jr, Tocker JE, et al. IL25 elicits a multipotent progenitor cell population that promotes T(H)2 cytokine responses. Nature. 2010;464(7293):1362–6.
Siracusa MC, Saenz SA, Wojno ED, Kim BS, Osborne LC, Ziegler CG, et al. Thymic stromal lymphopoietin-mediated extramedullary hematopoiesis promotes allergic inflammation. Immunity. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't]. 2013;39(6):1158–70.
Johnston LK, Hsu CL, Krier-Burris RA, Chhiba KD, Chien KB, McKenzie A, et al. IL-33 precedes IL-5 in regulating eosinophil commitment and is required for eosinophil homeostasis. Journal of immunology (Baltimore, Md : 1950). 2016;197(9):3445–53.
Kim YK, Uno M, Hamilos DL, Beck L, Bochner B, Schleimer R, et al. Immunolocalization of CD34 in nasal polyposis. Effect of topical corticosteroids. Am J Respir Cell Mol Biol. 1999;20(3):388–97.
Acknowledgements
Work in the Siracusa lab is supported by the NIH (RO1 AI123224 to M.C.S.) and by the New Jersey Health foundation. We would like to thank members of the Siracusa lab for their critical reading of this manuscript and Servier Medical Art for use of their graphics.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Immunology and Inflammation
Rights and permissions
About this article

Cite this article
Henry, E.K., Inclan-Rico, J.M. & Siracusa, M.C. Type 2 Cytokine Responses: Regulating Immunity to Helminth Parasites and Allergic Inflammation. Curr Pharmacol Rep 3, 346–359 (2017). https://doi.org/10.1007/s40495-017-0114-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40495-017-0114-1