
Abstract
Atypical hemolytic uremic syndrome is a thrombotic microangiopathy characterized by hemolysis, thrombocytopenia, and acute kidney injury, usually caused by alternative complement system overactivation due to pathogenic genetic variants or antibodies to components or regulatory factors in this pathway. Previously, a lack of effective treatment for this condition was associated with mortality, end-stage kidney disease, and the risk of disease recurrence after kidney transplantation. Plasma therapy has been used for atypical hemolytic uremic syndrome treatment with inconsistent results. Complement-blocking treatment changed the outcome and prognosis of patients with atypical hemolytic uremic syndrome. Early administration of eculizumab, a monoclonal C5 antibody, leads to improvements in hematologic, kidney, and systemic manifestations in patients with atypical hemolytic uremic syndrome, even with apparent dialysis dependency. Pre- and post-transplant use of eculizumab is effective in the prevention of atypical hemolytic uremic syndrome recurrence. Evidence on eculizumab use in secondary hemolytic uremic syndrome cases is controversial. Recent data favor the restrictive use of eculizumab in carefully selected atypical hemolytic uremic syndrome cases, but close monitoring for relapse after drug discontinuation is emphasized. Prophylaxis for meningococcal infection is important. The long-acting C5 monoclonal antibody ravulizumab is now approved for atypical hemolytic uremic syndrome treatment, enabling a reduction in the dosing frequency and improving the quality of life in patients with atypical hemolytic uremic syndrome. New strategies for additional and novel complement blockage medications in atypical hemolytic uremic syndrome are under investigation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Atypical HUS (aHUS) is a serious disease, which, if left untreated, may lead to multiple organ damage and end stage renal disease. |
Alternative complement pathway dysregulation plays a key role in the pathophysiology of the disease. Complement system inhibition is an effective treatment strategy, leading to normalization of clinical and laboratory parameters and improvement of renal outcome. |
Eculizumab is the first complement inhibitor available for effective aHUS treatment. Ravulizumab, a long-acting complement inhibitor, re-engineered from eculizumab, enables reduction in dosing frequency and improving the quality of life in patients with aHUS. |
New strategies for complement blockage in aHUS treatment are under investigation. |
1 Introduction
Hemolytic uremic syndrome (HUS) is a thrombotic microangiopathy (TMA) characterized by intravascular hemolysis, thrombocytopenia, and acute kidney injury. Independent of the primary etiologic factor, injury involves endothelial damage, leading to platelet activation [1], microthrombi formation, microangiopathic hemolytic anemia, complement cascade activation [2], and multiple end-organ damage, including kidney failure and extra-renal manifestations with neurological [3], cardiac [4], gastrointestinal [5], and respiratory [6] involvement. According to the current understanding of its pathophysiology, HUS is classified into infectious associated, atypical, and secondary [7]. This review concentrates on the diagnosis and management of atypical HUS (aHUS) in pediatric patients.
Most of the infectious-associated HUS cases are caused by Shiga toxin producing Escherichia coli (STEC) and appear after a period of colitis and bloody diarrhea. The Shiga toxin binds to cell membrane glycolipid Gb3, which is strongly expressed in kidney endothelia, especially in children [8,9,10]. Streptococcus pneumoniae and some other infections are less common causes of HUS [11, 12]. Recently, coronavirus disease 2019 was implicated as a cause [13] or a trigger for aHUS [14, 15].
In secondary HUS, a coexisting disease or health state such as autoimmunity, transplantation, cancer, and pregnancy or the use of certain cytotoxic drugs are associated with a similar disease manifestation [7]. Atypical HUS is usually caused by dysregulation of the alternative complement pathway, resulting in overactivation and excessive production of the terminal attack complex C5b-C9, leading to endothelial cell injury [16]. Dysregulation of the alternative complement system may be due to loss-of-function mutations in genes of regulatory proteins [complement factor H (CFH), membrane cofactor protein (or CD46), complement factor I and thrombomodulin], gain-of-function mutations in the genes of the C3 convertase components (complement factor B and C3), or anti-CFH inhibitory autoantibodies [17]. In addition, mutations in diacylglycerol kinase ɛ and cobalamin C, proteins not related to the complement system, have been described as a cause of aHUS [18, 19]. The absence of a pathogenic mutation does not exclude the diagnosis of aHUS.
The relative frequencies of secondary and primary TMA in adults were evaluated in a retrospective cohort of 564 patients with TMA diagnosed during 2009–16. Atypical HUS was found in only 18/564 patients (3%), whereas secondary TMA (pregnancy, malignancy, infections, drugs) was the most frequent diagnosis [20]. In contrast, Alfandary et al. reported on HUS etiologies in Israeli children (n = 75) [21] who were classified according to contemporary guidelines [7] into four major groups: infection associated, aHUS, secondary HUS, and unknown. Atypical HUS was relatively common (24%) and STEC HUS was a rare diagnosis (only 6.6%), most probably owing to the high prevalence of genetic diseases in this country, secondary to high rates of consanguineous marriages in selected populations. These reports emphasize the need to define the country- and age-specific prevalence of HUS subclasses.
2 Differential Diagnosis
In most Western countries, STEC HUS, diagnostically confirmed by STEC identification in a stool culture or by polymerase chain reaction, is ten times more common than aHUS [22]. Even in STEC-negative stool samples, serological studies did show evidence of recent STEC infection in many children with HUS [23].
The diagnosis of aHUS begins with the same biochemical and hematologic criteria as for typical HUS or any other TMA. Atypical HUS in children is usually suspected in patients aged younger than 6 months, or with a non-synchronous HUS family history, or in relapsing cases and in children without a history of bloody diarrhea. However, the presence of gastrointestinal symptoms may also reflect a microangiopathic ischemia of the intestinal tract and does not exclude the possibility of aHUS, as described for example in children and adolescents with anti-FH antibodies [24]. Therefore, the previous classification of diarrhea-positive and diarrhea-negative HUS is not relevant anymore. A short period of diarrhea or the concomitant appearance of diarrhea and HUS should raise the suspicion for aHUS versus STEC HUS, as the latter usually appears at the end of the gastrointestinal illness (4–5 days from diarrhea onset) [25]. Other aHUS features include a slower and insidious onset, sometimes preceded by vague and nonspecific signs and symptoms [26], a lower rate of diarrhea, a low prevalence of leukocytosis (under 16,000/μL), and a higher rate of consanguinity [21]. Shiga toxin producing E. coli infection can even trigger an aHUS episode in patients with complement pathogenic genetic variants [27]. Anti-CFH antibody-associated HUS was originally described by Dragon-Durey et al. [28]. Age of onset in this aHUS variant is later than aHUS because of pathogenic genetic variants, as well as an increase in gastrointestinal complaints prior to disease onset. Genetic variants in the CFH-related peptide and the pathogenesis of this association have been described [29].
An important differential diagnosis in patients presenting with aHUS is thrombotic thrombocytopenic purpura. This is a TMA that occurs secondary to congenital deficiency of the metalloprotease ADAMTS13 or acquired anti-ADAMTS13 antibodies [30]. ADAMTS13 is a critical factor that prevents the accumulation of von Willebrand factor multimers, secondary to platelet activation [31]. Therefore, determination of ADAMTS13 activity is mandatory before the diagnosis of aHUS. For more details on the diagnostic approach to TMA, readers are referred to recent publications (7).
2.1 Natural aHUS History
Atypical HUS is associated with increased mortality, depending on the underlying pathogenic genetic variant [32]. The risk of HUS recurrence is higher in patients with pathogenic genetic variants in CFH (31–55%), membrane cofactor protein (18–52%), and C3 (50%), and is 30% in patients without a known pathogenic genetic variant [27]. Previously, when an effective treatment was absent, a third of pediatric patients and half of adult patients with aHUS who survived the acute phase remained dialysis dependent. The risk of recurrent disease after kidney transplantation was as high as 80% [33, 34].
3 Management of aHUS
3.1 Supportive Treatment
Supportive treatment is based on general principles of AKI management such as correction/avoiding volume overload and electrolyte abnormalities, hypertension control, stopping nephrotoxic drugs, initiation of dialysis therapy if indicated, and provision of adequate nutrition. In severe anemia (Hb <7 g/dL), blood transfusions are indicated. Platelets transfusions should be avoided unless there is evidence of active bleeding or the need for surgical intervention.
3.2 Role of Plasma Therapy
Since the original description of HUS, numerous treatment options have been tested, including different types of anticoagulants and plasma therapy. Plasma therapy originally showed inconsistent results, mostly because of the mix of HUS cases of currently known different etiologies. While plasma therapy showed no efficacy in STEC HUS, its role in aHUS cases was inconsistent. As complement dysregulation was increasingly recognized as a key mechanism in aHUS, owing to the deficiency of a plasmatic factor (mainly CFH and CFI) or the presence of a pathogenic antibody (such as anti-FH antibodies), the use of plasma therapy with or without exchange became an alternative again [35,36,37]. Plasma exchange/plasma infusion has uncertain benefits and a high rate of technique-related complications in children. A multicenter study summarized the contemporary experience with plasma therapy in 71 children with aHUS (in the months prior to more widespread use of eculizumab) [38]. In 59 of them (83%), plasma was administered within the first 33 days of disease. Plasma exchange was the dominant technique. Complications of central venous catheters occurred in 31% of patients with a catheter in-situ. A hematological remission was obtained in 89% within a median period of 11.5 days. Twelve patients (17%) remained dialysis dependent at day 33. Thus, although some benefit could be shown by plasma therapy and or exchange, it was not consistent and associated with complications.
3.3 Anti-Complement Treatment
3.3.1 Eculizumab
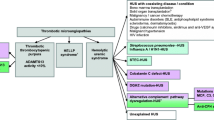
Eculizumab (Soliris®; Alexion Pharmaceutical Inc., Washington, DC, USA) is a humanized chimeric monoclonal antibody. Binding to complement C5, it prevents its cleavage to C5a and C5b, blocking terminal membrane attack complex formation (Fig. 1). The pharmacologic characteristics of eculizumab are similar to other monoclonal antibodies. Eculizumab is administered as an intravenous infusion and is primarily distributed in blood plasma [39]. Because of its molecular size, eculizumab is not excreted in urine, except in patients with heavy proteinuria [40].

Simplified alternative complement cascade and its regulation. Complement components: filled squares (basic: blue; activated: orange). Cofactors for this pathway activation: red ovals; regulators (FH, FI, membrane cofactor protein, DAF, CD59): green triangles. The active products of this cascade (C3a, C5a, and C5b-9): yellow circles. Pathogenic genetic variants and antibodies leading to its over-activation: green squares. Sites of pharmacologic complement inhibition for aHUS treatment: white squares. Activating factors: FB factor B, FD factor D, P properdin. Regulatory factors: anti-FH Ab-anti-factor H antibodies, DAF-decay-accelerating factor, FH factor H, FI factor I, MCP membrane cofactor protein
In 2009, the first reports on the efficacy of eculizumab in pediatric [41] and adult [42] patients with aHUS were published. Later, the efficacy and safety of eculizumab were shown across four prospective studies including a pediatric cohort trial [43,44,45,46], and eculizumab received US Food and Drug Administration approval and has subsequently been adopted as first-line therapy in patients with aHUS. Eculizumab treatment led to the normalization of hematologic parameters (platelet count and lactate dehydrogenase levels) in 82% of patients after 26 weeks, and kidney function improvement in 73% of patients (at least a 25% decrease is serum creatinine levels). All patients were able to discontinue plasma therapy and 82% of patients were able to discontinue dialysis with no resulting deaths or meningococcal infections [45]. Eculizumab can also improve central nervous system manifestations, ischemic cardiomyopathy, other ischemic manifestations, ophthalmologic involvement, and ulcero-necrotic skin lesions [7, 13]. Recommendations for clinical practice including eculizumab doses and regimens have been published [7]. Genetic screening results are not necessary prior to eculizumab treatment initiation, as patients with and without identified genetic variants in complement genes may have similar responses to eculizumab treatment, with the exception of diacylglycerol kinase ɛ and cobalamin C deficiency-associated HUS [47, 48].
3.3.1.1 Timing of Eculizumab Initiation
In adults, an earlier initiation of eculizumab after aHUS onset increases the odds of kidney function improvement [43], as well as other systemic manifestations. In pediatric patients with suspected aHUS, eculizumab is considered a first-line treatment and should be initiated as soon as possible. Early eculizumab initiation was associated with better kidney outcomes, based on open-label, single-arm, prospective clinical studies [49]. These studies included 97 patients, with a median age of 29 (0–80) years, 25 (26%) of them were younger than 18 years of age. A multivariate regression analysis demonstrated that a shorter time from aHUS manifestation to eculizumab treatment, younger age, higher baseline lactate dehydrogenase, and lower baseline hemoglobin were independent predictive factors of an estimated glomerular filtration rate change from baseline. If eculizumab is unavailable, plasma exchange with fresh frozen plasma (60 mL/kg/session or a daily plasma infusion of 10 mL/kg) should be initiated, with a switch to eculizumab when possible [45].
3.3.1.2 Monitoring of Effectiveness
Complement blockade is assessed using complement hemolytic activity (CH50) and is considered to be optimal when it is less than 10%. C5 function and alternative pathway hemolytic activity (AP50 or CH50) are also reported as markers of eculizumab effectiveness [50]. Checking CH50 before the next dose of eculizumab can be a practical approach to ensure complement blockade and adjustment of dose or intervals between treatments, if needed [51, 52]. The simultaneous testing of CH50 and eculizumab trough concentrations (if available through an investigative treatment protocol) can be used in patients without laboratory and clinical improvement after one or two eculizumab doses, or in patients experiencing a relapse under eculizumab. Resistance to eculizumab can be seen due to concurrent infection, inflammation or surgery, urinary leak of eculizumab in proteinuric patients, non-complement-related HUS (diacylglycerol kinase ɛ and CblC-HUS), or a variant in the eculizumab C5-binding site gene, mostly present in Asian individuals [53,54,55]. In a case series study that included 13 (34%) pediatric patients, Ardissino et al. have shown that high eculizumab trough concentrations associated with CH50 less than 10% indicated an overdosage of the drug and allowed spacing of the interval between doses, thus reducing treatment burden and cost [56]. The use of different biomarkers of complement activation (sC5b9, C3d, and C5a) is not validated and requires further studies [53]. A case series study by Galbusera et al. [57] that included 47 (32%) patients aged <18 years assessed the performance of an ex vivo kit to test C5b-9 levels within the endothelium, to distinguish active disease from remission, and to determine relapses during eculizumab dosage tapering or after its discontinuation. Among 121 study patients with primary and secondary aHUS, 96% had stable endothelial C5b-9 levels after 3–4 weeks of eculizumab treatment. Hence, the C5b-9 endothelial deposition assay might show further merit in the diagnosis and differentiation of active disease from remission.
It is unknown if a complete blockade is necessary to prevent disease progression in the acute and/or remission phases. In one cohort of patients with aHUS, no recurrences were observed during eculizumab treatment when a minimal CH50 of less than 30% was maintained, instead of complete suppression [56].
3.3.1.3 Infection Prophylaxis
The activated terminal complement pathway is required for efficient serum bactericidal activity against encapsulated bacteria, including Neisseria meningitidis. The risk of invasive meningococcal disease under eculizumab treatment is estimated to be at a >2000-fold increase compared with the normal population. Thus, patients receving eculizumab should receive a quadrivalent A,C,W,Y meningococcal conjugate vaccine and B meningococcal vaccine, as well as long-term antimicrobial prophylaxis with penicillin (or macrolides for penicillin-allergic patients) for the duration of eculizumab treatment. [58,59,60]
3.3.1.4 Limited Use and Treatment Discontinuation
After introduction of eculizumab, life-long therapy was suggested and the risk of eculizumab withdrawal was emphasized [61, 62]. Since then, an increasing number of case reports and small cohort studies have provided information on safe eculizumab withdrawal in selected patients [63,64,65,66,67]. Eculizumab discontinuation was assessed in a prospective multicenter open-label study in 55 patients (including 19 children) with aHUS, 51% of them had rare variants in complement genes (membrane cofactor protein: 22%, CFH: 11%, CFI: 10%). During the follow-up, 13 patients (23%; six of them children) experienced aHUS relapse. In a multivariable analysis, female sex, presence of a rare variant in a complement gene, and an increased sC5b-9 plasma level at eculizumab discontinuation were associated with an increased risk of aHUS relapse. All but two of the relapsed patients regained their baseline kidney function after eculizumab re-initiation [68]. Close monitoring of hematologic parameters is needed in case of eculizumab discontinuation for early recognition of relapse and immediate treatment re-initiation.
3.3.1.5 Eculizumab in Anti-CFH Antibody-Associated HUS
Eculizumab can also be efficacious in this form of aHUS as in other complement-aHUS [69]. However, alternative efficacious treatment with PE, followed by glucocorticoids and an immunosuppressive drug (such as mycophenolate or cyclophosphamide) is recommended, as shown in a study from India [70]. An earlier retrospective analysis from the same group described a similar initial response and a later relapse rate with the use of rituximab, in addition to plasma exchange and corticosteroids. This approach can even lead to a decrease of anti-CFH antibody titers, possibly allowing safe discontinuation of any treatment with time [71].
3.3.1.6 Eculizumab in Patients with aHUS Receiving Long-Term Dialysis
In one of the original studies on eculizumab in patients with aHUS and substantial renal damage, dialysis was discontinued in four of five patients (80%) who had required dialysis at the time of initiation of eculizumab [43]. Povey et al. reported on a case of full kidney recovery with eculizumab treatment in a young woman with aHUS receiving long-term dialysis. [72]. Haskin et al. described a boy with aHUS and complete anuria receiving dialysis for 4.5 months who weaned off dialysis after eculizumab therapy[73]. This highlights the importance of a treatment trial with eculizumab, even in patients already receiving long-term dialysis.
3.3.1.7 Eculizumab in Kidney Transplantation After aHUS
Prior to eculizumab, the outcome following isolated kidney transplantation was poor, with recurrence of aHUS post-transplant in 50–60% of patients [74]. Pre- and post-transplant use of eculizumab allowed favorable short- and long-term outcomes in patients with aHUS. A retrospective multicenter study from a large French nationwide registry of adult patients with atypical HUS showed a better graft survival in patients treated with prophylactic eculizumab therapy in comparison with those who received it after experiencing an aHUS recurrence, based on a pre-transplant risk for recurrence stratification [75]. These findings were confirmed in another observational study of 344 patients with aHUS, including 49 (14%) pediatric patients who underwent kidney transplantation [76].
3.3.1.8 Eculizumab in Secondary HUS
Thrombotic microangiopathy/HUS may be seen in different underlying diseases, such as autoimmune diseases (systemic lupus erythematosus, anti-phospholipid syndrome, scleroderma), malignant hypertension, medications, and cancer. The pathophysiology of TMA in many of these disorders is related to endothelial damage by different mechanisms, not always related to complement dysregulation. Data on eculizumab efficacy in secondary aHUS are controversial, based on adult studies [77, 78],.[79, 80]. In a retrospective study of 110 patients with secondary HUS, including eight (7%) patients under 18 years of age, the kidney outcome at 3 months of follow-up was not different in 38 patients treated with eculizumab and 38 matched patients not treated with eculizumab [81].
Transplant-associated TMA (TA-TMA) is a multifactorial disorder, associated with high mortality [82]. Use of calcineurin inhibitors, graft-versus-host disease, and viral infections are risk factors for TA-TMA [83]. Complement pathways may become activated in cases with TA-TMA resulting in tissue damage [84]. High-dose chemotherapy, calcineurin inhibitors, infection with different viruses, and graft-versus-host disease can lead to direct endothelial damage and classic or alternative complement pathway activation[85, 86]. Cyclosporine can reduce levels of ADAMTS13 by inhibiting its secretion or by releasing von Willebrand factor multimers that form complexes with ADAMTS13 [87]. Immune dysregulation after HSCT can be a reason for anti-CFH antibody formation [88].
Eculizumab has shown its benefits in the treatment of patients with TA-TMA [81, 89, 90], but the exact eculizumab dosing is not well established. For pediatric patients, the initial dose is based on body weight, and subsequent dose adjustments are based on maintaining suppressed CH50 levels. Induction therapy is administered weekly for 4 weeks, and then maintenance therapy every 2 weeks is continued [79]. A supplemental dose of 600 mg should be given before plasma infusion or within 1 hour after plasma exchange [43]. A phase II trial evaluating an early intervention with eculizumab to treat TMA/aHUS-associated multiple organ dysfunction syndrome in hematopoietic stem cell transplant recipients is now ongoing (NCT03518203).
3.3.2 Ravulizumab
Eculizumab requires a standard treatment regimen of intravenous infusions every 2–3 weeks, increasing the infusion burden and associated adverse events. To overcome this problem, ravulizumab (Alexion Pharmaceutical, Inc.) was re-engineered from eculizumab by a two amino-acid substitution. Ravulizunab targets the same epitope on C5 as eculizumab, but has a four times longer half-life because of an augmented endosomal dissociation of C5 and recycling to the vascular compartment via the neonatal Fc receptor pathway, providing an increased duration of terminal complement inhibition [91]. Ravulizumab has recently been approved for the treatment of aHUS in adults and children [92].
The efficacy and safety of ravulizumab were evaluated in two single-arm, multicenter, 26-week phase III studies in adult and pediatric patients with aHUS naive to complement inhibitor treatment or with previous eculizumab treatment. Ravulizumab resulted in stable kidney and hematologic parameters, with no unexpected safety concerns when administered every 4–8 weeks both in adult patients [93] and in pediatric patients [94, 95]. The most common treatment-related adverse events with ravulizumab in treatment-naive patients were headache, diarrhea, and vomiting. While ravulizumab may have similar efficacy and safety profiles to eculizumab, there are no head-to-head comparative studies in patients with aHUS. An indirect comparison of patient-level data from pivotal trials of ravulizumab and eculizumab indicates that there are no significant between-group differences in platelet count and kidney outcomes at 26 weeks, after adjustments for baseline characteristics [96]. A reduction in dosing frequency is an important element in improving the quality of life in patients with aHUS [87]. The frequency of infusions has been shown to be one of the most important factors contributing to the improved quality of life for patients switching to ravulizumab [97]. Ravulizumab has recently been approved for the treatment of patients with aHUS in several countries [98,99,100]. Further studies on the efficacy of ravulizumab for HSCT-associated HUS are currently underway (NCT04557735).
4 Potential Future Treatment Options
A number of potential treatments for aHUS are under investigation (Fig. 1). Crovalimab is an intravenous C5 antagonist that recognizes different epitopes on C5 than eculizumab. Its use would enable C5 neutralization in patients carrying certain genetic polymorphisms that prevent eculizumab binding [101]. The prevalence of these variants in Asian individuals is reported as high as 3.5% [49], justifying crovalimab-specific use in this population. A phase III, multicenter single-arm study evaluating the efficacy, safety, pharmacokinetics, and pharmacodynamics of crovalimab is currently being studied in 90 adult and adolescent patients with aHUS (NCT04861259), with an estimated study completion date in 2024.
Avacopan is an orally administered C5aR1 antagonist that inhibits the functions of C3a, C4a, and C5a. Its efficacy in ANCA-positive vasculitis has already been shown [102]. A phase II trial in patients with aHUS receiving dialysis has been completed (NCT02464891).
The combined C5 and leukotriene B4 inhibitor, nomacopan (Coversin; rVA576), was recently reported to be effective in patients with TMA [103].
Iptacopan (LNP023) is a first-in-class, orally administered, potent and highly selective factor B inhibitor of the alternative complement pathway [104]. A phase III multicenter, single-arm, open-label study to evaluate the efficacy and safety of iptacopan in adult patients with aHUS who are treatment naive to complement inhibitor therapy has been initiated (NCT04889430).
Small interfering RNA are a class of double-stranded RNAs that is able to silence its target genes through enzymatic cleavage of target messenger RNA [105]. Cemdisiran (ALN-CC5), a small interfering RNA complementary to C5 mRNA, conjugated with N-acetylgalactosamine for targeted delivery to hepatocytes has shown efficacy in inhibiting circulating C5 in healthy volunteers as well as patients with paroxysmal nocturnal hemoglobinuria [106] ]. Switching from eculizumab to cemdisiran in patients with aHUS aged 12 years and older was proposed for evaluation in a phase II clinical trial (NCT03999840), but the study was withdrawn because of a lack of economic support. A phase II study aimed at evaluating the safety, tolerability, and pharmacokinetics of Cemdisiran in adults with aHUS has also been terminated owing to a lack of enrollment (NCT03303313).
5 Conclusions
Acute hemolytic uremic syndrome is a serious disease, which, if left untreated, may lead to multiple organ damage and end-stage kidney disease. Among its different etiologies, pathogenic genetic variants in components of the alternative complement pathway are more frequent in pediatric patients. Alternative complement pathway dysregulation plays a key role in the pathophysiology of the disease. Complement system inhibition is an effective treatment strategy, leading to normalization of clinical and laboratory parameters and improvement of kidney outcomes. Eculizumab is the first complement inhibitor available for effective aHUS treatment. Ravulizumab, a long-acting complement inhibitor, re-engineered from eculizumab, enables a reduction in dosing frequency and improves the quality of life in patients with aHUS. New strategies for complement blockage in aHUS treatment are under investigation.
References
Atefi G, Aisiku O, Shapiro N, Hauser C, Dalle Lucca J, Flaumenhaft R, et al. Complement activation in trauma patients alters platelet function. Shock. 2016;46(3 Suppl. 1):83–8. https://doi.org/10.1097/SHK.0000000000000675.
Huber-Lang M, Ignatius A, Brenner RE. Role of complement on broken surfaces after trauma. Adv Exp Med Biol. 2015;865:43–55. https://doi.org/10.1007/978-3-319-18603-0_3.
Bauer A, Loos S, Wehrmann C, Horstmann D, Donnerstag F, Lemke J, et al. Neurological involvement in children with E. coli O104:H4-induced hemolytic uremic syndrome. Pediatr Nephrol. 2014;29(9):1607–15. https://doi.org/10.1007/s00467-014-2803-x. (Epub 2014 Mar 25).
Thayu M, Chandler WL, Jelacic S, Gordon CA, Rosenthal GL, Tarr PI. Cardiac ischemia during hemolytic uremic syndrome. Pediatr Nephrol. 2003;18(3):286–9. https://doi.org/10.1007/s00467-002-1039-3. (Epub 2003 Feb 7).
Grodinsky S, Telmesani A, Robson WL, Fick G, Scott RB. Gastrointestinal manifestations of hemolytic uremic syndrome: recognition of pancreatitis. J Pediatr Gastroenterol Nutr. 1990;11(4):518–24. https://doi.org/10.1097/00005176-199011000-00013.
Piastra M, Ruggiero A, Langer A, Caresta E, Chiaretti A, Pulitanò S, et al. Pulmonary hemorrhage complicating a typical hemolytic-uremic syndrome. Respiration. 2004;71(5):537–41. https://doi.org/10.1159/000080643.
Loirat C, Fakhouri F, Ariceta G, Besbas N, Bitzan M, Bjerre A, et al. HUS International. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol. 2016;31(1):15–39. https://doi.org/10.1007/s00467-015-3076-8.
Mukhopadhyay S, Linstedt AD. Manganese blocks intracellular trafficking of Shiga toxin and protects against Shiga toxicosis. Science. 2012;335(6066):332–5. https://doi.org/10.1126/science.1215930.
Amaral MM, Sacerdoti F, Jancic C, Repetto HA, Paton AW, Paton JC, et al. Action of shiga toxin type-2 and subtilase cytotoxin on human microvascular endothelial cells. PLoS ONE. 2013;8(7):e70431. https://doi.org/10.1371/journal.pone.0070431.
Chaisri U, Nagata M, Kurazono H, Horie H, Tongtawe P, Hayashi H, et al. Localization of Shiga toxins of enterohaemorrhagic Escherichia coli in kidneys of paediatric and geriatric patients with fatal haemolytic uraemic syndrome. Microb Pathog. 2001;31(2):59–67. https://doi.org/10.1006/mpat.2001.0447.
Szilágyi A, Kiss N, Bereczki C, Tálosi G, Rácz K, Túri S, et al. The role of complement in Streptococcus pneumoniae-associated haemolytic uraemic syndrome. Nephrol Dial Transpl. 2013;28(9):2237–45. https://doi.org/10.1093/ndt/gft198.
Allen U, Licht C. Pandemic H1N1 influenza A infection and (atypical) HUS: more than just another trigger? Pediatr Nephrol. 2011;26(1):3–5. https://doi.org/10.1007/s00467-010-1690-z.
Mahajan R, Lipton M, Broglie L, Jain NG, Uy NS. Eculizumab treatment for renal failure in a pediatric patient with COVID-19. J Nephrol. 2020;33(6):1373–6. https://doi.org/10.1007/s40620-020-00858-2.
Alizadeh F, O’Halloran A, Alghamdi A, Chen C, Trissal M, Traum A, et al. Toddler with new onset diabetes and atypical hemolytic-uremic syndrome in the setting of COVID-19. Pediatrics. 2021;147(2):e2020016774. https://doi.org/10.1542/peds.2020-016774.
Kaufeld J, Reinhardt M, Schröder C, Bräsen JH, Wiech T, Brylka P, et al. Atypical hemolytic and uremic syndrome triggered by infection with SARS-CoV2. Kidney Int Rep. 2021;6(10):2709–12. https://doi.org/10.1016/j.ekir.2021.07.004.
Nester CM, Barbour T, de Cordoba SR, Dragon-Durey MA, Fremeaux-Bacchi V, Goodship TH, et al. Atypical aHUS: state of the art. Mol Immunol. 2015;67(1):31–42. https://doi.org/10.1016/j.molimm.2015.03.246.
Fakhouri F, Zuber J, Frémeaux-Bacchi V, Loirat C. Haemolytic uraemic syndrome. Lancet. 2017;390(10095):681–96. https://doi.org/10.1016/S0140-6736(17)30062-4. (Erratum In: Lancet. 2017 Aug 12;390(10095):648).
Lemaire M, Frémeaux-Bacchi V, Schaefer F, Choi M, Tang WH, Le Quintrec M, et al. Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet. 2013;45(5):531–6. https://doi.org/10.1038/ng.2590.
Chen M, Zhuang J, Yang J, Wang D, Yang Q. Atypical hemolytic uremic syndrome induced by CblC subtype of methylmalonic academia: a case report and literature review. Medicine (Baltimore). 2017;96(43):e8284. https://doi.org/10.1097/MD.0000000000008284.
Bayer G, von Tokarski F, Thoreau B, Bauvois A, Barbet C, Cloarec S, et al. Etiology and outcomes of thrombotic microangiopathies. Clin J Am Soc Nephrol. 2019;14(4):557–66. https://doi.org/10.2215/CJN.11470918.
Alfandary H, Rinat C, Gurevich E, Eisenstein I, Goldberg O, Kropach N, et al. Hemolytic uremic syndrome: a contemporary pediatric experience. Nephron. 2020;144(3):109–17. https://doi.org/10.1159/000505401.
Ardissino G, Salardi S, Colombo E, Testa S, Borsa-Ghiringhelli N, Paglialonga F, et al. Epidemiology of haemolytic uremic syndrome in children: data from the North Italian HUS network. Eur J Pediatr. 2016;175(4):465–73. https://doi.org/10.1007/s00431-015-2642-1.
Banatvala N, Griffin PM, Greene KD, Barrett TJ, Bibb WF, Green JH, et al.; Hemolytic Uremic Syndrome Study Collaborators. The United States National Prospective Hemolytic Uremic Syndrome Study: microbiologic, serologic, clinical, and epidemiologic findings. J Infect Dis. 2001;183(7):1063–70. https://doi.org/10.1086/319269.
Dragon-Durey MA, Sethi SK, Bagga A, Blanc C, Blouin J, Ranchin B, et al. Clinical features of anti-factor H autoantibody-associated hemolytic uremic syndrome. J Am Soc Nephrol. 2010;21(12):2180–7. https://doi.org/10.1681/ASN.2010030315.
Ardissino G, Vignati C, Masia C, Capone V, Colombo R, Tel F, et al.; ItalKid-HUS Network. Bloody diarrhea and Shiga toxin-producing Escherichia coli hemolytic uremic syndrome in children: data from the ItalKid-HUS Network. J Pediatr. 2021;237:34–40.e1. https://doi.org/10.1016/j.jpeds.2021.06.048.
Sawai T, Nangaku M, Ashida A, Fujimaru R, Hataya H, Hidaka Y, et al. Diagnostic criteria for atypical hemolytic uremic syndrome proposed by the Joint Committee of the Japanese Society of Nephrology and the Japan Pediatric Society. Pediatr Int. 2014;56(1):1–5. https://doi.org/10.1111/ped.12274.
Fremeaux-Bacchi V, Fakhouri F, Garnier A, Bienaimé F, Dragon-Durey MA, Ngo S, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol. 2013;8(4):554–62. https://doi.org/10.2215/CJN.04760512.
Dragon-Durey MA, Loirat C, Cloarec S, Macher MA, Blouin J, Nivet H, et al. Anti-Factor H autoantibodies associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16(2):555–63. https://doi.org/10.1681/ASN.2004050380.
Skerka C, Józsi M, Zipfel PF, Dragon-Durey MA, Fremeaux-Bacchi V. Autoantibodies in haemolytic uraemic syndrome (HUS). Thromb Haemost. 2009;101(2):227–32.
Loirat C, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura in children. Curr Opin Pediatr. 2013;25(2):216–24. https://doi.org/10.1097/MOP.0b013e32835e7888.
von Furlan M. Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and hemolytic-uremic syndrome. Adv Nephrol Necker Hosp. 2000;30:71–81.
Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5(10):1844–59. https://doi.org/10.2215/CJN.02210310.
Verhave JC, Wetzels JF, van de Kar NC. Novel aspects of atypical haemolytic uraemic syndrome and the role of eculizumab. Nephrol Dial Transpl. 2014;29 Suppl 4:iv131–41. https://doi.org/10.1093/ndt/gfu235
Le Quintrec M, Zuber J, Moulin B, Kamar N, Jablonski M, Lionet A, et al. Complement genes strongly predict recurrence and graft outcome in adult renal transplant recipients with atypical hemolytic and uremic syndrome. Am J Transpl. 2013;13(3):663–75. https://doi.org/10.1111/ajt.12077.
Landau D, Shalev H, Levy-Finer G, Polonsky A, Segev Y, Katchko L. Familial hemolytic uremic syndrome associated with complement factor H deficiency. J Pediatr. 2001;138(3):412–7. https://doi.org/10.1067/mpd.2001.112649.
Taylor CM, Machin S, Wigmore SJ, et al. Clinical practice guidelines for the management of atypical haemolytic uraemic syndrome in the United Kingdom. Br J Haematol. 2010;148:37–47.
Waters AM, Licht C. aHUS caused by complement dysregulation: new therapies on the horizon. Pediatr Nephrol. 2011;26:41–57.
Johnson S, Stojanovic J, Ariceta G, Bitzan M, Besbas N, Frieling M, et al. An audit analysis of a guideline for the investigation and initial therapy of diarrhea negative (atypical) hemolytic uremic syndrome. Pediatr Nephrol. 2014;29(10):1967–78. https://doi.org/10.1007/s00467-014-2817-4.
European Medicines Agency (EMA). Assessment report eculizumab. London: European Medicines Agency; 2013.
Volokhina EB, van de Kar NC, Bergseth G, van der Velden TJ, Westra D, Wetzels JF, et al. Sensitive, reliable and easy-performed laboratory monitoring of eculizumab therapy in atypical hemolytic uremic syndrome. Clin Immunol. 2015;160(2):237–43. https://doi.org/10.1016/j.clim.2015.05.018.
Gruppo RA, Rother RP. Eculizumab for congenital atypical hemolytic-uremic syndrome. N Engl J Med. 2009;360(5):544–6. https://doi.org/10.1056/NEJMc0809959.:19179329.
Nürnberger J, Philipp T, Witzke O, Opazo Saez A, Vester U, Baba HA, et al. Eculizumab for atypical hemolytic-uremic syndrome. N Engl J Med. 2009;360(5):542–4. https://doi.org/10.1056/NEJMc0808527. (Erratum in: N Engl J Med. 2009 Jun 4;360(23):2487. Philipp, Thomas [added]).
Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368(23):2169–81. https://doi.org/10.1056/NEJMoa1208981.
Licht C, Greenbaum LA, Muus P, Babu S, Bedrosian CL, Cohen DJ, et al. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int. 2015;87(5):1061–73. https://doi.org/10.1038/ki.2014.423.
Greenbaum LA, Fila M, Ardissino G, Al-Akash SI, Evans J, Henning P, et al. Eculizumab is a safe and effective treatment in pediatric patients with atypical hemolytic uremic syndrome. Kidney Int. 2016;89(3):701–11. https://doi.org/10.1016/j.kint.2015.11.026.
Fakhouri F, Hourmant M, Campistol JM, Cataland SR, Espinosa M, Gaber AO, et al. Terminal complement inhibitor eculizumab in adult patients with atypical hemolytic uremic syndrome: a single-arm, open-label trial. Am J Kidney Dis. 2016;68(1):84–93. https://doi.org/10.1053/j.ajkd.2015.12.034. (Epub 2016 Mar 21).
Brocklebank V, Kumar G, Howie AJ, Chandar J, Milford DV, Craze J, et al. Long-term outcomes and response to treatment in diacylglycerol kinase epsilon nephropathy. Kidney Int. 2020;97(6):1260–74. https://doi.org/10.1016/j.kint.2020.01.045.
Beck BB, van Spronsen F, Diepstra A, Berger RM, Kömhoff M. Renal thrombotic microangiopathy in patients with cblC defect: review of an under-recognized entity. Pediatr Nephrol. 2017;32(5):733–41. https://doi.org/10.1007/s00467-016-3399-0.
Walle JV, Delmas Y, Ardissino G, Wang J, Kincaid JF, Haller H. Improved renal recovery in patients with atypical hemolytic uremic syndrome following rapid initiation of eculizumab treatment. J Nephrol. 2017;30(1):127–34. https://doi.org/10.1007/s40620-016-0288-3.
Willrich MAV, Andreguetto BD, Sridharan M, Fervenza FC, Tostrud LJ, Ladwig PM, et al. The impact of eculizumab on routine complement assays. J Immunol Methods. 2018;460:63–71. https://doi.org/10.1016/j.jim.2018.06.010.
Gatault P, Brachet G, Ternant D, Degenne D, Récipon G, Barbet C, et al. Therapeutic drug monitoring of eculizumab: rationale for an individualized dosing schedule. MAbs. 2015;7(6):1205–11. https://doi.org/10.1080/19420862.2015.1086049. (Epub 2015 Sep 4).
Cugno M, Gualtierotti R, Possenti I, Testa S, Tel F, Griffini S, et al. Complement functional tests for monitoring eculizumab treatment in patients with atypical hemolytic uremic syndrome. J Thromb Haemost. 2014;12(9):1440–8. https://doi.org/10.1111/jth.12615.
Wehling C, Amon O, Bommer M, Hoppe B, Kentouche K, Schalk G, et al. Monitoring of complement activation biomarkers and eculizumab in complement-mediated renal disorders. Clin Exp Immunol. 2017;187(2):304–15. https://doi.org/10.1111/cei.12890.
Harder MJ, Kuhn N, Schrezenmeier H, Höchsmann B, von Zabern I, Weinstock C, et al. Incomplete inhibition by eculizumab: mechanistic evidence for residual C5 activity during strong complement activation. Blood. 2017;129(8):970–80. https://doi.org/10.1182/blood-2016-08-732800.
Nishimura J, Yamamoto M, Hayashi S, Ohyashiki K, Ando K, Brodsky AL, et al. Genetic variants in C5 and poor response to eculizumab. N Engl J Med. 2014;370(7):632–9. https://doi.org/10.1056/NEJMoa1311084.
Ardissino G, Tel F, Sgarbanti M, Cresseri D, Giussani A, Griffini S, et al. Complement functional tests for monitoring eculizumab treatment in patients with atypical hemolytic uremic syndrome: an update. Pediatr Nephrol. 2018;33(3):457–61. https://doi.org/10.1007/s00467-017-3813-2.
Galbusera M, Noris M, Gastoldi S, Bresin E, Mele C, Breno M, et al. An ex vivo test of complement activation on endothelium for individualized eculizumab therapy in hemolytic uremic syndrome. Am J Kidney Dis. 2019;74(1):56–72. https://doi.org/10.1053/j.ajkd.2018.11.012.
McNamara LA, Topaz N, Wang X, Hariri S, Fox L, MacNeil JR. High risk for invasive meningococcal disease among patients receiving eculizumab (Soliris) despite receipt of meningococcal vaccine. MMWR Morb Mortal Wkly Rep. 2017;66(27):734–7. https://doi.org/10.15585/mmwr.mm6627e1.
Benamu E, Montoya JG. Infections associated with the use of eculizumab: recommendations for prevention and prophylaxis. Curr Opin Infect Dis. 2016;29(4):319–29. https://doi.org/10.1097/QCO.0000000000000279.
Haut Conseil de la santé publique. Avis: actualisation de l’avis relatif à l’antibioprophylaxie et la vaccination méningococcique des personnes traitées par eculizumab (Soliris 300 mg solution à diluer pour perfusion) [French]. Paris: Haut Conseil de la santé publique; 2017. http://www.hcsp.fr/Explore.cgi/avisrapportsdomaine?clefr=447. Accessed 31 Jan 2022.
Goodship TH, Cook HT, Fakhouri F, Fervenza FC, Frémeaux-Bacchi V, Kavanagh D, et al.; Conference Participants. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2017;91(3):539–51. https://doi.org/10.1016/j.kint.2016.10.005
US Food and Drug Administration. Approval package for Soliris. 2011.
Merrill SA, Brittingham ZD, Yuan X, Moliterno AR, Sperati CJ, Brodsky RA. Eculizumab cessation in atypical hemolytic uremic syndrome. Blood. 2017;130(3):368–72. https://doi.org/10.1182/blood-2017-02-770214. (Epub 2017 May 1).
Ardissino G, Possenti I, Tel F, Testa S, Salardi S, Ladisa V. Discontinuation of eculizumab treatment in atypical hemolytic uremic syndrome: an update. Am J Kidney Dis. 2015;66(1):172–3. https://doi.org/10.1053/j.ajkd.2015.04.010.
Ardissino G, Testa S, Possenti I, Tel F, Paglialonga F, Salardi S, et al. Discontinuation of eculizumab maintenance treatment for atypical hemolytic uremic syndrome: a report of 10 cases. Am J Kidney Dis. 2014;64(4):633–7. https://doi.org/10.1053/j.ajkd.2014.01.434.
Macia M, de Alvaro MF, Dutt T, Fehrman I, Hadaya K, Gasteyger C, et al. Current evidence on the discontinuation of eculizumab in patients with atypical haemolytic uraemic syndrome. Clin Kidney J. 2017;10(3):310–9. https://doi.org/10.1093/ckj/sfw115.
Wijnsma KL, Duineveld C, Wetzels JFM, van de Kar NCAJ. Eculizumab in atypical hemolytic uremic syndrome: strategies toward restrictive use. Pediatr Nephrol. 2019;34(11):2261–77. https://doi.org/10.1007/s00467-018-4091-3. Epub 2018 Nov 6. Erratum in: Pediatr Nephrol. 2019 Jan 15
Fakhouri F, Fila M, Hummel A, Ribes D, Sellier-Leclerc AL, Ville S, et al. Eculizumab discontinuation in children and adults with atypical hemolytic-uremic syndrome: a prospective multicenter study. Blood. 2021;137(18):2438–49. https://doi.org/10.1182/blood.2020009280.
Diamante Chiodini B, Davin JC, Corazza F, Khaldi K, Dahan K, Ismaili K, Adams B. Eculizumab in anti-factor h antibodies associated with atypical hemolytic uremic syndrome. Pediatrics. 2014;133(6):e1764–8. https://doi.org/10.1542/peds.2013-1594.
Bagga A, Khandelwal P, Mishra K, Thergaonkar R, Vasudevan A, Sharma J, et al. Indian Society of Pediatric Nephrology. Hemolytic uremic syndrome in a developing country: consensus guidelines. Pediatr Nephrol. 2019;34(8):1465–82. https://doi.org/10.1007/s00467-019-04233-7
Hackl A, Ehren R, Kirschfink M, Zipfel PF, Beck BB, Weber LT, et al. Successful discontinuation of eculizumab under immunosuppressive therapy in DEAP-HUS. Pediatr Nephrol. 2017;32(6):1081–7. https://doi.org/10.1007/s00467-017-3612-9.
Povey H, Vundru R, Junglee N, Jibani M. Renal recovery with eculizumab in atypical hemolytic uremic syndrome following prolonged dialysis. Clin Nephrol. 2014;82(5):326–31. https://doi.org/10.5414/CN107958.
Haskin O, Falush Y, Davidovits M. Is eculizumab indicated in patients with atypical hemolytic uremic syndrome already on prolonged dialysis? A case report and review of the literature. Pediatr Nephrol. 2019;34(12):2601–4. https://doi.org/10.1007/s00467-019-04341-4. (Epub 2019 Sep 13).
Loirat C, Fremeaux-Bacchi V. Hemolytic uremic syndrome recurrence after renal transplantation. Pediatr Transpl. 2008;12(6):619–29. https://doi.org/10.1111/j.1399-3046.2008.00910.x.
Zuber J, Frimat M, Caillard S, Kamar N, Gatault P, Petitprez F, et al. Use of highly individualized complement blockade has revolutionized clinical outcomes after kidney transplantation and renal epidemiology of atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2019;30(12):2449–63. https://doi.org/10.1681/ASN.2019040331.
Siedlecki AM, Isbel N, Van de Walle J, James Eggleston J, Cohen DJ; Global aHUS Registry. Eculizumab use for kidney transplantation in patients with a diagnosis of atypical hemolytic uremic syndrome. Kidney Int Rep. 2018;4(3):434–46. https://doi.org/10.1016/j.ekir.2018.11.010.
Cavero T, Rabasco C, López A, Román E, Ávila A, Sevillano Á, et al. Eculizumab in secondary atypical haemolytic uraemic syndrome. Nephrol Dial Transpl. 2017;32(3):466–74. https://doi.org/10.1093/ndt/gfw453.
Kello N, Khoury LE, Marder G, Furie R, Zapantis E, Horowitz DL. Secondary thrombotic microangiopathy in systemic lupus erythematosus and antiphospholipid syndrome, the role of complement and use of eculizumab: case series and review of literature. Semin Arthritis Rheum. 2019;49(1):74–83. https://doi.org/10.1016/j.semarthrit.2018.11.005. (Epub 2018 Dec 4).
Duineveld C, Wetzels JFM. Complement inhibitors are not useful in secondary hemolytic uremic syndromes. Kidney Int. 2019;96(4):829–33. https://doi.org/10.1016/j.kint.2019.08.001.
Brocklebank V, Kavanagh D. Complement C5-inhibiting therapy for the thrombotic microangiopathies: accumulating evidence, but not a panacea. Clin Kidney J. 2017;10(5):600–24. https://doi.org/10.1093/ckj/sfx081. (Epub 2017 May 8).
Le Clech A, Simon-Tillaux N, Provôt F, Delmas Y, Vieira-Martins P, Limou S, et al. Atypical and secondary hemolytic uremic syndromes have a distinct presentation and no common genetic risk factors. Kidney Int. 2019;95(6):1443–52. https://doi.org/10.1016/j.kint.2019.01.023.
Postalcioglu M, Kim HT, Obut F, Yilmam OA, Yang J, Byun BC, et al. Impact of thrombotic microangiopathy on renal outcomes and survival after hematopoietic stem cell transplantation. Biol Blood Marrow Transpl. 2018;24(11):2344–53. https://doi.org/10.1016/j.bbmt.2018.05.010.May11.
Jodele S, Davies SM, Lane A, Khoury J, Dandoy C, Goebel J, et al. Diagnostic and risk criteria for HSCT-associated thrombotic microangiopathy: a study in children and young adults. Blood. 2014;124(4):645–53. https://doi.org/10.1182/blood-2014-03-564997.
Jodele S, Fukuda T, Vinks A, Mizuno K, Laskin BL, Goebel J, et al. Eculizumab therapy in children with severe hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Biol Blood Marrow Transpl. 2014;20(4):518–25. https://doi.org/10.1016/j.bbmt.2013.12.565.
Laskin BL, Maisel J, Goebel J, Yin HJ, Luo G, Khoury JC. Renal arteriolar C4d deposition: a novel characteristic of hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Transplantation. 2013;96(2):217–23. https://doi.org/10.1097/TP.0b013e31829807aa.
Chapin J, Shore T, Forsberg P, Desman G, Van Besien K, Laurence J. Hematopoietic transplant-associated thrombotic microangiopathy: case report and review of diagnosis and treatments. Clin Adv Hematol Oncol. 2014;12(9):565–73.
Hershko K, Simhadri VL, Blaisdell A, Hunt RC, Newell J, Tseng SC, et al. Cyclosporin A impairs the secretion and activity of ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 repeat). J Biol Chem. 2012;287(53):44361–71. https://doi.org/10.1074/jbc.M112.383968.
Changsirikulchai S, Myerson D, Guthrie KA, McDonald GB, Alpers CE, Hingorani SR. Renal thrombotic microangiopathy after hematopoietic cell transplant: role of GVHD in pathogenesis. Clin J Am Soc Nephrol. 2009;4(2):345–53. https://doi.org/10.2215/CJN.02070508.
de Latour RP, Xhaard A, Fremeaux-Bacchi V, Coppo P, Fischer AM, Helley D, et al. Successful use of eculizumab in a patient with post-transplant thrombotic microangiopathy. Br J Haematol. 2013;161(2):279–80. https://doi.org/10.1111/bjh.12202.
Okano M, Sakata N, Ueda S, Takemura T. Recovery from life-threatening transplantation-associated thrombotic microangiopathy using eculizumab in a patient with very severe aplastic anemia. Bone Marrow Transpl. 2014;49(8):1116–8. https://doi.org/10.1038/bmt.2014.97.
Sheridan D, Yu ZX, Zhang Y, Patel R, Sun F, Lasaro MA, et al. Design and preclinical characterization of ALXN1210: A novel anti-C5 antibody with extended duration of action. PLoS One. 2018;13(4):e0195909. https://doi.org/10.1371/journal.pone.0195909.
Ravu (5. FDA (2018) Ravulizumab prescribing information. Alexion Pharmaceutical, Inc., Boston 16. EMA (2019) Ravulizumab summary of product characteristics. Alexion Europe SAS, Levalloise-Perret.
Rondeau E, Scully M, Ariceta G, Barbour T, Cataland S, Heyne N, et al.; 311 Study Group. The long-acting C5 inhibitor, ravulizumab, is effective and safe in adult patients with atypical hemolytic uremic syndrome naïve to complement inhibitor treatment. Kidney Int. 2020;97(6):1287–96. https://doi.org/10.1016/j.kint.2020.01.035(Erratum in: Kidney Int. 2020 Dec;98(6):1621. Erratum in: Kidney Int. 2021 May;99(5):1244).
Tanaka K, Adams B, Aris AM, Fujita N, Ogawa M, Ortiz S, et al. The long-acting C5 inhibitor, ravulizumab, is efficacious and safe in pediatric patients with atypical hemolytic uremic syndrome previously treated with eculizumab. Pediatr Nephrol. 2021;36(4):889–98. https://doi.org/10.1007/s00467-020-04774-2. (Erratum in: Pediatr Nephrol. 2020 Dec 9).
Ariceta G, Dixon BP, Kim SH, Kapur G, Mauch T, Ortiz S, et al.; 312 Study Group. The long-acting C5 inhibitor, ravulizumab, is effective and safe in pediatric patients with atypical hemolytic uremic syndrome naïve to complement inhibitor treatment. Kidney Int. 2021;100(1):225–37. https://doi.org/10.1016/j.kint.2020.10.046.
Tomazos I, Hatswell AJ, Cataland S, Chen P, Freemantle N, Lommele Å, et al. Comparative efficacy of ravulizumab and eculizumab in the treatment of atypical hemolytic uremic syndrome: an indirect comparison using clinical trial data. Clin Nephrol. 2022;97(5):261–72. https://doi.org/10.5414/CN110516.
Peipert JD, Kulasekararaj A, Gaya A, Langemeijer SMC, Yount S, Fernandez FA, et al. Patient preferences for the treatment of paroxysmal nocturnal hemoglobinuria: results of a patient survey of ravulizumab (alxn1210) and eculizumab. HemaSphere. 2019. https://doi.org/10.1097/01.HS9.0000561220.73976.40.
European Medicines Agency. Ultomiris 300 mg concentrate for solution for infusion: summary of product characteristics. 2020. https://www.ema.europa.eu/. Accessed 9 Nov 2020.
Alexion Pharmaceutical, Inc. UltomirisⓇ (ravulizumab-cwvz) injection, for intravenous use: US prescribing information. 2019. Available from: https://www.Ultomiris.com/. Accessed 9 Nov 2020.
Alexion Pharmaceutical, Inc. UltomirisⓇ for drip infusion 300 mg: Japanese prescribing information. 2019.
Fukuzawa T, Sampei Z, Haraya K, Ruike Y, Shida-Kawazoe M, Shimizu Y, et al. Long lasting neutralization of C5 by SKY59, a novel recycling antibody, is a potential therapy for complement-mediated diseases. Sci Rep. 2017;7(1):1080. https://doi.org/10.1038/s41598-017-01087-7.
Jayne DRW, Merkel PA, Schall TJ, Bekker P; ADVOCATE Study Group. Avacopan for the treatment of ANCA-associated vasculitis. N Engl J Med. 2021;384(7):599–609. https://doi.org/10.1056/NEJMoa2023386
Goodship THJ, Pinto F, Weston-Davies WH, Silva J, Nishimura JI, Nunn MA, et al. Use of the complement inhibitor coversin to treat HSCT-associated TMA. Blood Adv. 2017;1(16):1254–8. https://doi.org/10.1182/bloodadvances.2016002832.
Schubart A, Anderson K, Mainolfi N, Sellner H, Ehara T, Adams CM, et al. Small-molecule factor B inhibitor for the treatment of complement-mediated diseases. Proc Natl Acad Sci USA. 2019;116(16):7926–31. https://doi.org/10.1073/pnas.1820892116.
Bakhtiyari S, Haghani K, Basati G, Karimfar MH. siRNA therapeutics in the treatment of diseases. Ther Deliv. 2013;4(1):45–57. https://doi.org/10.4155/tde.12.136.
Badri P, Jiang X, Borodovsky A, Najafian N, Kim J, Clausen VA, et al. Pharmacokinetic and pharmacodynamic properties of cemdisiran, an RNAi therapeutic targeting complement component 5, in healthy subjects and patients with paroxysmal nocturnal hemoglobinuria. Clin Pharmacokinet. 2021;60(3):365–78. https://doi.org/10.1007/s40262-020-00940-9. (Erratum in: Clin Pharmacokinet. 2022 Jun; 61(6):919).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No sources of funding were received for the preparation of this article.
Conflicts of interest/competing interests
The authors have no conflicts of interest that are directly relevant to the content of this article. Daniel Landau participates as the Israeli delegate in the aHUS International Registry meetings, organized by Alexion Pharmaceutical, Inc.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Authors’ contributions
YG wrote the manuscript’s first draft. Both authors critically read and approved the manuscript’s last version.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gurevich, E., Landau, D. Pharmacological Management of Atypical Hemolytic Uremic Syndrome in Pediatric Patients: Current and Future. Pediatr Drugs 25, 193–202 (2023). https://doi.org/10.1007/s40272-022-00555-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40272-022-00555-6