
Abstract
The global burden of chronic disorders such as Parkinson’s disease (PD) has rapidly increased over recent decades. Despite an increasing understanding of PD pathophysiology, there are no effective therapies capable of stopping or slowing the progression of this neurological condition. It has been suggested that type 2 diabetes mellitus (T2DM) may be a risk factor for PD and comorbid T2DM may worsen PD symptoms, as well as accelerate neurodegeneration. In fact, the similar pathological mechanisms shared by PD and T2DM have inspired several studies on the therapeutic potential of T2DM drugs against PD, among which glucagon-like peptide-1 receptor (GLP-1R) agonists are promising candidates. Here, we highlight the mechanisms linking T2DM and PD, as well as the links between insulin resistance (IR) and PD patients’ risk of developing cognitive deficits. We also briefly review the effects of GLP-1R agonists on PD and discuss how the successful use of these substances in preclinical models of PD has paved the way for PD clinical trials. We further discuss how recent evidence on the beneficial effects of dulaglutide on cognitive function of T2DM patients may have important implications for PD drug repurposing.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Type 2 diabetes mellitus and Parkinson’s disease share similar pathological mechanisms, such as insulin resistance |
Insulin resistance may increase the risk of a PD patient developing cognitive impairment and dementia |
Preclinical and clinical data have shown that glucagon-like peptide-1 receptor agonists may provide promising therapy for Parkinson’s disease |
Emerging evidence suggests that dulaglutide may represent an attractive resource for drug repurposing in Parkinson’s disease |
1 Introduction
Parkinson’s disease (PD) is the fastest growing neurological disorder with regard to prevalence, disability, and death [1]. In fact, the prevalence of PD has more than doubled over recent decades and more than 12 million people are expected to suffer from PD by 2040 [1]. This neurological disorder results from the progressive and selective degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc) [2, 3]. The degeneration of SNpc dopamine neurons is likely caused by the formation of cytoplasmic inclusions known as Lewy bodies (LBs), which in turn results from the aggregation of misfolded α-synuclein (αSyn) protein into fibrils [4]. PD is typically diagnosed on the basis of motor signs, but it may also manifest with a wide spectrum of nonmotor symptoms [2].
Type 2 diabetes mellitus (T2DM) is a chronic metabolic disease characterized by consistently elevated levels of blood glucose, which result from a reduced whole-body sensitivity to the action of insulin known as insulin resistance (IR) [5]. The glucagon-like peptide-1 (GLP-1) is an incretin hormone produced both peripherally by small-intestinal L cells and centrally by pre-proglucagon neurons in the nucleus of the solitary tract (NST) that acts via GLP-1 receptor (GLP-1R) to modulate insulin signaling [6]. Insulin signal dysregulation may lead to the downregulation of phosphatidylinositol 3-kinase (PI3K)-protein kinase B (AKT) pathway, which targets several substrates that are essential for neuronal survival. The disruption of these signaling downstream events may contribute to the hyperphosphorylation and accumulation of αSyn, and consequently to PD [7,8,9]. Mitochondrial dysfunction, oxidative stress, and chronic inflammation are also common features in the pathophysiology of both T2DM and PD [7,8,9].
2 The Relationship Between Parkinson’s Disease and Type 2 Diabetes Mellitus
Epidemiological studies have suggested that T2DM may be a risk factor for PD. Findings from population-based studies showed that diabetes may increase PD risk by up to 38% [10, 11]. A recent record-linkage cohort study showed a significantly increased rate of PD in a cohort of 2,017,115 patients with T2DM compared to a cohort of 6,173,208 individuals without T2DM [12]. In addition, comorbid T2DM was associated with more severe motor and cognitive impairment [13,14,15], as well as with faster motor and cognitive decline [13].
Several studies have proposed that disruptions in shared pathogenic pathways may lead to IR and, ultimately, to T2DM and PD [7]. Impaired insulin signaling decreases PI3K-AKT activity, which in turn impairs the modulation of downstream targets, such as forkhead box protein O (FoxO), mechanistic target of rapamycin (mTOR), and glycogen synthase kinase 3β (GSK3β) [16]. These substrates control a variety of cellular processes involved in PD pathogenesis, including the degradation of αSyn, mitochondrial biogenesis, and the modulation of inflammatory signaling and oxidative stress [9, 16]. For example, findings from PD preclinical studies revealed that the inhibition of mTOR signaling led to neuronal cell death by suppressing AKT [17, 18], while an anti-aggregation gene therapy approach protected against the neurotoxic effects of αSyn aggregation by activating AKT [19]. In addition, abnormal activated GSK3β has been found to increase αSyn phosphorylation, and postmortem analysis of brain tissue from PD patients showed an increased expression of GSK3β within the halo of LBs in nigral neurons, which suggests that GSK3β may interact with substrates in LBs to play a role in PD pathogenesis [20]. Conversely, GSK3β inhibition was found to decrease αSyn phosphorylation and aggregation in an in vitro rotenone-model of PD [21].
Mounting evidence indicates that the expression of various transcription factors involved in glucose metabolism and insulin regulation is also altered in both PD and T2DM [16]. A recent proof-of-concept study showed that IR-associated diabetes promotes the development and progression of PD through aberrant expression of αSyn, impairment of mitochondrial function, upregulation of mitochondrial oxidative stress, and deregulation of the polo-like kinase-2 (PLK2) signaling [22]. It was also demonstrated that the upregulation of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α) expression suppressed IR-induced mitochondrial dysfunction and PLK2 hyperactivity, as well as downregulated αSyn overexpression [22]. In addition, a genome-wide meta-analysis identified that gene sets regulated by PGC-1α are downregulated in postmortem tissue from PD patients [23].
Increased inflammation has also been recognized as a link between PD and T2DM. Postmortem analysis revealed higher activated microglia levels in the SNpc, putamen, hippocampus, and cortex of brains from subjects with PD than those from healthy subjects [24], as well as higher inflammatory cytokine levels, such as interleukin (IL)-1β, tumor necrosis factor (TNF), and IL-6 [25, 26]. Increased peripheral concentrations of IL-1β, TNF, IL-6, and IL-2 were also found in patients with PD [27]. In fact, increased plasma concentration of IL-6 was prospectively associated with an increased risk of developing PD [28]. In addition, increased expression of the transcription factor nuclear factor kappa beta (NF-κB), which is known to modulate several genes involved in multiple aspects of inflammatory responses, has been found in dopaminergic neurons of PD brains [29, 30]. Conversely, the inhibition of this factor was shown to protect dopaminergic neurons against neurotoxin-induced cell death in PD animal models [31, 32].
3 The Role of Insulin Signaling in Parkinson’s Disease Cognitive Impairment
Up to 60% of PD patients with dementia were found to have IR [33], and a reduced glucose metabolism in multiple brain areas was found in PD patients with cognitive decline compared with those without cognitive impairment [34, 35]. PD patients with diabetes also had lower global cognitive z scores compared to those without diabetes, which suggests that comorbid T2DM may also be associated with a more severe cognitive dysfunction in PD [14].
The effects of insulin on cognition have been linked to the activation of PI3K-AKT pathway, and the dysfunctional PI3K-AKT signaling to downstream targets such as GSK3β appears to contribute to the development of cognitive impairment and dementia in PD patients [36, 37]. The activation of GSK3β increases tau protein phosphorylation, and the abnormal activation of GSK-3β by phosphorylated αSyn contributes to the hyperphosphorylation of tau; therefore, αSyn and tau have synergistic effects on neurodegeneration and cognitive decline. A longitudinal follow-up study suggested that a variation in the tau gene (MAPT) is a genetic risk factor for the early development of cognitive impairment and dementia in PD [38]. In addition, postmortem findings revealed the presence of tau pathology in brains of PD patients with dementia [39]. A similar interplay between phosphorylated αSyn and β-amyloid peptides (Aβ) has also been suggested to play a role in the development of cognitive impairment and dementia in PD [7,8,9]. For example, transgenic mice expressing both human αSyn and Aβ performed poorly on hippocampal-dependent learning and memory tasks, while transgenic mice expressing only human αSyn had a normal performance [40]. Additionally, decreased cerebrospinal fluid (CSF) concentration of the isoform Aβ42, which may reflect increased accumulation levels of Aβ in the cortex, has been associated with cognitive impairment in PD, and it has also been considered a risk factor for PD dementia [41,42,43,44].
Chronic inflammation has also been associated with cognitive decline in PD [16]. HLA-DR-positive microglia was found in the hippocampus of PD patients with dementia [45]. Cortical microglial activation is higher in PD patients with and without dementia than in healthy controls, although the spatial extent of the microglial activation is greater in PD patients with dementia [46]. The majority of PD patients with dementia also showed a reduction in glucose metabolism in the hippocampus [46]. The activation of the peroxisome proliferator-activated receptor gamma (PPAR-γ), which is known to increase insulin sensitization, protected a rat model of PD against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced memory impairment [47]. The neuroprotective effects of PPAR-γ activation was associated with the inhibition of NF-κB activation, suppression of inducible nitric oxide synthase (iNOS) induction, and decrease in microglial activation [48].
4 Therapeutic Potential of Glucagon-like Peptide-1 Receptor Agonists Against Parkinson’s Disease
GLP-1R agonists are among the most promising T2DM drugs against PD (Fig. 1) [7,8,9, 49]. The GLP-1R agonist exenatide has been found to protect neurons against 6-hydroxydopamine (6-OHDA)- and MPTP-induced toxicity, and to improve motor function in mouse models of PD [50,51,52]. The treatment with exenatide also increased striatal dopamine level and improved motor performance in diabetic rats with MPTP-induced PD [50]. These neuroprotective effects were associated with exenatide’s ability to increase the activity of antioxidant enzymes such as superoxide dismutase (SOD), catalase (CAT) and glutathione (GSH), as well as to suppress the expression of inflammatory cytokines, such as IL-1α and TNF-α [50]. An initial single-blind trial demonstrated that exenatide improved both motor and cognitive function in patients with PD [53]. These effects persisted over a year following exenatide exposure [54]. A further double-blind, placebo-controlled trial showed that individuals with PD receiving exenatide exhibited better motor function than PD individuals receiving placebo, and this beneficial effect was still detectable following a 12-week washout period [55]. A post hoc analysis also suggested that PD patients with IR or obesity might have better cognitive response to exenatide than other subgroups of PD patients [56].

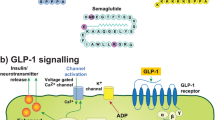
Potential mechanisms underlying the neuroprotective effects of glucagon-like peptide-1 receptor (GLP-1R) agonists on Parkinson’s disease (PD). Chronic insulin resistance (IR) may lead to the downregulation of phosphatidylinositol 3-kinase (PI3K)-protein kinase B (AKT) pathway, which may contribute to the pathogenesis of PD. GLP-1R agonists have been found to exert neuroprotective effects against PD by modulating PI3K-AKT signaling and its major downstream targets, such as forkhead box protein O (FoxO), mechanistic target of rapamycin (mTOR), glycogen synthase kinase 3β (GSK3β), and nuclear factor kappa beta (NF-κB). Activated AKT phosphorylates GSK3, which decreases the levels of hyperphosphorylation of tau, as well as α-synuclein (αSyn) and β-amyloid peptides (Aβ) aggregation. AKT-mediated phosphorylation of FoxO inhibits apoptosis-related factors and enhances mitochondrial function. Activation of mTORC signaling by AKT is important for synaptic plasticity and neuronal survival. Finally, AKT-mediated inhibition of NF-κB signaling downregulates pro-inflammatory cytokine release and microglial activation
Recently, it was found that PD subjects treated with exenatide had enhanced tyrosine phosphorylation of insulin receptor substrate 1 (IRS-1) compared with placebo-treated subjects (tyrosine phosphorylation of IRS-1 is needed for insulin-evoked responses) [57]. It was also found that neuronal-derived exosomes from exenatide-treated PD patients showed higher expression levels of PI3K-AKT and phosphorylated mTOR than those from placebo-treated patients. The activation of PI3K-AKT and mTOR cascades has been shown to regenerate nigrostriatal axons and prevent neurodegeneration in toxin-based models of PD [58, 59]. In addition, recent evidence suggests that the highly brain-penetrant NLY01, a pegylated form of exenatide, protected against dopaminergic neurodegeneration, ameliorated behavioral deficits, and decreased Lewy-like pathology in mouse models of PD [60]. These effects were associated with the inhibition of astrocyte conversion to the neurotoxic A1 phenotype by microglia as NLY01 inhibited the NF-κB pathway, as well as decreased the release of IL-1β, TNF-α and complement component 1, subcomponent q (C1q) from the αSyn-activated microglia [60]. These findings inspired the design of a double-blinded, placebo-controlled trial that will assess the safety, tolerability, and efficacy of NLY01 on motor signs and symptoms in individuals with early-untreated PD (clinical trial identifier NCT04154072).
Other GLP-1R agonists such as liraglutide, lixisenatide, and semaglutide have also demonstrated neuroprotective effects in animal models of PD [51, 61]. Both liraglutide and lixisenatide were found to be more effective than exenatide in preventing MPTP-induced decrease in nigral tyrosine hydroxylase (TH) levels and motor impairment in PD mice [51]. Conversely, once-weekly semaglutide was more effective than once-daily liraglutide in rescuing reduced TH levels in the SNpc and striatum of MPTP-treated mice [61]. An ongoing double-blinded, placebo-controlled trial will address the efficacy of once-daily liraglutide on motor, non-motor, and cognitive symptoms of PD (clinical trial identifier NCT02953665). In addition, a current double-blinded, placebo-controlled trial will evaluate the efficacy of once-daily lixisenatide on the progression of motor disability in patients with early PD (clinical trial identifier NCT03439943). A double-blinded, placebo-controlled study is also underway to evaluate the efficacy of semaglutide administered once weekly on motor function of newly diagnosed patents with PD (clinical trial identifier NCT03659682 study).
5 Glucagon-like Peptide-1 Receptor Agonist Dulaglutide as a Potential Candidate for Drug Repurposing in Parkinson’s Disease
A recent exploratory analysis of the REWIND trial showed that long-term treatment with the GLP-1R agonist dulaglutide reduced cognitive impairment in persons with T2DM [62]. This outcome persisted after accounting for incident clinical stroke, which was also previously demonstrated to be reduced by dulaglutide treatment [63]. Given that cognitive impairment is a common complication following stroke, the observed cognitive improvement may reflect a neuroprotective effect of dulaglutide rather than a simple manifestation of the beneficial effect of this drug on the incidence of clinically relevant strokes [62, 63]. In addition, a recent study demonstrated that dulaglutide improved cognitive function and memory in the streptozotocin-induced mouse model of Alzheimer's disease (AD) by enhancing PI3K-AKT-GSK3β signaling pathways, and thereby decreasing tau and neurofilament phosphorylation [64]. It was also found that intraperitoneally injected dulaglutide significantly increased the expression of GLP-1 and GLP-1R in the brain of AD mice [22].
In view of the emerging evidence that dulaglutide might have beneficial effects on cognitive function [62], the mechanistic assessment of dulaglutide efficacy in preclinical animal models of PD (and the potential translation of preclinical findings into clinical practice) is highly encouraged. In addition, dulaglutide may offer some advantages compared to other GLP-1R agonists. First, dulaglutide is a GLP-1/Fc fusion protein with a substantially prolonged circulation half-life, which supports convenient, once-weekly dosing [65]. This long half-life mainly results from a reduced renal clearance, in addition to being resistant to dipeptidyl peptidase 4 (DPP-4) degradation [65]. Although lixisenatide and liraglutide are also degradation-resistant, these peptides have shorter half-life than dulaglutide, and thus require daily injections [65]. Other once-weekly GLP-1 agonists such as exenatide once weekly, albiglutide, and semaglutide have also been approved for the treatment of T2DM, but several studies have shown that such analogs vary in efficacy and tolerability. For example, dulaglutide 1.5 mg showed a greater decrease in glycated hemoglobin (HbA1c) levels than albiglutide [66]. Dulaglutide also showed a favorable safety profile in patients with T2DM and moderate-to-severe chronic kidney disease [67]. In addition, the production of anti-drug antibodies was markedly higher in patients treated with once-weekly exenatide than those treated with dulaglutide [68, 69]. A head-to-head trial showed that nausea was reported by 23% of patients receiving semaglutide 0.5 mg and 13% of patients receiving dulaglutide 0.75 mg. These events contributed to a higher proportion of premature treatment discontinuation in semaglutide-treated groups than in dulaglutide-treated groups [70]. Second, clinical findings suggest that adherence to GLP-1R agonist therapy significantly improves with once-weekly products compared to once- or twice-daily formulations [71, 72]. In addition, a higher proportion of dulaglutide users were adherent to and persistent with their treatment, compared to subjects treated with either once-weekly exenatide or semaglutide [73]. GLP-1R agonist users have higher preference for dulaglutide than other once-weekly formulations because it is a single-dose, easy-to-use injectable pen device; while albiglutide and once-weekly exenatide need to be reconstituted before use, dulaglutide needs no reconstitution [74]. Given that polypharmacy and regimen complexity have been associated with medication non-adherence in PD, simpler regimens may lead to better treatment adherence, particularly in PD patients with cognitive impairment [75]. Third, dulaglutide has been found to prevent major adverse cardiovascular events in T2DM patients with and without previous cardiovascular disease [76]. This finding led FDA to approve for the first time a T2DM drug for reducing primary and secondary cardiovascular outcomes in T2DM patients. Therefore, PD patients, who are at a higher risk of developing cardiovascular diseases [77], would greatly benefit from the cardioprotective potential of dulaglutide therapy.
Although there is no evidence that dulaglutide is capable of crossing the blood–brain barrier (BBB) and thus playing a direct role in central GLP-1R modulation, peripherally administered dulaglutide could exert neuroprotective effects by several other mechanisms. First, dulaglutide may alter blood levels of other substances, which can then cross the BBB. For example, dulaglutide was found to decrease serum levels of TNF-α, IL-6, and C-reactive protein in T2DM patients [78, 79]. Given that these substances can get through the BBB to stimulate neuroinflammation, and that chronic neuroinflammation is one of the hallmarks of PD pathophysiology [80], it is possible to suggest that the beneficial effects of dulaglutide on peripheral inflammation might play a protective role in central neuroinflammation. Second, circulating dulaglutide may access the brain via leaky sites at the circumventricular organs (CVOs). Although intravenously administrated liraglutide and semaglutide have not been found to measurably cross the BBB in mice [81], previous studies suggested that these drugs interact with the brain by accumulating in CVOs and GLP-1R-expressing areas adjacent to the ventricles [82, 83]. In addition, specialized structures in the blood-CSF barrier of the ventricular wall have been found to control the transport of macromolecules between the blood and CSF compartments. For example, blood-borne leptin is transported from the periphery into the CSF by tanycytes, which are specialized ependymal cells located in the median eminence of the hypothalamus, a circumventricular organ at the bottom of the mediobasal hypothalamus. From the CSF, leptin can freely gain access to other leptin-sensitive areas, such as the hippocampus [84]. Third, peripherally acting GLP‐1R agonists may communicate with the brain by interacting with vagal afferent nerves at the nodose ganglion or hepatic portal region [85]. Infusion of GLP‐1 into the hepatic portal vein facilitates the release of insulin, whereas GLP-1-induced insulin secretion is greatly reduced in rats with selective hepatic vagotomy [86]. It was also found that the control of food intake and the regulation of glucose homeostasis were impaired in rats with a specific knockdown of GLP-1R expression in vagal afferents [87]. The effects of peripherally injected GLP-1 on food intake, gastric emptying, and insulin secretion were also impaired in truncally vagotomized subjects with pyloroplasty [88]. These findings highlight an important role for vagal afferent neurons in mediating GLP-1 function, and thus supporting the crosstalk between the periphery and CNS [85]. In addition, recent evidence suggests that different GLP-1R-positive neuronal circuits play distinct roles in controlling physiological versus pharmacological effects on food intake, glucose homeostasis, and gastric emptying [89]. The finding that key neuronal populations have central relevance to the transduction of gut-derived GLP-1R signals provides further evidence supporting the importance of the gut-brain axis for gut-brain GLP-1R signaling [89]. Given the pleiotropic effects of GLP‐1R agonists on the gut–brain axis, and that the gut-brain axis has been found to play a synergistic role in the pathogenesis of PD [90], therapeutic strategies targeting this pathway could potentially provide an effective treatment option for PD.
6 Conclusion
IR is among the multiple pathogenic mechanisms shared by PD and T2DM. Several animal model and clinical studies agree on the effectiveness of T2DM drugs such as GLP‐1R agonists against PD. In addition, the evidence of protective effects of dulaglutide against cognitive decline in T2DM patients potentially opens up a new opportunity for drug repurposing in PD, especially because disease-modifying therapy for this neurological disorder is currently unavailable. Dulaglutide is currently commercially exploited as a therapy for T2DM, thereby allowing fast-track assessment of its safety and efficacy in PD clinical trials. The exact mechanisms by which T2DM may contribute to PD risk remain unclear, and further efforts to determine how GLP‐1R agonists exert benefits for PD might help to identify novel drug targets for PD and related disorders.
References
Feigin VL, Abajobir AA, Abate KH, Abd-Allah F, Abdulle AM, Abera SF, et al. Global, regional, and national burden of neurological disorders during 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017;16:877–97. https://doi.org/10.1016/S1474-4422(17)30299-5.
Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, et al. Parkinson disease. Nat Rev Dis Prim. 2017;3:17013. https://doi.org/10.1038/nrdp.2017.13.
Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain. 1991;114:2283–301. https://doi.org/10.1093/brain/114.5.2283.
Lashuel HA. Do Lewy bodies contain alpha-synuclein fibrils? and Does it matter? A brief history and critical analysis of recent reports. Neurobiol Dis. 2020;141:104876. https://doi.org/10.1016/j.nbd.2020.104876.
Wu H, Deng X, Shi Y, Su Y, Wei J, Duan H. PGC-1α, glucose metabolism and type 2 diabetes mellitus. J Endocrinol. 2016;229:R99–115. https://doi.org/10.1530/JOE-16-0021.
Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–57. https://doi.org/10.1053/j.gastro.2007.03.054
Cheong JLY, de Pablo-Fernandez E, Foltynie T, Noyce AJ. The association between Type 2 diabetes mellitus and Parkinson’s disease. J Parkinsons Dis. 2020. https://doi.org/10.3233/JPD-191900.
Green H, Tsitsi P, Markaki I, Aarsland D, Svenningsson P. Novel treatment opportunities against cognitive impairment in Parkinson’s disease with an emphasis on diabetes-related pathways. CNS Drugs. 2019;33:143–60. https://doi.org/10.1007/s40263-018-0601-x.
Athauda D, Foltynie T. Insulin resistance and Parkinson’s disease: A new target for disease modification? Prog Neurobiol. 2016;145–146:98–120. https://doi.org/10.1016/j.pneurobio.2016.10.001.
Yang YW, Hsieh TF, Li CI, Liu CS, Lin WY, Chiang JH, et al. Increased risk of Parkinson disease with diabetes mellitus in a population-based study. Med (United States). 2017;96:e5921. https://doi.org/10.1097/MD.0000000000005921.
Yue X, Li H, Yan H, Zhang P, Chang L, Li T. Risk of Parkinson disease in diabetes mellitus: An updated meta-analysis of population-based cohort studies. Med (United States). 2016;95:e3549. https://doi.org/10.1097/MD.0000000000003549.
De Pablo-Fernandez E, Goldacre R, Pakpoor J, Noyce AJ, Warner TT. Association between diabetes and subsequent Parkinson disease: A record-linkage cohort study. Neurology. 2018;91:e139–42. https://doi.org/10.1212/WNL.0000000000005771.
Pagano G, Polychronis S, Wilson H, Giordano B, Ferrara N, Niccolini F, et al. Diabetes mellitus and Parkinson disease. Neurology. 2018;90:e1654–62. https://doi.org/10.1212/WNL.0000000000005475.
Bohnen NI, Kotagal V, Müller MLTM, Koeppe RA, Scott PJH, Albin RL, et al. Diabetes mellitus is independently associated with more severe cognitive impairment in Parkinson disease. Parkinsonism Relat Disord. 2014;20:1394–8. https://doi.org/10.1016/j.parkreldis.2014.10.008.
Giuntini M, Baldacci F, Del Prete E, Bonuccelli U, Ceravolo R. Diabetes is associated with postural and cognitive domains in Parkinson’s disease. Results from a single-center study. Parkinson Relat Disord. 2014;20:671–2. https://doi.org/10.1016/j.parkreldis.2014.02.016.
Santiago JA, Potashkin JA. Shared dysregulated pathways lead to Parkinson’s disease and diabetes. Trends Mol Med. 2013;19:176–86. https://doi.org/10.1016/j.molmed.2013.01.002.
Xu Y, Liu C, Chen S, Ye Y, Guo M, Ren Q, et al. Activation of AMPK and inactivation of Akt result in suppression of mTOR-mediated S6K1 and 4E-BP1 pathways leading to neuronal cell death in in vitro models of Parkinson’s disease. Cell Signal. 2014;26:1680–9. https://doi.org/10.1016/j.cellsig.2014.04.009.
Malagelada C, Zong HJ, Greene LA. RTP801 is induced in Parkinson’s disease and mediates neuron death by inhibiting Akt phosphorylation/activation. J Neurosci. 2008;28:14363–71. https://doi.org/10.1523/JNEUROSCI.3928-08.2008.
Hashimoto M, Rockenstein E, Mante M, Crews L, Bar-On P, Gage FH, et al. An antiaggregation gene therapy strategy for Lewy body disease utilizing β-synuclein lentivirus in a transgenic model. Gene Ther. 2004;11:1713–23. https://doi.org/10.1038/sj.gt.3302349.
Nagao M, Hayashi H. Glycogen synthase kinase-3beta is associated with Parkinson’s disease. Neurosci Lett. 2009;449:103–7. https://doi.org/10.1016/j.neulet.2008.10.104.
Yuan YH, Yan WF, Sun JD, Huang JY, Mu Z, Chen NH. The molecular mechanism of rotenone-induced α-synuclein aggregation: EMPHASIZING the role of the calcium/GSK3β pathway. Toxicol Lett. 2015;233:163–71. https://doi.org/10.1016/j.toxlet.2014.11.029.
Hong C-T, Chen K-Y, Wang W, Chiu J-Y, Wu D, Chao T-Y, et al. Insulin resistance promotes Parkinson’s disease through aberrant expression of α-synuclein, mitochondrial dysfunction, and deregulation of the polo-like kinase 2 signaling. Cells. 2020;9:740. https://doi.org/10.3390/cells9030740.
Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML, et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci Transl Med. 2010;2:52ra73. https://doi.org/10.1126/scitranslmed.3001059.
Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 2003;106:518–26. https://doi.org/10.1007/s00401-003-0766-2.
Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, et al. Interleukin-1β, interleukin-6, epidermal growth factor and transforming growth factor-α are elevated in the brain from Parkinsonian patients. Neurosci Lett. 1994;180:147–50. https://doi.org/10.1016/0304-3940(94)90508-8.
Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor necrosis factor-α (TNF-α) increases both in the brain and in the cerebrospinal fluid from Parkinsonian patients. Neurosci Lett. 1994;165:208–10. https://doi.org/10.1016/0304-3940(94)90746-3.
Qin XY, Zhang SP, Cao C, Loh YP, Cheng Y. Aberrations in peripheral inflammatory cytokine levels in Parkinson disease: A systematic review and meta-analysis. JAMA Neurol. 2016;73:1316–24. https://doi.org/10.1001/jamaneurol.2016.2742.
Chen H, O’Reilly EJ, Schwarzschild MA, Ascherio A. Peripheral inflammatory biomarkers and risk of Parkinson’s disease. Am J Epidemiol. 2008;167:90–5. https://doi.org/10.1093/aje/kwm260.
Mogi M, Kondo T, Mizuno Y, Nagatsu T. p53 protein, interferon-γ, and NF-κB levels are elevated in the parkinsonian brain. Neurosci Lett. 2007;414:94–7. https://doi.org/10.1016/j.neulet.2006.12.003.
Hunot S, Brugg B, Ricard D, Michel PP, Muriel MP, Ruberg M, et al. Nuclear translocation of NF-κb is increased in dopaminergic neurons of patients with Parkinson disease. Proc Natl Acad Sci USA. 1997;94:7531–6. https://doi.org/10.1073/pnas.94.14.7531.
Ghosh A, Roy A, Liu X, Kordower JH, Mufson EJ, Hartley DM, et al. Selective inhibition of NF-κB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson’s disease. Proc Natl Acad Sci USA. 2007;104:18754–9. https://doi.org/10.1073/pnas.0704908104.
Zhang F, Shi JS, Zhou H, Wilson B, Hong JS, Gao HM. Resveratrol protects dopamine neurons against lipopolysaccharide-induced neurotoxicity through its anti-inflammatory actions. Mol Pharmacol. 2010;78:466–77. https://doi.org/10.1124/mol.110.064535.
Bosco D, Plastino M, Cristiano D, Colica C, Ermio C, De Bartolo M, et al. Dementia is associated with insulin resistance in patients with Parkinson’s Disease. J Neurol Sci. 2012;315:39–43. https://doi.org/10.1016/j.jns.2011.12.008.
Garcia-Garcia D, Clavero P, Salas CG, Lamet I, Arbizu J, Gonzalez-Redondo R, et al. Posterior parietooccipital hypometabolism may differentiate mild cognitive impairment from dementia in Parkinson’s disease. Eur J Nucl Med Mol Imaging. 2012;39:1767–77. https://doi.org/10.1007/s00259-012-2198-5.
Hosokai Y, Nishio Y, Hirayama K, Takeda A, Ishioka T, Sawada Y, et al. Distinct patterns of regional cerebral glucose metabolism in Parkinson’s disease with and without mild cognitive impairment. Mov Disord. 2009;24:854–62. https://doi.org/10.1002/mds.22444.
Wang Y, Liu W, He X, Zhou F. Parkinson’s disease-associated Dj-1 mutations increase abnormal phosphorylation of tau protein through Akt/Gsk-3β pathways. J Mol Neurosci. 2013;51:911–8. https://doi.org/10.1007/s12031-013-0099-0.
McNay EC, Ong CT, McCrimmon RJ, Cresswell J, Bogan JS, Sherwin RS. Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol Learn Mem. 2010;93:546–53. https://doi.org/10.1016/j.nlm.2010.02.002.
Goris A, Williams-Gray CH, Clark GR, Foltynie T, Lewis SJG, Brown J, et al. Tau and α-synuclein in susceptibility to, and dementia in, Parkinson’s disease. Ann Neurol. 2007;62:145–53. https://doi.org/10.1002/ana.21192.
Wills J, Jones J, Haggerty T, Duka V, Joyce JN, Sidhu A. Elevated tauopathy and alpha-synuclein pathology in postmortem Parkinson’s disease brains with and without dementia. Exp Neurol. 2010;225:210–8. https://doi.org/10.1016/j.expneurol.2010.06.017.
Masliah E, Rockenstein E, Veinbergs I, Sagara Y, Mallory M, Hashimoto M, et al. β-Amyloid peptides enhance α-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc Natl Acad Sci USA. 2001;98:12245–50. https://doi.org/10.1073/pnas.211412398.
Terrelonge M, Marder KS, Weintraub D, Alcalay RN. CSF β-Amyloid 1–42 predicts progression to cognitive impairment in newly diagnosed Parkinson disease. J Mol Neurosci. 2016;58:88–92. https://doi.org/10.1007/s12031-015-0647-x.
Parnetti L, Farotti L, Eusebi P, Chiasserini D, De Carlo C, Giannandrea D, et al. Differential role of CSF alpha-synuclein species, tau, and Aβ42 in Parkinson’s disease. Front Aging Neurosci. 2014;6:53. https://doi.org/10.3389/fnagi.2014.00053.
Alves G, Lange J, Blennow K, Zetterberg H, Andreasson U, Førland MG, et al. CSF Aβ42 predicts early-onset dementia in Parkinson disease. Neurology. 2014;82:1784–90. https://doi.org/10.1212/WNL.0000000000000425.
Siderowf A, Xie SX, Hurtig H, Weintraub D, Duda J, Chen-Plotkin A, et al. CSF amyloid β 1–42 predicts cognitive decline in Parkinson disease. Neurology. 2010;75:1055–61. https://doi.org/10.1212/WNL.0b013e3181f39a78.
McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the: Substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38:1285–91. https://doi.org/10.1212/wnl.38.8.1285.
Edison P, Ahmed I, Fan Z, Hinz R, Gelosa G, Ray Chaudhuri K, et al. Microglia, amyloid, and glucose metabolism in parkinson’s disease with and without dementia. Neuropsychopharmacology. 2013;38:938–49. https://doi.org/10.1038/npp.2012.255.
Barbiero JK, Santiago RM, Persike DS, da Silva Fernandes MJ, Tonin FS, da Cunha C, et al. Neuroprotective effects of peroxisome proliferator-activated receptor alpha and gamma agonists in model of parkinsonism induced by intranigral 1-methyl-4-phenyl-1,2,3,6-tetrahyropyridine. Behav Brain Res. 2014;274:390–9. https://doi.org/10.1016/j.bbr.2014.08.014.
Dehmer T, Heneka MT, Sastre M, Dichgans J, Schulz JB. Protection by pioglitazone in the MPTP model of Parkinson’s disease correlates with IκBα induction and block of NFκB and iNOS activation. J Neurochem. 2004;88:494–501. https://doi.org/10.1046/j.1471-4159.2003.02210.x.
Markaki I, Winther K, Catrina SB, Svenningsson P. Repurposing GLP1 agonists for neurodegenerative diseases. Int Rev Neurobiol. 2020;155:91–112. https://doi.org/10.1016/bs.irn.2020.02.007.
Elbassuoni EA, Ahmed RF. Mechanism of the neuroprotective effect of GLP-1 in a rat model of Parkinson’s with pre-existing diabetes. Neurochem Int. 2019;131:104583. https://doi.org/10.1016/j.neuint.2019.104583.
Liu W, Jalewa J, Sharma M, Li G, Li L, Hölscher C. Neuroprotective effects of lixisenatide and liraglutide in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Neuroscience. 2015;303:42–50. https://doi.org/10.1016/j.neuroscience.2015.06.054.
Li Y, Perry T, Kindy MS, Harvey BK, Tweedie D, Holloway HW, et al. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc Natl Acad Sci. 2009;106:1285–90. https://doi.org/10.1073/pnas.0806720106.
Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Ell P, Soderlund T, et al. Exenatide and the treatment of patients with Parkinson’s disease. J Clin Invest. 2013;123:2730–6. https://doi.org/10.1172/JCI68295.
Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Kahan J, Ell P, et al. Motor and Cognitive Advantages Persist 12 Months After Exenatide Exposure in Parkinson’s Disease. J Parkinsons Dis. 2014;4:337–44. https://www.medra.org/servlet/aliasResolver?alias=iospress&, https://doi.org/10.3233/JPD-140364
Athauda D, Maclagan K, Skene SS, Bajwa-Joseph M, Letchford D, Chowdhury K, et al. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390:1664–75. https://doi.org/10.1016/S0140-6736(17)31585-4.
Athauda D, Maclagan K, Budnik N, Zampedri L, Hibbert S, Aviles-Olmos I, et al. Post hoc analysis of the Exenatide-PD trial-Factors that predict response. Eur J Neurosci. 2019;49:410–21. https://doi.org/10.1111/ejn.14096.
Athauda D, Gulyani S, Karnati HK, Li Y, Tweedie D, Mustapic M, et al. Utility of neuronal-derived exosomes to examine molecular mechanisms that affect motor function in patients with Parkinson disease. JAMA Neurol. 2019;76:420. http://archneur.jamanetwork.com/article.aspx?, https://doi.org/10.1001/jamaneurol.2018.4304
Kim SR, Chen X, Oo TF, Kareva T, Yarygina O, Wang C, et al. Dopaminergic pathway reconstruction by Akt/Rheb-induced axon regeneration. Ann Neurol. 2011;70:110–20. https://doi.org/10.1002/ana.22383.
Zhou Q, Liu C, Liu W, Zhang H, Zhang R, Liu J, et al. Rotenone induction of hydrogen peroxide inhibits mTOR-mediated S6K1 and 4E-BP1/eIF4E pathways, leading to neuronal apoptosis. Toxicol Sci. 2015;143:81–96. https://doi.org/10.1093/toxsci/kfu211.
Yun SP, Kam TI, Panicker N, Kim S, Oh Y, Park JS, et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med. 2018;24:931–8. https://doi.org/10.1038/s41591-018-0051-5.
Zhang L, Zhang L, Li L, Hölscher C. Neuroprotective effects of the novel GLP-1 long acting analogue semaglutide in the MPTP Parkinson’s disease mouse model. Neuropeptides. 2018;71:70–80. https://linkinghub.elsevier.com/retrieve/pii/S0143417918300684
Cukierman-Yaffe T, Gerstein HC, Colhoun HM, Diaz R, García-Pérez L-E, Lakshmanan M, et al. Effect of dulaglutide on cognitive impairment in type 2 diabetes: an exploratory analysis of the REWIND trial. Lancet Neurol. 2020;19:582–90. https://doi.org/10.1016/S1474-4422(20)30173-3.
Gerstein HC, Hart R, Colhoun HM, Diaz R, Lakshmanan M, Botros FT, et al. The effect of dulaglutide on stroke: an exploratory analysis of the REWIND trial. Lancet Diabetes Endocrinol. 2020;8:106–14. https://doi.org/10.1016/S2213-8587(19)30423-1.
Zhou M, Chen S, Peng P, Gu Z, Yu J, Zhao G, et al. Dulaglutide ameliorates STZ induced AD-like impairment of learning and memory ability by modulating hyperphosphorylation of tau and NFs through GSK3β. Biochem Biophys Res Commun. 2019;511:154–60. https://doi.org/10.1016/j.bbrc.2019.01.103.
Geiser JS, Heathman MA, Cui X, Martin J, Loghin C, Chien JY, et al. Clinical pharmacokinetics of dulaglutide in patients with type 2 diabetes: analyses of data from clinical trials. Clin Pharmacokinet. 2016;55:625–34. https://doi.org/10.1007/s40262-015-0338-3.
Zaccardi F, Htike ZZ, Webb DR, Khunti K, Davies MJ. Benefits and harms of once-weekly glucagon-like peptide-1 receptor agonist treatments a systematic review and network meta-analysis comparative clinical profiles of once-weekly GLP-1RAs. Ann Intern Med. 2015;164:102–13. https://doi.org/10.7326/M15-1432.
Tuttle KR, Lakshmanan MC, Rayner B, Busch RS, Zimmermann AG, Woodward DB, et al. Dulaglutide versus insulin glargine in patients with type 2 diabetes and moderate-to-severe chronic kidney disease (AWARD-7): a multicentre, open-label, randomised trial. Lancet Diabetes Endocrinol. 2018;6:605–17. https://doi.org/10.1016/S2213-8587(18)30104-9.
Milicevic Z, Anglin G, Harper K, Konrad RJ, Skrivanek Z, Glaesner W, et al. Low incidence of anti-drug antibodies in patients with type 2 diabetes treated with once-weekly glucagon-like peptide-1 receptor agonist dulaglutide. Diabetes Obes Metab. 2016;18:533–6. https://doi.org/10.1111/dom.12640.
Fineman MS, Mace KF, Diamant M, Darsow T, Cirincione BB, Booker Porter TK, et al. Clinical relevance of anti-exenatide antibodies: Safety, efficacy and cross-reactivity with long-term treatment. Diabetes Obes Metab. 2012;14:546–54. https://doi.org/10.1111/j.1463-1326.2012.01561.x.
Pratley RE, Aroda VR, Lingvay I, Lüdemann J, Andreassen C, Navarria A, et al. Semaglutide versus dulaglutide once weekly in patients with type 2 diabetes (SUSTAIN 7): a randomised, open-label, phase 3b trial. Lancet Diabetes Endocrinol. 2018;6:275–86. https://doi.org/10.1016/S2213-8587(18)30024-X.
Nguyen H, Dufour R, Caldwell-Tarr A. Glucagon-like peptide-1 receptor agonist (GLP-1RA) therapy adherence for patients with type 2 diabetes in a medicare population. Adv Ther. 2017;34:658–73. https://doi.org/10.1007/s12325-016-0470-y.
Kostev K, Ouwens M, Grandy S, Johnsson KM, Qiao Q. Adherence to GLP-1 receptor agonist therapy administered by once-daily or once-weekly injection in patients with type 2 diabetes in Germany. Diabetes Metab Syndr Obes Targets Ther. 2016;9:201–5. https://doi.org/10.2147/DMSO.S99732.
Mody R, Yu M, Nepal B, Konig M, Grabner M. Adherence and persistence among patients with type 2 diabetes initiating dulaglutide compared with semaglutide and exenatide BCise: 6-month follow-up from US real-world data. Diabetes Obes Metab. 2020. https://doi.org/10.1111/dom.14195.
Zhou AY, Trujillo JM. Comparison of usability, accuracy, preference, and satisfaction among three once-weekly GLP-1 receptor agonist pen devices. Diabetes Spectr. 2018;31:359–66. https://doi.org/10.2337/ds17-0048.
Daley DJ, Myint PK, Gray RJ, Deane KHOL. Systematic review on factors associated with medication non-adherence in Parkinson’s disease. Parkinsonism Relat Disord. 2012;18:1053–61. https://doi.org/10.1016/j.parkreldis.2012.09.004.
Gerstein HC, Colhoun HM, Dagenais GR, Diaz R, Lakshmanan M, Pais P, et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): a double-blind, randomised placebo-controlled trial. Lancet. 2019;394:121–30. https://doi.org/10.1016/S0140-6736(19)31149-3.
Park JH, Kim DH, Park YG, Kwon DY, Choi M, Jung JH, et al. Association of parkinson disease with risk of cardiovascular disease and all-cause mortality, A nationwide Population-Based Cohort Study. Circulation. 2020;141:1205–7. https://doi.org/10.1161/CIRCULATIONAHA.119.044948.
Li H, Xu X, Wang J, Kong X, Chen M, Jing T, et al. A randomized study to compare the effects of once-weekly dulaglutide injection and once-daily glimepiride on glucose fluctuation of type 2 diabetes mellitus patients: A 26-week follow-up. J Diabetes Res. 2019. https://doi.org/10.1155/2019/6423987.
Ferdinand KC, White WB, Calhoun DA, Lonn EM, Sager PT, Brunelle R, et al. Effects of the once-weekly glucagon-like peptide-1 receptor agonist dulaglutide on ambulatory blood pressure and heart rate in patients with type 2 diabetes mellitus. Hypertension. 2014;64:731–7. https://doi.org/10.1161/HYPERTENSIONAHA.114.03062.
Wang Q, Liu Y, Zhou J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl Neurodegener. 2015;4:19. https://doi.org/10.1186/s40035-015-0042-0.
Salameh TS, Rhea EM, Talbot K, Banks WA. Brain uptake pharmacokinetics of incretin receptor agonists showing promise as Alzheimer’s and Parkinson’s disease therapeutics. Biochem Pharmacol. 2020;180:114187. https://doi.org/10.1016/j.bcp.2020.114187.
Gabery S, Salinas CG, Paulsen SJ, Ahnfelt-Rønne J, Alanentalo T, Baquero AF, et al. Semaglutide lowers body weight in rodents via distributed neural pathways. JCI Insight. 2020;5:e133429. https://doi.org/10.1172/jci.insight.133429.
Secher A, Jelsing J, Baquero AF, Hecksher-Sørensen J, Cowley MA, Dalbøge LS, et al. The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight loss. J Clin Invest. 2014;124:4473–88. https://doi.org/10.1172/JCI75276.
Balland E, Dam J, Langlet F, Caron E, Steculorum S, Messina A, et al. Hypothalamic tanycytes are an ERK-gated conduit for leptin into the brain. Cell Metab. 2014;19:293–301. https://doi.org/10.1016/j.cmet.2013.12.015.
Muscogiuri G, DeFronzo RA, Gastaldelli A, Holst JJ. Glucagon-like peptide-1 and the central/peripheral nervous system: crosstalk in diabetes. Trends Endocrinol Metab. 2017;28:88–103. https://doi.org/10.1016/j.tem.2016.10.001.
Nishizawa M, Nakabayashi H, Uehara K, Nakagawa A, Uchida K, Koya D. Intraportal GLP-1 stimulates insulin secretion predominantly through the hepatoportal-pancreatic vagal reflex pathways. Am J Physiol Endocrinol Metab. 2013;305:E376–87. https://doi.org/10.1152/ajpendo.00565.2012.
Krieger JP, Arnold M, Pettersen KG, Lossel P, Langhans W, Lee SJ. Knockdown of GLP-1 receptors in vagal afferents affects normal food intake and glycemia. Diabetes. 2016;65:34–43. https://doi.org/10.2337/db15-0973.
Plamboeck A, Veedfald S, Deacon CF, Hartmann B, Wettergren A, Svendsen LB, et al. The effect of exogenous GLP-1 on food intake is lost in male truncally vagotomized subjects with pyloroplasty. Am J Physiol Gastrointest Liver Physiol. 2013;304:G1117–27. https://doi.org/10.1152/ajpgi.00035.2013.
Varin EM, Mulvihill EE, Baggio LL, Koehler JA, Cao X, Seeley RJ, et al. Distinct neural sites of GLP-1R expression mediate physiological versus pharmacological control of incretin action. Cell Rep. 2019;27(3371–84):e3. https://doi.org/10.1016/j.celrep.2019.05.055.
Kim DS, Choi H-I, Wang Y, Luo Y, Hoffer BJ, Greig NH. A new treatment strategy for Parkinson’s disease through the gut-brain axis. Cell Transplant. 2017;26:1560–71. https://doi.org/10.1177/0963689717721234.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Daniella Balduino Victorino has received funding from São Paulo Research Foundation (FAPESP #2016/17746-3). Fulvio Alexandre Scorza has received funding from São Paulo Research Foundation (FAPESP #2018/18568-7) and The National Council for Scientific and Technological Development (CNPq #405811/2018-7). Carla Alessandra Scorza has received funding from The National Council for Scientific and Technological Development (CNPq #302583/2017-3).
Conflicts of interest/Competing interests
None of the authors has conflict of interest to declare.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Authors' contributions
The first draft of the manuscript was written by DBV. MN and MGM contributed to the writing. FAS and CAS provided critical review. All authors critically reviewed drafts of the manuscript and approved the final version.
Rights and permissions
About this article

Cite this article
Victorino, D.B., Nejm, M., Guimarães-Marques, M. et al. Repurposing GLP-1 Receptor Agonists for Parkinson’s Disease: Current Evidence and Future Opportunities. Pharm Med 35, 11–19 (2021). https://doi.org/10.1007/s40290-020-00374-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40290-020-00374-5