
Abstract
The current treatment options for neurodegenerative diseases in older adults rely mainly on providing symptomatic relief. Yet, it remains imperative to identify agents that slow or halt disease progression to avoid the most disabling features often associated with advanced disease stages. A potential overlap between the pathological processes involved in diabetes and neurodegeneration has been established, raising the question of whether incretin-based therapies for diabetes may also be useful in treating neurodegenerative diseases in older adults. Here, we review the different agents that belong to this class of drugs (GLP-1 receptor agonists, dual/triple receptor agonists, DPP-4 inhibitors) and describe the data supporting their potential role in treating neurodegenerative conditions including Parkinson’s disease and Alzheimer’s disease. We further discuss whether there are any distinctive properties among them, particularly in the context of safety or tolerability and CNS penetration, that might facilitate their successful repurposing as disease-modifying drugs. Proof-of-efficacy data will obviously be of the greatest importance, and this is most likely to be demonstrable in agents that reach the central nervous system and impact on neuronal GLP-1 receptors. Additionally, however, the long-term safety and tolerability (including gastrointestinal side effects and unwanted weight loss) as well as the route of administration of this class of agents may also ultimately determine success and these aspects should be considered in prioritising which approaches to subject to formal clinical trial evaluations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
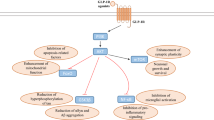
There is a potential overlap between the pathological processes involved in diabetes and neurodegeneration. |
Incretin-based therapies may provide a novel way of treating neurodegenerative diseases in older adults by slowing down or halting disease progression. |
Several candidates for drug repurposing are currently being investigated. |
1 Neurodegeneration and Type 2 Diabetes Mellitus (T2DM)
The classification of neurodegenerative diseases takes into account their varying clinical presentations, which reflect closely the site and distribution of the pathology but also, perhaps more importantly, relies on the pathological appearance of the brain according to aggregation of different proteins and protein isoforms. While the specific aggregating protein is the main factor determining the pathological classification of each disease, independent studies have confirmed that multiple overlapping pathways can all contribute to the pathophysiology of several types of neurodegenerative diseases in older adults [1]. Furthermore, the presence of only one aggregated protein is the exception rather than the rule. Most patients demonstrate multiple pathologies at post-mortem, which may reflect interaction (i.e. one protein misfolding pathway triggering another) or alternatively that similar dysfunctional processes result in misfolding of several different proteins.
While the greatest risk factor for all forms of neurodegeneration is undoubtedly ageing, discoveries in the field of genetics have uncovered key pathways that lead to an increased risk of neurodegeneration even in the apparently sporadic forms of these diseases. As well as protein aggregation, it has become clear that neuroinflammation, lysosomal dysfunction and mitochondrial dysfunction all contribute to the risk of neurodegeneration and are therefore potential targets for therapeutic intervention.
It has also become clear that type 2 diabetes mellitus (T2DM) is at least a modest risk factor for neurodegeneration. This association appears to be strongest among those with the earliest onset of T2DM and the most severe stages of the disease (i.e. has a dose-dependent relationship) [2]. The co-occurrence of T2DM and neurodegeneration also appears to accelerate the rate of clinical progression of the neurodegenerative disease [3, 4]. Further, T2DM has a clear mechanistic overlap with neurodegeneration in that in this condition there is protein aggregation (human islet amyloid polypeptide, known as amylin), mitochondrial dysfunction and inflammation in the beta islet cells of the pancreas [5, 6]. Finally, there has been recent recognition of the role of insulin resistance in the brain as a contributory factor for neurodegeneration [7]. In contrast to peripheral insulin resistance that leads to the lack of glucose uptake and consequent hyperglycaemia, insulin resistance in the brain is now understood to lead to a cascade of processes that include protein aggregation, neuroinflammation, mitochondrial dysfunction and apoptosis [8]. The apparent overlap between the pathological processes of T2DM and neurodegeneration raises the question whether T2DM treatments may have a useful function in neurodegenerative disease [9,10,11].
There is thus growing interest in the role of incretin-based therapies as potential treatment options in older adults with neurodegenerative diseases. In this review, we describe the different therapies that belong to this class, briefly summarise the data supporting their potential role in neurodegeneration and discuss whether there are any distinctive properties among them that might facilitate the successful repurposing of one or more of these agents. While incretin-based approaches are also being investigated for the treatment of several neurological conditions (e.g. traumatic brain injury [12], stroke [13], Huntington’s disease [14] and amyotrophic lateral sclerosis [15]), for the purposes of this review we will primarily focus on Alzheimer’s disease and Parkinsonism.
2 Incretin-Based Therapies in T2DM
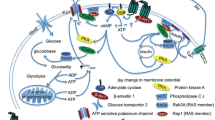
Glucagon-like peptide-1 (GLP-1) is an endogenous hormone released from intestinal L-cells in response to food intake [16]. GLP-1 is responsible for the incretin effect whereby a greater level of insulin is released due to enteral glucose levels than is released in response to an equivalent intravenous glucose load. GLP-1 circulates in the bloodstream and binds to GLP-1 receptors found on pancreatic beta islet cells. Under hyperglycaemic conditions, this stimulates insulin secretion while reducing glucagon secretion. Circulating GLP-1 is rapidly degraded by dipeptidyl peptidase-4 (DPP-4), which results in a short half-life and a brief duration of action. The discovery of agonists for the GLP-1 receptor that resist degradation by DPP-4, and therefore have a longer-lasting effect on blood glucose control, has rapidly led to the accumulation of clinical trial data confirming the usefulness of this class of drugs for treating T2DM. GLP-1 receptor agonists (incretin mimetics) and DPP-4 inhibitors (incretin enhancers) have thus emerged as effective glucose-lowering drugs, reducing glycated haemoglobin (HbA1c) and body weight while keeping the risk of hypoglycaemia low [17]. Their potential mechanisms of action in neurodegenerative processes have been recently reviewed in detail [51].
Beyond glycaemic control, GLP-1 receptor stimulation improves overall cell function by protecting pancreatic beta cells from apoptosis, reducing oxidative stress and regulating autophagy, in addition to eliciting anti-inflammatory signalling [18, 19]. GLP-1 receptor stimulation has also been shown to improve mitochondrial function in pancreatic islet cells [20]. These latter properties have clear potential relevance for neurodegenerative diseases that occur in older adults.
2.1 What is the Relative Potency of Incretin-Based Therapies in T2DM?
2.1.1 GLP-1 Receptor Agonists
There are currently six injectable GLP-1 receptor agonists approved for use in T2DM. Two of these, exenatide twice daily and lixisenatide once daily, are classified as short-acting agents. The remaining agents are long acting and include exenatide once weekly, liraglutide once daily, albiglutide once weekly, dulaglutide once weekly and semaglutide once weekly. There are substantial pharmacodynamic and pharmacokinetic differences between these drugs, and this is reflected in their varying levels of efficacy and tolerability (Table 1).
Exenatide twice daily and lixisenatide are synthetic derivatives of exendin-4, isolated from the salivary secretions of the Gila monster lizard. They significantly reduce HbA1c, in addition to reducing postprandial glucose by slowing the rate of gastric emptying [21,22,23,24,25]. These effects occur in conjunction with a concurrent increase in insulin production and decrease in glucagon secretion [26]. An extended-release formulation of exenatide, however, appears to show superior results in T2DM than the short-acting agents. Exenatide once weekly reaches therapeutic levels after 2 weeks, and after 6 weeks the drug attains a maximum concentration higher than that attained by a single injection of exenatide twice daily. Significantly greater reductions in HbA1c were noted with exenatide once weekly in comparison with the twice-daily formulation, and the percentage of patients achieving HbA1c ≤ 7% was greater with exenatide once weekly [27] (Table 2). Greater improvements in lipid profile, total cholesterol and triglycerides were also noted, as was better fasting glucose reductions and weight loss [28].
Liraglutide improves biphasic insulin secretion during hyperglycaemia and has been shown to reduce HbA1c more than both preparations of exenatide [29, 30]. While overall weight loss was comparable between liraglutide and exenatide twice daily, patients receiving liraglutide lost more weight than those receiving exenatide once weekly. Liraglutide has also been compared with albiglutide once weekly, showing a superior reduction in HbA1c, fasting blood glucose and weight loss [31]. Liraglutide thus appears to have superior effects on glycaemic control compared with other GLP-1 receptor agonists. However, this trend was not seen when liraglutide was compared with dulaglutide in metformin co-treated patients [32]. Dulaglutide was associated with a greater change in HbA1c from baseline, although this was deemed statistically noninferior. Dulaglutide was also superior to exenatide twice daily in reducing HbA1c and body weight, with a greater proportion of patients achieving HbA1c ≤ 7% [33].
The newest GLP-1 receptor agonist is semaglutide; treatment results in significantly larger reductions in HbA1c and weight compared with placebo, oral antidiabetic drugs (sitagliptin, sodium glucose cotransporter-2 inhibitors) and other GLP-1 receptor agonists (exenatide once weekly, liraglutide, dulaglutide). The rather high and constant levels of semaglutide potentially contribute to its efficacy, in addition to the amount of receptor activation that results from full DPP-4 protection and improved linker function. Due to its proven clinical efficacy, an oral formulation of semaglutide has been developed and could provide a suitable alternative for patients who are unable or unwilling to self-administer an injectable agent. It is non-covalently associated with sodium N-[8-(2-hydroxybenzoyl) amino] caprylate to improve bioavailability and diffusion across the intestinal membrane, enabling semaglutide to reach systemic circulation intact [34]. This preparation has shown comparable results to the injectable version in reducing HbA1c and body weight, with better results noted with higher doses. Oral semaglutide also has similar glycaemic efficacy to liraglutide, but results in greater weight loss [35]. The 14-mg dose has also been shown to have positive effects on cardiovascular mortality, whereby patients experienced a 51% relative risk reduction compared with placebo [36]. To date, there are no real-world studies assessing adherence rates between the oral and injectable preparations of semaglutide.
An important property of GLP-1 receptor agonists compared with other anti-diabetic agents is their relative safety regarding hypoglycaemia because of their glucose-level-dependent mechanism of action. For example, the rate of episodes of hypoglycaemia did not exceed 0.8% in patients who received liraglutide monotherapy. Both liraglutide and lixisenatide are also known to evoke fewer hypoglycaemic events compared with exenatide twice daily. Indeed, a direct comparison between lixisenatide and exenatide showed that 2.5% versus 7.9% of patients experienced symptomatic hypoglycaemia, respectively [24]. Similar findings have been reported for head-to-head comparisons between liraglutide and exenatide twice daily [30]. Frequency of hypoglycaemia increases slightly when these agents are used in combination with sulfonylureas.
These data suggest that semaglutide and liraglutide have greater potency than exenatide and lixisenatide at the GLP-1 receptor, but with important consequences of greater weight reduction, which while an advantage in patients with diabetes, might be a cause for concern if used in patients with neurodegeneration.
2.1.2 Dual and Triple Agonists
Glucose-dependent insulinotropic polypeptide (GIP) is a second incretin hormone. It is also released from the cells of the small intestine and stimulates GIP receptors on the beta islet cells to enhance insulin release in a glucose-level-dependent manner. Like GLP-1, GIP is also broken down by DPP-4, and has both GIP receptors in brain tissue and trophic effects on pancreatic tissue.
Dual receptor (GLP-1 and GIP) agonists reflect a further innovative class of glycaemic agents. Combining GLP-1 and GIP agonist infusions has superiority in enhancing insulin secretion compared with either treatment alone [37]. Several novel compounds employing this strategy have subsequently been developed and trialled in T2DM [38]. An acetylated form of a dual-incretin agonist (RG7697-NNC0090-2746), administered subcutaneously once daily, has been shown to significantly decrease HbA1c, body weight and both fasting and postprandial glucose in 56 patients with T2DM [39]. Higher doses were associated with a significant improvement in insulin resistance, as assessed by a reduction in homeostatic model assessment (HOMA) insulin-resistance index values. This effect in combination with a reduction in weight is thought to have caused the reduction in HbA1c, rather than this agent simply stimulating the secretion of insulin. While these are promising results, the performance of this dual agonist does not differ from that of liraglutide, as shown in a study that used liraglutide as an open-label reference [40].
Tirzepatide (LY3298176, Eli Lilly) has been developed as a once-weekly subcutaneous dual agonist injection. Preclinical studies with mice have shown that acute administration improves glucose-dependent insulin secretion and glucose tolerance [41]. Chronic administration decreased body weight and food intake in these animals, and the effects were significantly greater compared with dulaglutide. Similar findings were echoed in studies with healthy participants and diabetic patients, in which tirzepatide significantly reduced HbA1c compared with placebo, as well as fasting glucose and fasting insulin levels [41]. A greater number of patients achieved HbA1c < 6.5% when compared with dulaglutide or placebo [42]. Further, tirzepatide caused greater weight loss than dulaglutide, which may be explained by suppression of calorie intake and a slight but significant increase in energy expenditure [41]. The actions of GIP and GLP-1 receptors possibly occur at the level of the CNS. It is also hypothesised that the beneficial effects of tirzepatide on weight loss could be attributed to its greater potency at the GIP receptors. This is in contrast to other dual agents, such as RG7697-NNC0090-2746, which display balanced activity at the receptors [40]. Tirzepatide thus improved insulin resistance, suggesting a potential insulin-sensitising effect secondary to visceral fat reduction. While greater efficacy on HbA1c resulting from agonist actions at both GLP-1 and GIP receptors is clearly desirable in T2DM, greater weight loss may again be a concern when using these drugs in neurodegenerative diseases.
In a similar fashion, triple-acting agonists that activate receptors at GLP-1, GIP and glucagon have been evaluated in pre-clinical models of T2DM [43]. These compounds have been shown to have weight-reducing and anti-diabetic properties in mice [44], and perform similarly to clinical standard agents such as exendin-4 in glucose tolerance tests. They also have similar potency for cAMP stimulation in receptor-transfected cells [45]. However, there are reports of triagonists that have little or no effect on body weight despite glycaemia improvement in high-fat-fed mice [46]. While these results suggest unbalanced agonism or even submaximal potency, they could be of potential value in conditions where weight loss is not desired.
2.1.3 DPP-4 Inhibitors (Gliptins)
Formulated as oral drugs to be taken daily, DPP-4 inhibitors minimise the rapid cleavage of GLP-1 and GIP to enhance their anti-glycaemic effects in patients with diabetes. They also affect other gastrointestinal substrates including peptide tyrosine tyrosine (PYY) and oxyntomodulin by qualitatively altering their biological receptor activity (as opposed to inactivating them completely) [47]. Both of these peptides have anorectic effects, which are significantly reduced by DPP-4 inhibition and potentially explain why these agents are not associated with weight loss [47].
There are currently five DPP-4 inhibitors available—sitagliptin, saxagliptin, linagliptin, alogliptin (in the USA and Europe), and vildagliptin (only in Europe) (Table 3). Although they differ in terms of absorption and metabolism, as well as potency and duration of action, all approved gliptins have similar and modest anti-glycaemic effects [48]. Corroborating this view, a mixed treatment meta-analysis demonstrated no differences between various DPP-4 inhibitors in mean change from baseline in HbA1c and body weight [49]. There were no differences in the number of patients achieving HbA1c ≤ 7% with these agents except from those using alogliptin plus metformin; these individuals achieved HbA1c ≤ 7% more frequently than patients treated with saxagliptin plus metformin. Risk of hypoglycaemia with DPP-4 inhibitors is also low given their GLP-1 mediated glucose-dependent mechanism. Unlike GLP-1 receptor agonists, DPP-4 inhibitors do not lower postprandial glucose by altering gastric emptying or the rate at which ingested glucose enters the systemic circulation [50]. DPP-4 inhibitors do not reduce appetite or cause weight loss.
3 Prospects for Incretin-Based Therapies in Neurogenerative Diseases
The main purpose of this review is to review the data supporting the potential role of incretin-based therapies in neurodegeneration and to consider the properties of the different incretin approaches and the potential relevance of these differences with respect to the likelihood for success. A detailed review of the neuronal cellular processes that are engaged following GLP-1 receptor stimulation has been previously published [51,52,53].
3.1 Alzheimer’s Disease
Alzheimer’s disease (AD) is the most common neurodegenerative disease and manifests with progressive worsening of cognition. Pathological features of AD include aggregation of amyloid beta alongside neurofibrillary tangles, formed by hyperphosphorylated tau [54].
There is rapid growth in the literature pointing toward insulin deficiency and insulin resistance as mediators of AD-type neurodegeneration. Post-mortem ex-vivo stimulation using Western blotting and quantitative immunohistochemistry in AD cases without diabetes has shown that the hippocampal formation and cerebellar cortex exhibit reduced expression of insulin signalling in the IR-IRS1-PI3K pathway and insulin-like growth factor 1 in the IGF1R-IRS2-PI3K pathway. Basal activation states of insulin signalling were also closely related to cognitive ability [55]. Within this vein, feasibility studies have investigated the efficacy and safety of intranasal insulin infusions for 12 months in patients with mild cognitive impairment or AD dementia [56]. No differences were observed, however, between the placebo and insulin arms in terms of cognitive or functional outcomes and CSF biomarkers. While these findings are negative, interpretation of the study results are confounded by the fact that the delivery device was changed mid-trial. Indeed, other pilot studies using a different device have reported improvements in both cognition and cerebral glucose metabolism [57, 58]. Furthermore, in a mouse model of genetically induced AD, peripheral glucose intolerance was observed. Treatment with pioglitazone (a peroxisome proliferator-activated receptor gamma agonist that increases insulin sensitivity), however, significantly improved cognitive impairment in these mice, perhaps indicating a neurotrophic role of insulin [59].
GLP-1 receptor agonists have also shown neuroprotective effects in several preclinical studies of AD [60]. In the 12-month-old female APP/PS1/tau transgenic mouse, administration of lixisenatide was associated with a marked reduction in both neurofibrillary tangles and amyloid plaques within the hippocampi [61]. Lixisenatide also prevented synaptic damage induced by aggregated beta amyloid peptide accumulation in a rat model of AD, and additionally strengthened spatial memory by affecting the signalling pathways involving AKT and phosphatidylinositol 3-kinase (PI3K). In a similar manner, exenatide has been shown to reduce neuroinflammation by suppressing tumour necrosis factor (TNF)-α levels in rats and preventing the loss of hippocampal neurons, with an associated improvement in memory impairment [62]. These findings have been replicated in studies of liraglutide, including its ability to increase neuronal progenitor cells within the dentate nucleus and enhance long-term potentiation in both the hippocampus and cortex [63]. While there are reports of liraglutide also reducing amyloid plaque load, these findings are inconsistent [64, 65].
Dual and triple receptor agonists have shown promising results in animal models of AD. A GLP-1/GIP dual agonist, DA5-CH, strengthened working and long-term spatial memory in 9-month-old transgenic mice. This behavioural change was accompanied by a reduction in hippocampal amyloid senile plaques and phosphorylated tau proteins [66]. DA-JC4, another dual agonist, similarly decreased phosphorylated tau levels in the rat cerebral cortex and hippocampus and prevented spatial learning difficulties. It also reduced apoptosis, attenuated chronic inflammation and reactivated insulin signalling pathways [67]. Agonists activating GLP-1, GIP and glucagon receptors (triple agonists) have also been investigated in mouse models of AD and show similar beneficial effects on both memory ability and reducing the aberrant mechanisms contributing to Alzheimer’s pathology [68]. These dual and triple agents may also have superior effects compared with GLP-1 receptor agonists. While liraglutide and the dual receptor agonist DA-JC1 were equally efficient in stimulating neurogenesis, DA-JC1 was better at decreasing inflammatory markers such as reactive astrocytes in the hippocampus [69].
Despite the large amount of supporting evidence, human studies of GLP-1 receptor agonists in AD patients are scant and inconclusive. Pilot investigations of exenatide twice daily in AD found no significant effects on clinical or cognitive measures, in addition to imaging or CSF biomarkers [70]. Yet, a reduction of amyloid plaques in plasma neuronal extracellular vesicles was noted. These results are difficult to evaluate, however, as the study terminated early due to insufficient patient recruitment. Similarly, a 26-week, randomised, double-blind trial of liraglutide was shown to improve glucose consumption in the brains of people with AD compared with placebo controls. No effect on the accumulation of neurofibrillary amyloid plaques, or an improvement in cognition, was found. The authors suggest that the size of the cohort and the duration of the study could have precluded definite clinical conclusions. As with other GLP-1 receptor agonists, the most common side effects were gastrointestinal (i.e. nausea) and transient in nature. Weight loss was also seen, but this abated after 2–3 months of treatment. The effects of liraglutide on Alzheimer’s neurodegeneration have been further investigated using a multicentre and randomised, double-blind, placebo-control design over 12 months (ClinicalTrials.gov identifier: NCT01843075), with conference results recently announced indicating that while the trial failed to meet the primary outcome (cerebral glucose metabolic rate), there was nevertheless an advantage in hippocampal volume and executive function [71].
While not directly studied in the AD population, there are also reports to indicate that the hazard of substantive cognitive impairment was reduced by 14% in diabetic patients treated long term with dulaglutide [72]. Similarly, the pooled post-hoc analysis from three large cardiovascular outcome trials (LEADER, SUSTAIN-6, PIONEER-6), which utilised liraglutide or semaglutide in T2DM, has indicated that dementia was significantly reduced by 53% in favour of this GLP-1 receptor agonist compared with placebo [73]. These promising findings have encouraged Novo Nordisk to recently announce that they will enter phase III development in AD with oral semaglutide 14 mg, aiming to recruit 3700 people in the early disease stages for a 2-year period. This will be the largest study of its kind and will hopefully provide more conclusive evidence with regards to the efficacy and safety of repurposing GLP-1 receptor agonists in neurodegeneration.
Gliptins can protect neurons against amyloid beta-induced cytotoxicity and prevent the activation of glycogen synthase kinase and tau hyperphosphorylation by restoring insulin downstream signalling pathways. Animal models of AD have provided evidence to support these claims. Saxagliptin elevated hippocampal GLP-1 levels, increased beta amyloid and tau protein clearance rate and improved the global neuroinflammatory profile [74]. Linagliptin increased brain incretin levels and dampened both amyloid burden and tau phosphorylation. Chronic administration of sitagliptin in triple transgenic AD mice was also associated with increased levels of brain GLP-1 and dose-dependent reductions in inflammatory biomarkers, amyloid precursor protein levels and amyloid beta deposition. The effectiveness of these compounds was related to their ability to rescue insulin cascade.
3.2 Parkinson’s Disease
Parkinson’s disease (PD) is a progressive and chronic disorder of the nervous system. Cardinal motor manifestations comprise resting tremor, increased muscular tone (rigidity) and slowed imprecise movement (bradykinesia), alongside non-motor symptoms such as cognitive decline, constipation and anosmia [75]. The disease is hallmarked by progressive damage to dopaminergic neurons within the substantia nigra as part of a more widespread pathological process affecting multiple brain cell types as well as non-neural tissues, and the concomitant formation of intracellular Lewy bodies (abnormal aggregates of alpha synuclein) thought to be responsible for initiating the processes of cellular toxicity [76]. The main current treatment options for PD, including dopamine replacement therapies and deep brain stimulation, are entirely symptomatic and have little impact on the progression of the underlying disease. Patients will develop dopamine refractory problems or worsening of symptoms over time, causing detriment to their quality of life [77]. There is therefore a clear need for treatments that slow down, stop or reverse the condition [78]. There are growing data to support the view that re-purposing incretin-based therapies may have therapeutic potential in PD [79].
Population-based longitudinal cohort studies have found a lower incidence of PD among people with T2DM using GLP-1 receptor agonists or DPP-4 inhibitors [80]. A recent UK study used propensity scores to take into account the potential bias associated with differences between T2DM patients that may influence the choice of anti-diabetic treatment used, and still found a major reduction in the risk of PD among T2DM patients using GLP-1 receptor agonists [81]. The study did not have sufficient power nor duration of follow up to discriminate between the different GLP-1 receptor agonists or DPP4 inhibitors.
3.2.1 GLP-1 Receptor Agonists and Dual Agonists
To study therapeutics that may ameliorate disease symptoms and progression, traditional animal models utilise toxins methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) or 6-hydroxydopamine (6-OHDA). These are toxic to dopaminergic cells by inhibiting mitochondrial activity and evoking a heightened inflammatory response within the brain. The administration of GLP-1 receptor agonists protects against these toxic insults [82, 83].
Exenatide, liraglutide and lixisenatide prevent motor dysfunction in 6-OHDA models of PD, while liraglutide and lixisenatide induced a marked increase in anti-apoptotic pathways compared with exenatide [84]. However, post-lesioning treatment with exenatide protected and increased striatal tissue dopamine concentrations, in addition to the number of nigral TH neurons [85]. These 6-OHDA models further show that exenatide normalised both abnormal behaviours including apomorphine and amphetamine-induced rotations [82, 85]. In a rotenone-induced PD model, liraglutide in combination with sitagliptin increased striatal dopamine and tyrosine hydroxylase (TH) protein levels. Neuroinflammation and neuronal loss was also reversed [86]. Comparable findings have been found in MPTP mouse models; exenatide increased the number of viable dopaminergic neurons [87], in addition to increasing TH-positive neurons and concentrations of both dopamine and its metabolites [88]. Semaglutide demonstrates similar results in addition to improving motor impairment and reducing alpha synuclein aggregation, a finding that was not observed with other agents [89, 90]. Together, these findings suggest GLP-1 receptor agonists have neuroprotective effects against dopaminergic toxins.
A limitation of these animal models is that the neurotoxins tend to cause a fixed neurological deficit rather than a progressive form of neurodegeneration. Newer animal models that are more representative of the human disease have been developed using stereotactic injections of alpha synuclein preformed fibrils in healthy or alpha synuclein transgenic rodents. In these models, a progressive neurodegenerative process is observed including aggregation of alpha synuclein and a motor phenotype reminiscent of human PD [91]. A pegylated form of exenatide (NLY01) has been shown to have neuroprotective effects in an alpha synuclein transgenic model of alpha-synucleinopathy-induced neurodegeneration. NLY01 protects against the loss of dopamine neurons and behavioural deficits [92]. On the basis of these positive results, a phase II, multicentre clinical study with 240 de novo (untreated) PD patients is currently underway in Northern America (ClinicalTrials.gov identifier: NCT04154072). The treatment arms comprise NLY01 2.5 mg, NLY01 5.0 mg or placebo subcutaneous injections, and the primary outcome measure will assess the change in both motor experiences of daily living and motor symptom severity.
Dual GLP-1/GIP agonists have been reported to show superior effects to single GLP-1 receptor agonists, although it is not clear whether equivalent optimal doses were used in the experiments published to date. In an MPTP mouse model of PD, the novel dual agonist DA3-CH was compared with liraglutide [93]. Motor coordination and grip strength was significantly improved by both agents, but more so by DA3-CH. Levels of TH expressed in substantia nigra neurons and striatal axon fibres were also increased in both treatment groups, yet DA-CH3 was better at reversing MPTP toxicity. Inflammation and microgliosis was reduced largely in DA3-CH-treated animals than in those receiving liraglutide, while glial cell-derived neurotrophic factor (GDNF) levels were higher. Similar findings have been reported with other dual GLP-1/GIP receptor agonists (e.g. DA-JC4, DA-JC5, DA-CH5), demonstrating an enhanced level of protective growth factors and reduction in pro-inflammatory cytokines compared with liraglutide [94, 95].
Oxyntomodulin is a natural dual agonist, activating both GLP-1 and glucagon receptors. Its analogue (d-Ser2-oxyntomodulin) shows protective effects in MPTP mouse models whereby treatment prevented or reversed motor impairment and normalised the MPTP-induced reduction in TH-positive neurons within the substantia nigra and striatum [96]. While promising, further work is needed to fully evaluate these effects compared with other single and dual agonists.
Based on these encouraging preclinical data, two human clinical trials of exenatide in patients with moderate stage PD have been conducted. A small proof-of-concept open-label trial evaluating the safety and efficacy of exenatide 10 μg twice daily in PD showed significant improvement in motor scores and cognitive efficiency at 12 months [97], which was maintained even 1 year after stopping the drug [98]. Weight loss was the most commonly reported adverse event and prevented trial completion in one participant. Other gastrointestinal symptoms included constipation and nausea, neither of which compromised trial participation. These were similar to trials of exenatide in T2DM, and weight loss was fully reversible on cessation of the drug (Table 4).
A limitation of this study, however, was that it was an open-label design with a relatively small sample size, which may therefore have been influenced by placebo effects. Consequently, a double-blind clinical trial in 62 patients with moderate stage PD was conducted thereafter, with patients randomised to receiving exenatide 2 mg once weekly or placebo [99]. At 60 weeks, motor ability in the off-medication state was significantly better in patients using exenatide compared with placebo. Post-hoc analyses also showed that non-motor symptoms such as mood and emotional well-being also improved with exenatide use. These demonstrated effects were later associated with augmented brain insulin signalling as evidenced by tyrosine phosphorylation of IRS-1 and activated downstream Akt pathways [100]. As with the previous study, similar adverse events were seen in both trial arms, including injection-site reactions and gastrointestinal symptoms. Six serious adverse events occurred in the exenatide group and two in the placebo group, although it was concluded that none of these were related to the study interventions. A phase III trial of exenatide once weekly is currently being conducted across multiple centres within the UK over 96 weeks to fully evaluate whether the drug has effects that accumulate with prolonged exposure (ClinicalTrials.gov: NCT04232969), with additional trials in Sweden (ClinicalTrials.gov: NCT04305002) and South Korea (ClinicalTrials.gov: NCT04269642).
In a similar manner, liraglutide is the subject of a clinical trial in 57 patients with PD in California (ClinicalTrials.gov: NCT02953665). The primary outcome will include an assessment of motor function, non-motor symptoms and cognition. It is estimated that this study will be completed in December 2021. Lixisenatide is also under trial in France, where 158 early-stage PD patients (< 3 years since diagnosis) have been randomised to receive lixisenatide injections once daily or placebo for 12 months, followed by a 2-month washout period. As with other studies, the primary outcome will be a comparison of motor function at the end of the treatment period (ClinicalTrials.gov: NCT03439943).
3.2.2 DDP-4 Inhibitors
The administration of several DPP-4 inhibitors (sitagliptin, saxagliptin, vildagliptin) in the rotenone neurotoxic animal model of PD has been associated with marked improvements in both cognitive and motor abilities and resilience to dopaminergic cell loss in the substantia nigra pars compacta and striatal terminals [86, 101, 102]. While saxagliptin also decreased oxidative stress, it did not improve cognitive or motor deficits in 6-OHDA toxin rodents nor did it restore dopaminergic neurons in the substantia nigra [103]. Similarly, rats acutely or chronically pre-treated with supramaximal doses of sitagliptin were not protected against MPTP-induced striatal dopaminergic degeneration. Despite these discrepancies, DPP-4 inhibitors appear to have intrinsic anti-inflammatory and anti-apoptotic abilities, and further enhance neurotrophic factors. As yet, DPP-4 inhibitors are yet to be tested in patients with PD, but alogliptin is the subject of a multi-arm trial of disease-modifying drugs to be opened in Australia (https://theapm.org.au/clinical-trials).
3.2.3 Multiple System Atrophy
Multiple system atrophy (MSA) is a rare adult-onset neurodegenerative disease characterised by a variable combination of parkinsonism, cerebellar impairment and autonomic dysfunction [104]. Its pathological hallmark comprises accumulation of alpha synuclein aggregates in oligodendrocytes, forming glial cytoplasmic inclusions [105].
There are no treatments that have been shown to slow down the rate of clinical deterioration of MSA, with survival prognosis estimated at approximately 6–9 years from the time of diagnosis. As with PD, increasing evidence suggests impaired peripheral insulin/insulin-like growth factor-1 (IGF-1) signalling in MSA, as shown by increased insulin and IGF-1 plasma concentrations in MSA patients and reduced IGF-1 brain levels in transgenic mouse models of MSA [106, 107]. In a recent study, the serine phosphorylation (at serine sites 312 and 616) of insulin receptor substrate-1 (IRS-1), a marker of neuronal insulin resistance, was also increased in neurons and oligodendrocytes within the putamen of MSA patients compared with healthy controls [108]. The same study showed that mouse models of MSA have elevated serine [312] IRS-1 expression levels in the striatum compared with wild-type littermates. Treatment with exenatide decreased the expression of these markers, facilitated the preservation of dopaminergic neurons within the substantia nigra and reduced monomeric alpha synuclein load in the striatum.
To our current knowledge, there is only one human phase II clinical trial that aims to investigate the effects of GLP-1 receptor agonists in MSA (ClinicalTrials.gov: NCT04431713). Fifty patients with a probable or possible diagnosis of MSA (including both cerebellar and Parkinsonian phenotypes) will be randomised to receive exenatide once weekly for 48 weeks or to act as controls in an open-label design. The primary endpoint will be the difference in the total Unified Multiple System Atrophy Rating Scale (UMSARS) score (Part I and II), a scale designed to measure disease progression in MSA.
4 Factors Relevant to Incretin-Based Therapies for Neurodegeneration
4.1 Tolerability and Safety
4.1.1 Delayed Gastric Emptying
Gastrointestinal adverse events are associated with the use of all GLP-1 receptor agonists and are usually mild to moderate in severity. Nausea is the most common side effect reported across all agents, with up to 50% of patients being affected. It is dose dependent and tends to resolve with ongoing treatment. A meta-analysis of 35 studies showed that exenatide twice daily (10 μg) had a higher probability of producing nausea compared with long-acting exenatide and liraglutide. On the other hand, higher doses of dulaglutide were associated with an increased incidence of vomiting compared with exenatide [32]. Albiglutide and lixisenatide also cause nausea, but the rate of occurrence is much smaller compared with treatment with liraglutide or exenatide. Semaglutide carries similar gastrointestinal adverse events; nausea was reported in 20–24% and 11–24% of patients receiving injectable or oral semaglutide, respectively [109, 110]. Vomiting and diarrhoea was noted in a smaller proportion of patients receiving either preparation, and these events occurred more frequently with oral semaglutide than liraglutide [111]. Liraglutide, instead, was highly associated with constipation [111].
Similarly, dual GLP-1/GIP receptor agonists are associated with gastrointestinal adverse events. In the study investigating tirzepatide, there was a high incidence of vomiting, particularly with higher doses that were not titrated gradually. Importantly, these compounds are not related to an increase in gastrointestinal-related side effects compared with single GLP-1 receptor agonists; a similar proportion of patients receiving RG7697-NNC0090-2746 or liraglutide reported at least one event; in fact, adverse events were slightly higher with liraglutide use.
Nausea and vomiting result from dose-dependent delayed gastric emptying that is mediated by action on both central and peripheral receptors. While nausea is reported by users of both short- and long-acting agents, it is attenuated more quickly with long-acting agents because of their relatively reduced effects on gastric emptying. Another possible mechanism is the activation of centres involved in appetite regulation and nausea during peak GLP-1 plasma concentrations. The mechanisms causing diarrhoea induced by GLP-1 receptor use are less clear, although some studies suggest that these drugs may accelerate colonic transit or affect the physiological secretion of bile acids [112]. The consequence of delayed gastric emptying may have profound effects on patients who are reliant on the regular and predictable absorption of medication, as is the case in PD. Delay of levodopa absorption or complete dose failures after oral intake can lead to profound and disabling OFF periods during which patients may struggle to walk, or experience pain, stiffness or severe tremor. Short-acting exenatide and lixisenatide slow gastric emptying markedly compared with longer-acting agents [25, 113], and should therefore be used with caution in patients who already suffer substantially with delayed gastric emptying.
4.1.2 Weight Loss
Patients with T2DM are typically overweight, and thus benefit from weight loss associated with GLP-1 receptor agonist use. However, patients with neurodegenerative diseases may already be under-weight due to several factors including reduced appetite, depression, and excessive calorie expenditure from tremor or dyskinesia. Weight loss would therefore be undesirable in this cohort, and could cause further issues (e.g., increased risk of fractures from falls). Poorer prognostic outcomes comprising low quality of life and higher mortality rates have also been associated with weight loss in PD and AD, while the development of dyskinesia has further been correlated with both lower initial body weight and weight loss in PD [114]. From this perspective, albiglutide and dulaglutide could perhaps be considered as better treatment options as they are known to cause less weight loss in comparison with liraglutide, exenatide or semaglutide (however, see section on CNS penetration).
It should also be noted that weight loss is not simply related to direct gastrointestinal effects. The central effects of the incretins lead to both the loss of appetite and taste, which (as well as affecting weight and nutritional status) can affect the ability of patients to enjoy food. This is a superadded issue in PD, as patients may already have a degree of anosmia (loss of sense of smell) as part of their neurodegenerative condition.
4.1.3 Injection-Site Reactions
Injection-site reactions (e.g. nodules, itching, redness) are common with GIP/GLP-1 receptor agonist use, and this is particularly true for longer-acting agents. Only 5.1% of T2DM patients receiving exenatide twice daily reported skin side effects, while this increased to 16% of those receiving exenatide once weekly and 15% of those receiving albiglutide. This is attributed to the known properties of polymeric microspheres that enable the slow release of agents like exenatide. Although this may be considered a negative, reactions are most often transient (resolving in 4–8 weeks) and patients remain asymptomatic. Optimisation of administration practises, including good hygiene and selection of the best site and direction/angle of administration, are known to alleviate skin reactions in PD patients receiving apomorphine infusions and could therefore be applied here. Still, an oral approach may be highly favoured by patients with neurodegeneration. Compliance may also be higher with treatments that are less invasive and adherence to treatment with weekly injections is significantly better compared with daily injections in patients with T2DM [115]. Chronic compliance with incretin-based therapies is extremely important in the context of neurodegeneration as it is hypothesised that prolonged use may have cumulative effects on disease modification. Interestingly, a once-yearly exenatide implant device is currently being developed (Intarcia) that would remove the issue of nodule formation and greatly facilitate compliance.
4.1.4 Pancreatic Safety
There was some initial concern regarding the association of GLP-1 receptor agonist treatment with pancreatitis or pancreatic cancer. These were based on early observational data that identified an increased risk for both of these adverse effects [116], leading to an FDA warning. Two subsequent studies with exenatide and liraglutide in rodents further corroborated these findings, showing an elevation in pancreatic enzymes [117, 118]. However, several meta-analyses and retrospective cohort studies have ultimately failed to show any significant association between GLP-1 mimetic therapy and pancreatic safety [119, 120]. Despite these data, GLP-1 receptor agonists have a black box warning of pancreatitis, as well as a risk for thyroid C-cell tumours, although this is purely based on toxicity studies in animal models.
Examining clinical trials, the prevalence of these serious adverse reactions is very low. Asymptomatic increases in pancreatic enzymes were observed in five subjects receiving exenatide once weekly, compared with patients receiving insulin glargine [121]. Increased pancreatic enzymes were further noted in one patient receiving the dual GLP-1/GIP receptor agonist tirzepatide, who discontinued the study [41]. Pancreatitis has not been reported in patients using semaglutide (oral and injectable), although cholelithiasis was seen in a small number of patients receiving injectable semaglutide compared with placebo [109].
Similarly, there is no association between DPP-4 inhibitors and pancreatic cancer, but a small risk for acute pancreatitis has been reported in one study [122]. This increased incidence has been observed in patients receiving either sitagliptin or linagliptin. It is also recommended that renal function and liver function should be monitored when using sitagliptin or vildagliptin, respectively. Despite these potential biochemical adverse events, DDP-4 inhibitors are well tolerated.
4.1.5 Hypoglycaemic Events
One of the major benefits of incretin-based therapies is that they rarely cause hypoglycaemic events unless used in combination with sulfonylureas [123]. Crucially, this suggests non-diabetic patients with adult-onset neurodegenerative diseases could potentially use these drugs without the risk of developing low blood sugar [124].
4.2 Penetration of the CNS
Intuitively, incretin-based agents that are hypothesised to have effects on neuronal survival in the brain must be able to reach the CNS to engage with the receptors on the target tissue. However, the concept of the ‘gut–brain axis’ [125] may mean that peripheral effects mediated by GLP-1/GIP receptor stimulation can be associated with beneficial brain effects mediated by changes in pro or anti-inflammatory agents, changes in bile acid composition or changes in short-chain fatty acid signalling [126]. From the literature discussed here, it becomes clear that CNS penetration through blood–brain barrier (BBB) crossing is a key aspect in the potency of neuroprotection provided by different agents. As such, it would seem wise to place greater priority on those that have been shown to access the CNS and influence central GLP-1 receptors.
4.2.1 Incretin Mimetics
Animal models with liraglutide and lixisenatide have demonstrated that both agents are able to cross the BBB [127]. For lixisenatide, there is as much detected in the brain when administered with the lowest dose as with the highest dose [127]. There was also a 1.8-fold increase in cell proliferation and the level of cyclic adenosine monophosphate (cAMP) was enhanced post-injection with lixisenatide [127]. Further, liraglutide directly targets mouse hypothalamic GLP-1 receptors located on arcuate nucleus neurons, and these neurons are likely mediators of liraglutide-induced weight loss [128]. In the rodent brain, liraglutide uptake has also been observed in the paraventricular nucleus of the hypothalamus, medial eminence of the hypothalamus and area postrema (vomiting centre) in the hindbrain. Together, these regions form important autonomic control centres in the brain and contribute to widespread processes including endocrinological activities [129, 130]. While the animal data appears promising, transfer of liraglutide from blood to CSF is minimal in human patients with T2DM [131]. The authors of this paper suggest that weight loss by GLP-1 receptor agonist occurs without the agent entering the CSF. Instead, they may interact with sensory vagal afferents and circumventricular organs of the brain—both of which in rodent models are readily accessible to circulating GLP-1 receptor agonists, express GLP-1 receptors and have neuronal projections to hypothalamic nuclei [131].
Exenatide reaches the mouse brain intact, with almost 90% reaching the parenchyma [85]. The agent penetrates the brain even more efficiently than native GLP-1, without depending on circumventricular uptake. These findings have been replicated in human trials, which showed a neuroprotective effect in PD [99]; indeed, exenatide crossed the BBB and was detectable in CSF at concentrations equivalent to those found in preclinical animal models (approximately 1.5–2.0%) [132]. These findings are also in line with levels found in CSF in a pilot evaluation of exenatide in AD [70]. The entry rate from blood, however, is limited when high doses of exenatide are administered with the peptide showing weak self-inhibition [85]. Exenatide is therefore compatible with a transport system of limited capacity, which has practical implications when considering its therapeutic potential since it might limit the effectiveness of very high doses. In any case, exenatide may also support and preserve the integrity of the BBB as evidenced from stroke studies with mice [133]. It ameliorates BBB breakdown and reduces inflammation from cerebral ischaemia, potentially via reducing the oxygen–glucose deprivation-induced astrocyte-derived vascular endothelial growth factor [133]. This is of considerable importance for AD, where cerebral blood flow reductions and breakdown of the BBB contributes to cognitive decline.
Dual GLP-1/GIP receptor agonists, which have neuroprotective abilities, are also able to penetrate the CNS in a significant manner. DA5-CH, known to reduce tau phosphorylation and intracerebroventricular streptozocin-induced insulin desensitisation in rat models of AD, crosses the BBB at a higher rate compared with acetylated dual agonist DA1-JC and single GLP-1 receptor agonists (exenatide and liraglutide) [134]. Using DA5-CH, one study further demonstrated that transactivator of transcription (TAT) sequence modification enhanced penetration of the BBB significantly compared with dual receptor agonist (DA3-CH), which was a pegylated version [95]. This sequence is well recognised as a ligand to cell membrane receptor binding site, facilitating cell reuptake and BBB penetration.
Semaglutide exhibits limited brain access following peripheral administration in mice [135]. It can directly access the brainstem (area postrema and nucleus tractus solitarius), septal nucleus and hypothalamus. However, it cannot cross the BBB and instead interacts with the brain through circumventricular uptake. Within the arcuate nucleus, semaglutide stimulates anorexigenic cocaine-and-amphetamine-regulated transcript and proopiomelanocortin (CART/POMC) neurons, and further inhibits neuropeptide Y/agouti-related peptide (NPY/AgRP). These mechanisms work to mediate food intake and weight loss, and similarly occur when liraglutide is administered. Similarly, the larger albumin-based molecule sizes of albiglutide and dulaglutide hinders their transport across the BBB. It is difficult for either to diffuse into the brain at the area postrema or hypothalamus where there is a breakdown in the BBB. This could account for the relative lack of difference in weight loss with both albiglutide and dulaglutide compared with liraglutide [136].
Despite these results, there is still uncertainty whether GLP-1 receptor agonists can penetrate the BBB as some authors have argued that brain capillary binding or sequestration may not have been considered. To address this issue, a recent study has compared nine different agents in adult CD-1 mice [137]. They found that non-acylated and non-pegylated GLP-1 receptor agonists (exenatide and lixisenatide) had significant rates of blood-to-brain influx, as did dual GLP-1/GIP receptor agonists (DA3-CH and DA-JC4). However, acylated GLP-1 receptor agonists (liraglutide and semaglutide) did not measurably cross the BBB, even though both have been found to ameliorate many forebrain and midbrain pathologies in mouse models of AD and PD. It is possible that these agents may instead exert their neuroprotective effects by influencing the levels of another substance that can cross the BBB, by binding to brain endothelial cells and triggering release of an abluminal substance, or by influencing afferent nerve transmission [137]. The authors, however, suggest that liraglutide and semaglutide affect brain function by accumulating in brain regions outside of the BBB rather than being transported across it. This obviously has clear implications when evaluating whether an agent is suitable for disease modification, and future studies should aim to comprehensively compare the level of clinical improvement with the amount of CNS penetration.
4.2.2 Incretin Enhancers
Approved DPP-4 inhibitors are unable to penetrate the BBB. Their neuroprotective effects are thought to be peripheral relating to increases in circulating GLP-1 levels rather than directly within the CNS, as demonstrated by studies with linagliptin. While the GLP-1 receptor is expressed in both neurons and glia, in addition to being widely distributed throughout the CNS [138], neuroprotection from linagliptin is thought to occur at the neuronal level. A recent study in rodents, however, showed BBB crossing of an oral once-weekly DPP-4 inhibitor (omarigliptin) compared with trelagliptin [139], which the authors attribute to its low molecular weight and lipophilic properties. Intranasal administration of omarigliptin further showed a significantly higher brain/plasma ratio by 3.3-fold compared with the oral group, which was accompanied by a 2.6-fold increase in brain GLP-1 concentration.
While the site of action of GLP-1 stimulation that results in beneficial effects may not be exclusively on CNS neurons, it seems likely that CNS penetration would be a desirable property in the development of an incretin-based approach. Agents such as semaglutide fail to cross the BBB in rodent models but whether this extends to humans with neurodegeneration needs clarification. If it can be demonstrated that CNS penetration is not necessary to have equivalent neuroprotective effects, then agents restricted to the periphery may be favoured assuming that some of the adverse effects of the GLP-1 receptor agonists such as weight loss, while generally very desirable in people with T2DM, may be less well tolerated in AD or PD.
5 Conclusion
There are mounting data to suggest a role for brain insulin resistance, as well as neuroinflammation, either with or separately from T2DM, that may both contribute to the risk of and progression of neurodegenerative diseases. As such, GLP-1 analogues, dual/triple receptor agonists and DPP-4 inhibitors are emerging as promising therapeutic agents to slow down, stop or reverse progression of the neurodegenerative processes. They may exert their effects through multiple mechanisms that involve insulin-like growth factors, IRS-1 phosphorylation and insulin signalling via AKT pathways, or as anti-inflammatory agents. Maximising the translational potential of this approach is thus crucial. Significant pharmacokinetic differences exist between the different drug classes and compounds. This is reflected by the extent to which they exert glycaemic control and their tolerability, but also in the penetration of CNS that is potentially of considerable importance when evaluating putative disease-modifying effects on the brain. To this end, some agents may be more useful in treating neurodegenerative conditions. However, comparable data in the context of neurodegenerative models are sparse and more studies are needed to fully elucidate which agent, if any, has greater neuroprotective effects. A key question relates to whether CNS penetration is essential, as this route is also the source of potential adverse effects such as nausea and weight loss. As the number of agents entering human clinical trials rises, an optimistic view is that their effect sizes and tolerability may be compared between studies and including agents that do not penetrate the CNS. However, in the existing competitive commercial climate, it is unlikely that any direct head-to-head comparisons of incretin-based agents will be performed, unless driven forward in academic institutions rather than in the commercial sector. Despite the enthusiasm for these approaches based on existing laboratory data, epidemiological evidence and proof-of-concept clinical trials, the definitive evidence of efficacy of any incretin-based approach in the field of neurodegeneration is still awaited. It must be hoped that further positive results from formal efficacy trials add sufficient momentum to clinical research in this field to formally address which of the members of this class of drugs offers the best balance between efficacy and side effects in older adults with neurodegenerative disease.
References
Ahmed RM, Devenney EM, Irish M, Ittner A, Naismith S, Ittner LM, et al. Neuronal network disintegration: common pathways linking neurodegenerative diseases. J Neurol Neurosurg Psychiatry. 2016;87:1234–41.
Ott A, Stolk RP, van Harskamp F, Pols HAP, Hofman A, Breteler MMB. Diabetes mellitus and the risk of dementia: the Rotterdam Study. Neurology. 1999;53:1937–1937.
Sandyk R. The relationship between diabetes mellitus and Parkinson’s disease. Int J Neurosci. 1993;69:125–30.
Xu W, Caracciolo B, Wang H-X, Winblad B, Backman L, Qiu C, et al. Accelerated progression from mild cognitive impairment to dementia in people with diabetes. Diabetes. 2010;59:2928–35.
Mukherjee A, Morales-Scheihing D, Butler PC, Soto C. Type 2 diabetes as a protein misfolding disease. Trends Mol Med. 2015;21:439–49.
Kim J, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res. 2008;102:401–14.
Talbot K, Wang H-Y, Kazi H, Han L-Y, Bakshi KP, Stucky A, et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Investig. 2012;122:1316–38.
Foltynie T, Athauda D. Repurposing anti-diabetic drugs for the treatment of Parkinson’s disease: rationale and clinical experience. 1st ed. Amsterdam: Elsevier; 2020.
Perry TA, Greig NH. The glucagon-like peptides: a new genre in therapeutic targets for intervention in Alzheimer’s disease. J Alzheimer’s Dis. 2002.
Perry T, Haughey NJ, Mattson MP, Egan JM, Greig NH. Protection and reversal of excitotoxic neuronal damage by glucagon-like peptide-1 and exendin-4. J Pharmacol Exp Ther. 2002;302:881–8.
Perry T, Lahiri DK, Chen D, Zhou J, Shaw KTY, Egan JM, et al. A novel neurotrophic property of glucagon-like peptide 1: a promoter of nerve growth factor-mediated differentiation in PC12 cells. J Pharmacol Exp Ther. 2002;300:958–66.
Combs CK. Are GLP-1 receptor agonists useful against traumatic brain injury? J Neurochem. 2015.
Maskery MP, Holscher C, Jones SP, Price ci, Strain WD, Watkins CL, et al. Glucagon-like peptide-1 receptor agonists as neuroprotective agents for ischemic stroke: a systematic scoping review. J Cereb Blood Flow Metab. 2021;41:14.
Martin B, Golden E, Carlson OD, Pistell P, Zhou J, Kim W, et al. Exendin-4 improves glycemic control, ameliorates brain and pancreatic pathologies, and extends survival in a mouse Model of Huntington’s disease. Diabetes. 2009.
Li Y, Chigurupati S, Holloway HW, Mughal M, Tweedie D, Bruestle DA, et al. Exendin-4 ameliorates motor neuron degeneration in cellular and animal models of amyotrophic lateral sclerosis. PLoS One. 2012.
Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–57.
Gentilella R, Pechtner V, Corcos A, Consoli A. Glucagon-like peptide-1 receptor agonists in type 2 diabetes treatment: are they all the same? Diabetes Metab Res Rev. 2019;35:1–21.
Egan JM, Bulotta A, Hui H, Perfetti R. GLP-1 receptor agonists are growth and differentiation factors for pancreatic islet beta cells. Diabetes Metab Res Rev. 2003;19:115–23.
Ekkelund Petersen K, Rakipovski G, Raun K, Lykkesfeldt J. Does glucagon-like peptide-1 ameliorate oxidative stress in diabetes? Evidence based on experimental and clinical studies. Curr Diabetes Rev. 2016;12:331–58.
Fan R, Li X, Gu X, Chan JCN, Xu G. Exendin-4 protects pancreatic beta cells from human islet amyloid polypeptide-induced cell damage: potential involvement of AKT and mitochondria biogenesis. Diabetes Obes Metab. 2010;12:815–24.
Buse JB, Henry RR, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in sulfonylurea-treated patients with type 2 diabetes. Diabetes Care. 2004;27:2628–35.
DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care. 2005;28:1092–100.
Kendall DM, Riddle MC, Rosenstock J, Zhuang D, Kim DD, Fineman MS, et al. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in patients with type 2 diabetes treated with metformin and a sulfonylurea. Diabetes Care. 2005;28:1083–91.
Rosenstock J, Raccah D, Koranyi L, Maffei L, Boka G, Miossec P, et al. Efficacy and safety of lixisenatide once daily versus exenatide twice daily in type 2 diabetes inadequately controlled on metformin: a 24-week, randomized, open-label, active-controlled study (GetGoal-X). Diabetes Care. 2013;36:2945–51.
Linnebjerg H, Park S, Kothare PA, Trautmann ME, Mace K, Fineman M, et al. Effect of exenatide on gastric emptying and relationship to postprandial glycemia in type 2 diabetes. Regul Pept. 2008;151:123–9.
DeFronzo RA, Okerson T, Viswanathan P, Guan X, Holcombe JH, MacConell L. Effects of exenatide versus sitagliptin on postprandial glucose, insulin and glucagon secretion, gastric emptying, and caloric intake: a randomized, cross-over study. Curr Med Res Opin. 2008;24:2943–52.
Drucker DJ, Buse JB, Taylor K, Kendall DM, Trautmann M, Zhuang D, et al. Exenatide once weekly versus twice daily for the treatment of type 2 diabetes: a randomised, open-label, non-inferiority study. Lancet. 2008;372:1240–50.
Blevins T, Pullman J, Malloy J, Yan P, Taylor K, Schulteis C, et al. DURATION-5: Exenatide once weekly resulted in greater improvements in glycemic control compared with exenatide twice daily in patients with type 2 diabetes. J Clin Endocrinol Metab. 2011;96:1301–10.
Buse JB, Rosenstock J, Sesti G, Schmidt WE, Montanya E, Brett JH, et al. Liraglutide once a day versus exenatide twice a day for type 2 diabetes: a 26-week randomised, parallel-group, multinational, open-label trial (LEAD-6). Lancet. 2009;374:39–47.
Buse JB, Nauck M, Forst T, Sheu WHH, Shenouda SK, Heilmann CR, et al. Exenatide once weekly versus liraglutide once daily in patients with type 2 diabetes (DURATION-6): a randomised, open-label study. Lancet. 2013;381:117–24.
Pratley RE, Nauck MA, Barnett AH, Feinglos MN, Ovalle F, Harman-Boehm I, et al. Once-weekly albiglutide versus once-daily liraglutide in patients with type 2 diabetes inadequately controlled on oral drugs (HARMONY 7): a randomised, open-label, multicentre, non-inferiority phase 3 study. Lancet Diabetes Endocrinol. 2014;2:289–97.
Dungan KM, Povedano ST, Forst T, González JGG, Atisso C, Sealls W, et al. Once-weekly dulaglutide versus once-daily liraglutide in metformin-treated patients with type 2 diabetes (AWARD-6): a randomised, open-label, phase 3, non-inferiority trial. Lancet. 2014;384:1349–57.
Wysham C, Blevins T, Arakaki R, Colon G, Garcia P, Atisso C, et al. Efficacy and safety of dulaglutide added onto pioglitazone and metformin versus exenatide in type 2 diabetes in a randomized controlled trial (AWARD-1). Diabetes Care. 2014;37:2159–67.
Aroda VR, Rosenstock J, Terauchi Y, Altuntas Y, Lalic NM, Morales Villegas EC, et al. PIONEER 1: Randomized clinical trial of the efficacy and safety of oral semaglutide monotherapy in comparison with placebo in patients with type 2 diabetes. Diabetes Care. 2019;42:1724–32.
Pratley R, Amod A, Hoff ST, Kadowaki T, Lingvay I, Nauck M, et al. Oral semaglutide versus subcutaneous liraglutide and placebo in type 2 diabetes (PIONEER 4): a randomised, double-blind, phase 3a trial. Lancet. 2019;394:39–50.
Husain M, Birkenfeld AL, Donsmark M, Dungan K, Eliaschewitz FG, Franco DR, et al. Oral semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2019;381:841–51.
Nauck MA, Bartels E, Orskov C, Ebert R, Creutzfeldt W. Additive insulinotropic effects of exogenous synthetic human gastric inhibitory polypeptide and glucagon-like peptide-1-(7–36) amide infused at near-physiological insulinotropic hormone and glucose concentrations. J Clin Endocrinol Metab. 1993;76:912–7.
Bastin M, Andreelli F. Dual GIP–GLP1-receptor agonists in the treatment of type 2 diabetes: a short review on emerging data and therapeutic potential. Diabetes Metab Syndr Obes Targets Ther. 2019;12:1973–85.
Schmitt C, Portron A, Jadidi S, Sarkar N, DiMarchi R. Pharmacodynamics, pharmacokinetics and safety of multiple ascending doses of the novel dual glucose-dependent insulinotropic polypeptide/glucagon-like peptide-1 agonist RG7697 in people with type 2 diabetes mellitus. Diabetes Obes Metab. 2017;19:1436–45.
Frias JP, Bastyr EJ, Vignati L, Tschöp MH, Schmitt C, Owen K, et al. The sustained effects of a dual GIP/GLP-1 receptor agonist, NNC0090-2746, in patients with type 2 diabetes. Cell Metab. 2017;26:343–52.
Coskun T, Sloop KW, Loghin C, Alsina-Fernandez J, Urva S, Bokvist KB, et al. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: from discovery to clinical proof of concept. Mol Metab. 2018;18:3–14.
Frias JP, Nauck MA, Van J, Kutner ME, Cui X, Benson C, et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet. 2018;392:2180–93.
Capozzi ME, DiMarchi RD, Tschöp MH, Finan B, Campbell JE. Targeting the incretin/glucagon system with triagonists to treat diabetes. Endocr Rev. 2018;39:519.
Bhat VK, Kerr BD, Flatt PR, Gault VA. A novel GIP-oxyntomodulin hybrid peptide acting through GIP, glucagon and GLP-1 receptors exhibits weight reducing and anti-diabetic properties. Biochem Pharmacol. 2013.
Gault VA, Bhat VK, Irwin N, Flatt PR. A novel glucagon-like peptide-1 (GLP-1)/glucagon hybrid peptide with triple-acting agonist activity at glucose-dependent insulinotropic polypeptide, GLP-1, and glucagon receptors and therapeutic potential in high fat-fed Mice. J Biol Chem. 2013.
Bhat VK, Kerr BD, Vasu S, Flatt PR, Gault VA. A DPP-IV-resistant triple-acting agonist of GIP, GLP-1 and glucagon receptors with potent glucose-lowering and insulinotropic actions in high-fat-fed mice. Diabetologia. 2013.
Deacon CF. Physiology and pharmacology of DPP-4 in glucose homeostasis and the treatment of type 2 diabetes. Front Endocrinol (Lausanne). 2019;10.
Deacon CF. Dipeptidyl peptidase-4 inhibitors in the treatment of type 2 diabetes: a comparative review. Diabetes Obes Metab. 2011;13:7–18.
Craddy P, Palin H-J, Johnson KI. Comparative effectiveness of dipeptidylpeptidase-4 inhibitors in type 2 diabetes: a systematic review and mixed treatment comparison. Diabetes Ther. 2014;5:1–41.
Vella A, Bock G, Giesler PD, Burton DB, Serra DB, Saylan ML, et al. Effects of dipeptidyl peptidase-4 inhibition on gastrointestinal function, meal appearance, and glucose metabolism in type 2 diabetes. Diabetes. 2007;56:1475–80.
Athauda D, Foltynie T. Insulin resistance and Parkinson’s disease: a new target for disease modification? Prog Neurobiol. 2016;145:98–120.
Markaki I, Winther K, Catrina SB, Svenningsson P. Repurposing GLP1 agonists for neurodegenerative diseases. Int Rev Neurobiol. 2020.
Glotfelty EJ, Olson L, Karlsson TE, Li Y, Greig NH. Glucagon-like peptide-1 (GLP-1)-based receptor agonists as a treatment for Parkinson’s disease. Expert Opin Investig Drugs. 2020;29:595–602.
DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener. 2019;14:32.
Talbot K, Wang H, Kazi H, Han L, Bakshi KP, Stucky A, et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients. J Clin Investig. 2012;122:1316–38.
Craft S, Raman R, Chow TW, Rafii MS, Sun C-K, Rissman RA, et al. Safety, efficacy, and feasibility of intranasal insulin for the treatment of mild cognitive impairment and Alzheimer disease dementia. JAMA Neurol. 2020;77:1099.
Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol. 2012.
Craft S, Claxton A, Baker LD, Hanson AJ, Cholerton B, Trittschuh EH, et al. Effects of regular and long-acting insulin on cognition and Alzheimer’s disease biomarkers: a pilot clinical trial. J Alzheimer’s Dis. 2017.
Masciopinto F, Di Pietro N, Corona C, Bomba M, Pipino C, Curcio M, et al. Effects of long-term treatment with pioglitazone on cognition and glucose metabolism of PS1-KI, 3xTg-AD, and wild-type mice. Cell Death Dis. 2012;3:e448–e448.
Perry T, Lahiri DK, Sambamurti K, Chen D, Mattson MP, Egan JM, et al. Glucagon-like peptide-1 decreases endogenous amyloid-β peptide (Aβ) levels and protects hippocampal neurons from death induced by Aβ and iron. J Neurosci Res. 2003;72:603–12.
Cai H-Y, Yang J-T, Wang Z-J, Zhang J, Yang W, Wu M-N, et al. Lixisenatide reduces amyloid plaques, neurofibrillary tangles and neuroinflammation in an APP/PS1/tau mouse model of Alzheimer’s disease. Biochem Biophys Res Commun. 2018;495:1034–40.
Solmaz V, Çınar BP, Yiğittürk G, Çavuşoğlu T, Taşkıran D, Erbaş O. Exenatide reduces TNF-α expression and improves hippocampal neuron numbers and memory in streptozotocin treated rats. Eur J Pharmacol. 2015;765:482–7.
McClean PL, Hölscher C. Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer’s disease. Neuropharmacology. 2014;76:57–67.
McClean PL, Jalewa J, Hölscher C. Prophylactic liraglutide treatment prevents amyloid plaque deposition, chronic inflammation and memory impairment in APP/PS1 mice. Behav Brain Res. 2015;293:96–106.
Hansen HH, Fabricius K, Barkholt P, Kongsbak-Wismann P, Schlumberger C, Jelsing J, et al. Long-term treatment with liraglutide, a glucagon-like peptide-1 (GLP-1) receptor agonist, has no effect on β-amyloid plaque load in two transgenic APP/PS1 mouse models of Alzheimer’s disease. PLoS ONE. 2016;11:e0158205.
Cao Y, Hölscher C, Hu M-M, Wang T, Zhao F, Bai Y, et al. DA5-CH, a novel GLP-1/GIP dual agonist, effectively ameliorates the cognitive impairments and pathology in the APP/PS1 mouse model of Alzheimer’s disease. Eur J Pharmacol. 2018;827:215–26.
Shi L, Zhang Z, Li L, Hölscher C. A novel dual GLP-1/GIP receptor agonist alleviates cognitive decline by re-sensitizing insulin signaling in the Alzheimer ICV. STZ rat model. Behav Brain Res. 2017;327:65–74.
Tai J, Liu W, Li Y, Li L, Hölscher C. Neuroprotective effects of a triple GLP-1/GIP/glucagon receptor agonist in the APP/PS1 transgenic mouse model of Alzheimer’s disease. Brain Res. 2018;1678:64–74.
Salles GN, Calió ML, Hölscher C, Pacheco-Soares C, Porcionatto M, Lobo AO. Neuroprotective and restorative properties of the GLP-1/GIP dual agonist DA-JC1 compared with a GLP-1 single agonist in Alzheimer’s disease. Neuropharmacology. 2020;162:107813.
Mullins RJ, Mustapic M, Chia CW, Carlson O, Gulyani S, Tran J, et al. A pilot study of exenatide actions in Alzheimer’s disease. Curr Alzheimer Res. 2019;16:741–52.
Cairns E. CTAD 2020—Elad fails, but GLP-1s could still have a future in Alzheimer’s [Internet]. Evaluate. 2020. https://www.evaluate.com/vantage/articles/news/snippets/ctad-2020-elad-fails-glp-1s-could-still-have-future-alzheimers.
Cukierman-Yaffe T, Gerstein HC, Colhoun HM, Diaz R, García-Pérez L-E, Lakshmanan M, et al. Effect of dulaglutide on cognitive impairment in type 2 diabetes: an exploratory analysis of the REWIND trial. Lancet Neurol. 2020;19:582–90.
Ballard C, Nørgaard CH, Friedrich S, Mørch LS, Gerds T, Møller DV, et al. Liraglutide and semaglutide: pooled post hoc analysis to evaluate risk of dementia in patients with type 2 diabetes. Alzheimer’s Dement. 2020;16:1–2.
Kosaraju J, Gali CC, Khatwal RB, Dubala A, Chinni S, Holsinger RMD, et al. Saxagliptin: a dipeptidyl peptidase-4 inhibitor ameliorates streptozotocin induced Alzheimer’s disease. Neuropharmacology. 2013;72:291–300.
Chaudhuri KR, Naidu Y. Early Parkinson’s disease and non-motor issues. J Neurol. 2008;255:33–8.
Gibb WRG, Lees AJ. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1988;51:745–52.
Vijiaratnam N, Foltynie T. Therapeutic strategies to treat or prevent off episodes in adults with Parkinson’s disease. Drugs. 2020;80:775–96.
Athauda D, Foltynie T. The ongoing pursuit of neuroprotective therapies in Parkinson disease. Nat Rev Neurol. 2015;11:25–40.
Athauda D, Foltynie T. Drug repurposing in Parkinson’s disease. CNS Drugs. 2018;32:747–61.
Svenningsson P, Wirdefeldt K, Yin L, Fang F, Markaki I, Efendic S, et al. Reduced incidence of Parkinson’s disease after dipeptidyl peptidase-4 inhibitors—A nationwide case-control study. Mov Disord. 2016;31:1422–3.
Brauer R, Wei L, Ma T, Athauda D, Girges C, Vijiaratnam N, et al. Diabetes medications and risk of Parkinson’s disease: a cohort study of patients with diabetes. Brain. 2020;143:3067–76.
Harkavyi A, Abuirmeileh A, Lever R, Kingsbury AE, Biggs CS, Whitton PS. Glucagon-like peptide 1 receptor stimulation by exendin-4 reverses key deficits in distinct rodent models of Parkinson’s disease. J Neuroinflamm. 2008;5:19.
Bertilsson G, Patrone C, Zachrisson O, Andersson A, Dannaeus K, Heidrich J, et al. Peptide hormone exendin-4 stimulates subventricular zone neurogenesis in the adult rodent brain and induces recovery in an animal model of Parkinson’s disease. J Neurosci Res. 2008;86:326–38.
Liu W, Jalewa J, Sharma M, Li G, Li L, Hölscher C. Neuroprotective effects of lixisenatide and liraglutide in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Neuroscience. 2015;303:42–50.
Kastin AJ, Akerstrom V. Entry of exendin-4 into brain is rapid but may be limited at high doses. Int J Obes. 2003;27:313–8.
Badawi GA, Abd El Fattah MA, Zaki HF, El Sayed MI. Sitagliptin and liraglutide reversed nigrostriatal degeneration of rodent brain in rotenone-induced Parkinson’s disease. Inflammopharmacology. 2017;25:369–822.
Kim S, Moon M, Park S. Exendin-4 protects dopaminergic neurons by inhibition of microglial activation and matrix metalloproteinase-3 expression in an animal model of Parkinson’s disease. J Endocrinol. 2009;202:431–9.
Li Y, Perry T, Kindy MS, Harvey BK, Tweedie D, Holloway HW, et al. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc Natl Acad Sci. 2009;106:1285–90.
Zhang L, Zhang L, Li L, Hölscher C. Neuroprotective effects of the novel GLP-1 long acting analogue semaglutide in the MPTP Parkinson’s disease mouse model. Neuropeptides. 2018;71:70–80.
Zhang L, Zhang L, Li L, Hölscher C. Semaglutide is neuroprotective and reduces α-synuclein levels in the chronic MPTP mouse model of Parkinson’s disease. J Parkinsons Dis. 2019;9:157–71.
Chung HK, Ho H-A, Pérez-Acuña D, Lee S-J. Modeling α-synuclein propagation with preformed fibril injections. J Mov Disord. 2019;12:139–51.
Yun SP, Kam T-I, Panicker N, Kim S, Oh Y, Park J-S, et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med. 2018;24:931–8.
Yuan Z, Li D, Feng P, Xue G, Ji C, Li G, et al. A novel GLP-1/GIP dual agonist is more effective than liraglutide in reducing inflammation and enhancing GDNF release in the MPTP mouse model of Parkinson’s disease. Eur J Pharmacol. 2017;812:82–90.
Feng P, Zhang X, Li D, Ji C, Yuan Z, Wang R, et al. Two novel dual GLP-1/GIP receptor agonists are neuroprotective in the MPTP mouse model of Parkinson’s disease. Neuropharmacology. 2018;133:385–94.
Zhang L, Zhang L, Li Y, Li L, Melchiorsen JU, Rosenkilde M, et al. The novel dual GLP-1/GIP receptor agonist DA-CH5 is superior to single GLP-1 receptor agonists in the MPTP model of Parkinson’s disease. J Parkinsons Dis. 2020;10:523–42.
Liu W, Li Y, Jalewa J, Saunders-Wood T, Li L, Hölscher C. Neuroprotective effects of an oxyntomodulin analogue in the MPTP mouse model of Parkinson’s disease. Eur J Pharmacol. 2015;765:284–90.
Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Ell P, Soderlund T, et al. Exenatide and the treatment of patients with Parkinson’s disease. J Clin Investig. 2013;123:2730–6.
Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Kahan J, Ell P, et al. Motor and cognitive advantages persist 12 months after exenatide exposure in Parkinson’s disease. J Parkinsons Dis. 2015;4:337–44.
Athauda D, Maclagan K, Skene SS, Bajwa-Joseph M, Letchford D, Chowdhury K, et al. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390:1664–75.
Athauda D, Gulyani S, Karnati HK, Li Y, Tweedie D, Mustapic M, et al. Utility of neuronal-derived exosomes to examine molecular mechanisms that affect motor function in patients with Parkinson disease: a secondary analysis of the exenatide-PD trial. JAMA Neurol. 2019;76:420–9.
Abdelsalam RM, Safar MM. Neuroprotective effects of vildagliptin in rat rotenone Parkinson’s disease model: role of RAGE-NFκB and Nrf2-antioxidant signaling pathways. J Neurochem. 2015;133:700–7.
Nassar NN, Al-Shorbagy MY, Arab HH, Abdallah DM. Saxagliptin: a novel anti-Parkinsonian approach. Neuropharmacology. 2015;89:308–17.
de Turnes JM, Bassani TB, Souza LC, Vital MABF. Ineffectiveness of saxagliptin as a neuroprotective drug in 6-OHDA-lesioned rats. J Pharm Pharmacol. 2018;70:1059–68.
Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71:670–6.
Papp MI, Kahn JE, Lantos PL. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome). J Neurol Sci. 1989;94:79–100.
Pellecchia MT, Pivonello R, Longo K, Manfredi M, Tessitore A, Amboni M, et al. Multiple system atrophy is associated with changes in peripheral insulin-like growth factor system. Mov Disord. 2010;25:2621–6.
Ubhi K, Rockenstein E, Mante M, Inglis C, Adame A, Patrick C, et al. Neurodegeneration in a transgenic mouse model of multiple system atrophy is associated with altered expression of oligodendroglial-derived neurotrophic factors. J Neurosci. 2010;30:6236–46.
Bassil F, Canron MH, Vital A, Bezard E, Li Y, Greig NH, et al. Insulin resistance and exendin-4 treatment for multiple system atrophy. Brain. 2017;140:1420–36.
Sorli C, Harashima S, Tsoukas GM, Unger J, Karsbøl JD, Hansen T, et al. Efficacy and safety of once-weekly semaglutide monotherapy versus placebo in patients with type 2 diabetes (SUSTAIN 1): a double-blind, randomised, placebo-controlled, parallel-group, multinational, multicentre phase 3a trial. Lancet Diabetes Endocrinol. 2017;5:251–60.
Zinman B, Aroda VR, Buse JB, Cariou B, Harris SB, Hoff ST, et al. Efficacy, safety, and tolerability of oral semaglutide versus placebo added to insulin with or without metformin in patients with type 2 diabetes: the PIONEER 8 trial. Diabetes Care. 2019;42:2262–71.
Yamada Y, Katagiri H, Hamamoto Y, Deenadayalan S, Navarria A, Nishijima K, et al. Dose-response, efficacy, and safety of oral semaglutide monotherapy in Japanese patients with type 2 diabetes (PIONEER 9): a 52-week, phase 2/3a, randomised, controlled trial. Lancet Diabetes Endocrinol. 2020;8:377–91.
Sun F, Yu K, Yang Z, Wu S, Zhang Y, Shi L, et al. Impact of GLP-1 receptor agonists on major gastrointestinal disorders for type 2 diabetes mellitus: a mixed treatment comparison meta-analysis. Exp Diabetes Res. 2012;2012:1–14.
Nauck MA, Kemmeries G, Holst JJ, Meier JJ. Rapid tachyphylaxis of the glucagon-like peptide 1-induced deceleration of gastric emptying in humans. Diabetes. 2011;60:1561–5.
Sharma JC, Vassallo M. Prognostic significance of weight changes in Parkinson’s disease: the Park–weight phenotype. Neurodegener Dis Manag. 2014;4:309–16.
Kostev K, Ouwens M, Grandy S, Johnsson KM, Qiao Q. Adherence to GLP-1 receptor agonist therapy administered by once-daily or once-weekly injection in patients with type 2 diabetes in Germany. Diabetes Metab Syndr Obes Targets Ther. 2016;9:201–5.
Elashoff M, Matveyenko AV, Gier B, Elashoff R, Butler PC. Pancreatitis, pancreatic, and thyroid cancer with glucagon-like peptide-1-based therapies. Gastroenterology. 2011;141:150–6.
Vrang N, Jelsing J, Simonsen L, Jensen AE, Thorup I, Søeborg H, et al. The effects of 13 weeks of liraglutide treatment on endocrine and exocrine pancreas in male and female ZDF rats: a quantitative and qualitative analysis revealing no evidence of drug-induced pancreatitis. Am J Physiol Metab. 2012;303:E253–64.
Tatarkiewicz K, Belanger P, Gu G, Parkes D, Roy D. No evidence of drug-induced pancreatitis in rats treated with exenatide for 13 weeks. Diabetes Obes Metab. 2013;15:417–26.
Romley JA, Goldman DP, Solomon M, McFadden D, Peters AL. Exenatide therapy and the risk of pancreatitis and pancreatic cancer in a privately insured population. Diabetes Technol Ther. 2012;14:904–11.
Pinto LC, Falcetta MR, Rados DV, Leitão CB, Gross JL. Glucagon-like peptide-1 receptor agonists and pancreatic cancer: a meta-analysis with trial sequential analysis. Sci Rep. 2019;9:2375.
Gentilella R, Pechtner V, Corcos A, Consoli A. Glucagon-like peptide-1 receptor agonists in type 2 diabetes treatment: are they all the same? Diabetes Metab Res Rev. 2019;35:e3070.
Pinto LC, Rados DV, Barkan SS, Leitão CB, Gross JL. Dipeptidyl peptidase-4 inhibitors, pancreatic cancer and acute pancreatitis: a meta-analysis with trial sequential analysis. Sci Rep. 2018;8:782.
Nauck MA, Vilsboll T, Gallwitz B, Garber A, Madsbad S. Incretin-based therapies: viewpoints on the way to consensus. Diabetes Care. 2009;32:S223–31.
Onoviran OF, Li D, Toombs Smith S, Raji MA. Effects of glucagon-like peptide 1 receptor agonists on comorbidities in older patients with diabetes mellitus. Ther Adv Chronic Dis. 2019;10:204062231986269.
Breen DP, Halliday GM, Lang AE. Gut–brain axis and the spread of α-synuclein pathology: vagal highway or dead end? Mov Disord. 2019;34:307–16.
Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell. 2016;167:1469–80.
Hunter K, Hölscher C. Drugs developed to treat diabetes, liraglutide and lixisenatide, cross the blood brain barrier and enhance neurogenesis. BMC Neurosci. 2012;13:33.
Secher A, Jelsing J, Baquero AF, Hecksher-Sørensen J, Cowley MA, Dalbøge LS, et al. The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight loss. J Clin Investig. 2014;124:4473–88.
Qin C, Li J, Tang K. The paraventricular nucleus of the hypothalamus: development, function, and human diseases. Endocrinology. 2018;159:3458–72.
Yin W, Gore AC. The hypothalamic median eminence and its role in reproductive aging. Ann N Y Acad Sci. 2010;1204:113–22.
Christensen M, Sparre-Ulrich AH, Hartmann B, Grevstad U, Rosenkilde MM, Holst JJ, et al. Transfer of liraglutide from blood to cerebrospinal fluid is minimal in patients with type 2 diabetes. Int J Obes. 2015;39:1651–4.
Chen S, Yu SJ, Li Y, Lecca D, Glotfelty E, Kim HK, et al. Post-treatment with PT302, a long-acting exendin-4 sustained release formulation, reduces dopaminergic neurodegeneration in a 6-hydroxydopamine rat model of Parkinson’s disease. Sci Rep. 2018.
Shan Y, Tan S, Lin Y, Liao S, Zhang B, Chen X, et al. The glucagon-like peptide-1 receptor agonist reduces inflammation and blood-brain barrier breakdown in an astrocyte-dependent manner in experimental stroke. J Neuroinflamm. 2019;16:242.
Li C, Liu W, Li X, Zhang Z, Qi H, Liu S, et al. The novel GLP-1/GIP analogue DA5-CH reduces tau phosphorylation and normalizes theta rhythm in the icv. STZ rat model of AD. Brain Behav. 2020;10:e01505.
Gabery S, Salinas CG, Paulsen SJ, Ahnfelt-Rønne J, Alanentalo T, Baquero AF, et al. Semaglutide lowers body weight in rodents via distributed neural pathways. JCI Insight. 2020;5.
Nauck MA, Stewart MW, Perkins C, Jones-Leone A, Yang F, Perry C, et al. Efficacy and safety of once-weekly GLP-1 receptor agonist albiglutide (HARMONY 2): 52 week primary endpoint results from a randomised, placebo-controlled trial in patients with type 2 diabetes mellitus inadequately controlled with diet and exercise. Diabetologia. 2016;59:266–74.
Salameh TS, Rhea EM, Talbot K, Banks WA. Brain uptake pharmacokinetics of incretin receptor agonists showing promise as Alzheimer’s and Parkinson’s disease therapeutics. Biochem Pharmacol. 2020;180:114187.
Chowen JA, de Fonseca FR, Alvarez E, Navarro M, Garcı́a-egura LM, Blázquez E. Increased glucagon-like peptide-1 receptor expression in glia after mechanical lesion of the rat brain. Neuropeptides. 1999;33:212–5.
Ayoub BM, Mowaka S, Safar MM, Ashoush N, Arafa MG, Michel HE, et al. Repositioning of Omarigliptin as a once-weekly intranasal anti-Parkinsonian Agent. Sci Rep. 2018;8:8959.
Acknowledgements
None to declare.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was received specifically for the publication of this review.
Conflict of interest
CG reports no conflicts of interest. NV has received unconditional educational grants from IPSEN and Biogen, travel grants from IPSEN, AbbVie and The International Parkinson’s Disease and Movement Disorders Society, speaker’s honorarium from AbbVie and STADA and served on advisory boards for Abbvie and Brittania outside of the submitted work. DA has received travel grants from Bial. GA reports no conflicts of interest. SG reports no conflicts of interest. TF has received grants from National Institute of Health Research, Michael J Fox Foundation, John Black Charitable Foundation, Cure Parkinson’s Trust, Innovate UK, Van Andel Research Institute and Defeat MSA. He has served on Advisory Boards for Voyager Therapeutics, Handl therapeutics, Living Cell Technologies, Bial, and Profie Pharma. He has received honoraria for talks sponsored by Bial, Profile Pharma, and Boston Scientific.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Authors’ Contributions
CG: Review of literature, preparation of manuscript, critical revision for intellectual content; NV: Interpretation of data, critical revision of manuscript for intellectual content; DA: Interpretation of data, critical revision of manuscript for intellectual content; GA: Critical revision of manuscript for intellectual content; SG: Critical revision of manuscript for intellectual content; TF: Article concept and design, interpretation of data, critical revision of manuscript for intellectual content.
Rights and permissions
About this article

Cite this article
Girges, C., Vijiaratnam, N., Athauda, D. et al. The Future of Incretin-Based Approaches for Neurodegenerative Diseases in Older Adults: Which to Choose? A Review of their Potential Efficacy and Suitability. Drugs Aging 38, 355–373 (2021). https://doi.org/10.1007/s40266-021-00853-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40266-021-00853-7