
Abstract
Among the potential adverse effects of breast cancer treatment, chemotherapy-related cognitive impairment (CRCI) has gained increased attention in the past years. In this review, we provide an overview of the literature regarding CRCI in breast cancer, focusing on three main aspects. The first aspect relates to the molecular mechanisms linking individual drugs commonly used to treat breast cancer and CRCI, which include oxidative stress and inflammation, reduced neurogenesis, reduced levels of specific neurotransmitters, alterations in neuronal dendrites and spines, and impairment in myelin production. The second aspect is related to the clinical characteristics of CRCI in patients with breast cancer treated with different drug combinations. Data suggest the incidence rates of CRCI in breast cancer vary considerably, and may affect more than 50% of treated patients. Both chemotherapy regimens with or without anthracyclines have been associated with CRCI manifestations. While cross-sectional studies suggest the presence of symptoms up to 20 years after treatment, longitudinal studies confirm cognitive impairments lasting for at most 4 years after the end of chemotherapy. The third and final aspect is related to possible therapeutic interventions. Although there is still no standard of care to treat CRCI, several pharmacological and non-pharmacological approaches have shown interesting results. In summary, even if cognitive impairments derived from chemotherapy resolve with time, awareness of CRCI is crucial to provide patients with a better understanding of the syndrome and to offer them the best care directed at improving quality of life.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Mechanisms behind chemotherapy-related cognitive impairment development likely include oxidative stress and inflammation, reduced neurogenesis, reduced levels of specific neurotransmitters, alterations in neuronal dendrites and spines, and impairment in myelin production. |
Cognitive impairment before chemotherapy is usually reported in approximately 20–30% of patients with cancer, and chemotherapy-related cognitive impairment can affect up to 75% of patients after treatment. |
There is still no standard treatment for patients with chemotherapy-related cognitive impairment, and the clinical approach consists mainly of symptom management using pharmacological and non-pharmacological approaches, such as cognitive training and rehabilitation and physical activity. |
1 Introduction
Cancer chemotherapy has significantly improved the survival of patients with breast cancer (BC) over the past decades [1]. Nevertheless, although successful, this therapeutic approach may also significantly affect the long-term quality of life of survivors [2]. Among the potential adverse effects of BC chemotherapy, cognitive dysfunction, or the impairment of different cognitive domains, has gained increased attention in the last 20 years. Initially named as “chemobrain” or “chemofog”, the syndrome of cognitive disorders developed from chemotherapy is now commonly referred to as chemotherapy-related cognitive impairment (CRCI), although other terms can also be used [3]. Chemotherapy-related cognitive impairment includes symptoms of mental fogginess, slowed thinking, memory problems, inability to multi-task, and anxiety [4]. Originally thought to be associated with diagnosis-related and treatment-related depression, these symptoms are now recognized as critical neurological consequences derived from certain anti-cancer therapies.
Chemotherapy-related cognitive impairment has been widely studied in BC, as this is a common cancer type and patients usually have high survival rates [5]. Early-stage BC deserves even more attention owing to the successful and widespread use of adjuvant therapies that significantly decrease mortality [6]. Although data regarding incidence rates of cognitive decline vary substantially [4], previous reports, including cross-sectional and longitudinal studies, have shown that up to 75–78% of patients treated for BC develop symptoms of CRCI [4, 7]. A more recent meta-analysis focusing on longitudinal studies only, however, suggests that around 24% of patients with BC present with cognitive decline after treatment [8]. Deleterious effects of chemotherapy over different cognitive domains may start during or shortly after neo/adjuvant treatment completion. Although long-term or late effects are less well established, longitudinal studies have revealed that CRCI may persist or even develop months or years after the end of treatment [9,10,11].
In the clinic, wide variations in the cognitive assessment methodology, in the cut-offs used to classify the results, in the specific domains evaluated, and the controls used (either healthy subjects or those with cancer not treated with chemotherapy), as well as in the cultural and biological heterogeneity of the patients being analyzed have hindered efforts to determine a direct association between specific cognitive impairments and the underlying pathophysiological mechanisms [12, 13]. Patient age and cognitive reserve before chemotherapy are established factors influencing cognitive decline with treatment [14]. Menopausal status at diagnosis may also be relevant [15].
The most common chemotherapeutic agents used to treat early-stage BC include anthracyclines, alkylating agents, taxanes, antimetabolites, and platinum-based compounds. In this narrative review, we present an updated discussion about CRCI in BC with special attention to how these common chemotherapeutic agents are potentially linked to cognitive impairments, both in terms of molecular mechanisms and clinical manifestation patterns.
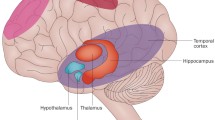
Some of the mechanisms linking these drugs to CRCI have been uncovered in preclinical studies, which revealed oxidative stress and inflammation as important players to cognitive deficits [16,17,18,19,20,21,22,23,24]. Other complex processes such as reduced neurogenesis [25,26,27,28], reduced levels of specific neurotransmitters [29, 30], alterations in neuronal dendrites and spines [31, 32], and impairment in myelin production [33] have also been implicated (Fig. 1). Clinical studies, however, have explored CRCI manifestations in patients treated with chemotherapy combinations, and have provided information on CRCI incidence and duration, as well as on the involved cognitive domains.

Possible mechanisms underlying chemotherapy-related cognitive impairment, including high levels of oxidative stress and inflammation, reduced levels of neurotransmitters, reduced neurogenesis, altered dendrites and spines in neurons, and impairment in myelin production. BDNF brain-derived neurotrophic factor, IL-1β interleukin-1β, IL-6 interleukin-6, Ox oxidized species, Red reduced species, TNFα tumor necrosis factor-α
A broad literature survey on the theme of CRCI was carried out in the Web of Science database. Additional documents selected from reference lists of articles, as well as from author searches were also included. Search details are available in the Electronic Supplementary Material (ESM). A full list of articles and a summary of their main characteristics are also shown in the ESM [5, 7, 9,10,11, 16,17,18,19,20,21,22,23, 25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80, 80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116, 116,117,118,119,120,121,122,123,124,125,126,127].
2 Mechanisms of CRCI Associated with Single Chemotherapeutic Agents
2.1 Anthracyclines
Anthracyclines are one of the most used chemotherapeutic classes in the treatment of BC. They exert cytotoxic effects mainly through DNA intercalation and topoisomerase II inhibition, causing DNA strand breaks [128]. Chemotherapy-related cognitive impairment animal models (mainly mice and rats) revealed that the main affected brain areas by anthracycline exposure are the prefrontal cortex and the hippocampus, with deficits especially observed in learning and memory, but also in exploratory behavior [19, 20, 25, 26, 37, 47, 48, 62, 82, 83]. In addition, the anthracycline doxorubicin (DOX) was shown to cause a reduction in glucose consumption in the pre-frontal cortex [62, 82] and hippocampus [82], and increase blood vessel density in these brain regions [80]. However, the passage of DOX across the blood–brain barrier (BBB) is considered restricted [16, 129]. Therefore, the neurological effects exerted by DOX might be caused either by the small amounts of the drug that reach the brain or by the occurrence of an indirect mechanism that incites central neurotoxicity.
Once in the tissues, DOX undergoes redox cycling, leading to the production of reactive oxygen species (ROS) [46]. As a consequence, this process leads to significant oxidative stress levels, with increases in protein oxidation and lipid peroxidation and an imbalance in cell antioxidant defense. The oxidative properties of DOX have been confirmed in several studies [17, 18, 20, 37, 47, 59, 72, 83, 130,131,132], showing both increases in pro-oxidative markers, such as lipid peroxidation, and reductions in levels of main antioxidant enzymes, including glutathione, superoxide dismutase (SOD), and catalase in the brain of animals. Nitrosative stress has also been described as a consequence of DOX treatment, further enhancing protein modifications and hampering intracellular signaling in brain tissue [17].
The brain is particularly susceptible to oxidative stress because of its high-energy requirements, limited antioxidant defenses, and limited capacity of anaerobic respiration. Therefore, DOX-mediated oxidative stress is closely related to the induction of cognitive damage [22]. In vitro studies using rat embryo-derived primary neurons or neuroblastoma cell lines have revealed a remarkable decrease in cell viability after treatment with DOX [18, 31, 47, 56, 59] with frequent associations between oxidative stress and apoptosis [17, 18, 20, 59], as well as with evidence of mitochondrial alterations [56]. Similar findings were observed for microglial cells treated with epirubicin (EPI) [133]. High intracellular oxidative stress can trigger apoptosis especially due to mitochondrial dysfunction. The main functions of mitochondria in neurons involve control of redox signaling and calcium homeostasis, developmental and synaptic plasticity, and control of cell survival and death [26, 134, 135]. After DOX treatment, mitochondrial dysfunction in the hippocampus [26] and brain cortex [83] was observed, with alterations in mitochondrial biogenesis and dynamics (fusion/fission), decreased oxidative phosphorylation levels, and a higher susceptibility to permeability transition pore opening, which can lead to apoptosis [26, 83].
Doxorubicin and its pegylated liposomal formulation are also capable of disrupting cellular autophagy [74]. Doxorubicin is a membrane-permeable drug that easily reaches the cytoplasm. Once it enters intracellular acid compartments, such as the lysosomes, DOX gets protonated and retained, increasing the pH inside lysosomes, which prevents them from fusing with autophagosomes and degrading their cargo [74]. As a consequence, autophagosomes accumulate in the cytoplasm along with damaged organelles including mitochondria [74] and peroxisomes, producing high levels of ROS [63]. All these intracellular disturbances can trigger neuronal cell death, potentially contributing to CRCI (although cognitive performance was not specifically assessed in these studies).
Oxidative stress is also related to the induction of inflammatory responses, and thought to be one of the main indirect mechanisms through which systemic DOX could affect the brain even without crossing the BBB. In several in vivo studies, DOX was shown to enhance both oxidative stress markers and the levels of inflammation. Reactive oxygen species generated from DOX redox cycling can lead to the activation of the nuclear factor-kappa B, mainly by inducing the inhibitor IκB decoupling and facilitating its targeting to the nucleus [136, 137]. Nuclear factor-kappa B regulates the expression of multiple genes involved in inflammation in immune cells, such as tumor necrosis factor-α (TNF-α), interleukin (IL)-1β, and IL-6 [138].
Tumor necrosis factor-α was one of the first molecules associated with DOX-mediated central nervous system (CNS) toxicity [16], activating other inflammatory cytokines and apoptotic factors in the brain that cause an increase in ROS and mitochondrial dysfunction [139, 140]. Higher levels of TNF-α have been frequently related to CRCI. In animals, TNF-α starts to rise in the circulation 1 hour after DOX administration [16], and it increases not only in the blood but also in the brain, after crossing the BBB [16, 17, 141, 142]. In patients, DOX-based chemotherapeutic regimens were shown to cause an increment in the plasma concentrations of TNF-α 6 hours after the first chemotherapy administration [90]. In addition to TNF-α, DOX can also increase other inflammatory molecules, such as prostaglandin E2 and cyclooxygenase 2, in the hippocampus [18, 20, 37] and frontal cortex [18] of different species of rodents.
Another link to the increase of TNF-α and other inflammatory mediators in plasma after DOX treatment is the oxidation of the apolipoprotein A1 [110]. Apolipoprotein A1, a multifunctional apolipoprotein that plays several roles in the human body including cholesterol transport and regulation of inflammation [143], is involved in the regulation of inflammatory responses by depressing the production of inflammatory cytokines, particularly TNF-α. Oxidation of apolipoprotein A1 leads to elevated peripheral TNF-α levels that can then cross the BBB and contribute to neuroinflammation and neuronal death [143, 144]. Interestingly, studies have shown that one of the alleles of the apolipoprotein E (APOE) gene, the APOE4, is one of the most reproducible genetic risk factors for CRCI [5, 93]. After treatment with DOX, greater deficiencies in spatial learning and memory along with reduced levels of gray matter in the frontal cortex were observed for APOE4 young and old mice compared with untreated animals [34, 48]. Although the mechanisms linking APOE4 and susceptibility to CRCI are not completely understood, an increased predisposition to inflammation may be a possible factor [34, 48].
After entering the brain, TNF-α binds to TNF receptors in microglia and astrocytes and activates these cells, amplifying the inflammatory signals [19]. Doxorubicin significantly increases the number of activated microglia (the first cells of the CNS parenchyma to become activated in response to inflammation, infection, and trauma), and consequently the levels of proinflammatory cytokines (IL-1β, IL-4, IL-6, and others) in the hippocampus of mice [19]. Doxorubicin also increases the numbers of reactive astrocytes in the hippocampus [20]. Reactive astrocytes (usually activated by reactive astrogliosis) release inflammatory mediators, including TNF-α, contributing to this proinflammatory signaling state that can culminate in neuronal injury or death [140].
In addition to oxidative stress and inflammation, DOX-induced cognitive impairment can also be associated with alterations in the levels of neurotransmitters [145]. In an animal model of CRCI, DOX significantly reduced the levels of serotonin and dopamine, neurotransmitters closely associated with cognitive functions [29]. In another study, DOX greatly reduced dopamine and its metabolite (3-methoxytiramine) in the hippocampus of rats; however, it did not affect serotonin and noradrenaline [30]. The effects of DOX on acetylcholine are also unclear. Some in vivo studies show DOX-mediated increase in acetylcholinesterase activity in the brain [20], while others demonstrate no alteration in this parameter [18, 59].
In addition, preclinical experiments have demonstrated that DOX treatment can affect the dynamics of glutamate in the synaptic cleft of mice brain, decreasing glutamate clearance with a decline in the uptake rate constant in the frontal cortex and delayed clearance in the dentate gyrus of the hippocampus [65]. The induced overflow of glutamate in the hippocampus was also higher for DOX-treated animals than saline controls [65]. Interestingly, brain TNF-α induced by DOX treatment has been shown to inhibit glutamate clearance through a similar mechanism as the glutamate uptake inhibitors [146]. Additionally, the activation of astrocytes induced by TNF-α can trigger a massive release of glutamate in the synaptic cleft, which can bind to N-methyl-D-aspartate receptor, lead to calcium-dependent excitotoxicity, and inhibit the synthesis of brain-derived neurotrophic factor (BDNF) [36]. Brain-derived neurotrophic factor plays a key role in neuronal survival and neurogenesis, affecting cognition and memory. In rats, DOX was shown to reduce the levels of BDNF and its receptor tropomyosin-related kinase B, as well as the amount of neural precursor cells in the hippocampus, all associated with a reduction in neurogenesis [26]. A remarkable neurogenesis reduction of 80–90% was observed after DOX administration to rats compared with a saline control [25]. Histological analyses also revealed morphological changes in neurons, with neurite loss or decreased neurite density [18, 31, 59, 147].
2.2 Alkylating Agents
Alkylating agents can interact with DNA bases (mainly guanine and adenine) generating covalent adducts that cause DNA lesions, which can be mutagenic and/or block essential biological processes, such as DNA replication and transcription, leading to cell death [148]. Cyclophosphamide (CP) is the most common alkylating agent used to treat BC. There are several reports associating CP treatment with cognitive dysfunction [21, 25, 27, 32, 78, 82]. Cyclophosphamide-related cognitive impairment, evaluated in studies using mice and rats, include mainly the domains of learning and memory. The mechanisms by which CP may cause cognitive dysfunction include oxidative stress, inflammation, neurogenesis inhibition, and structural changes affecting the morphology and possibly physiology of neurons. Cyclophosphamide is a prodrug that metabolizes into phosphoramide mustard (anticancer moiety) and acrolein (responsible for toxicity) [21]. Acrolein was shown to interfere with the antioxidant defense system and can be mutagenic to mammalian cells [149, 150]. Cyclophosphamide was shown to cause neuronal oxidative stress in animal models, increasing lipid peroxidation and reducing the levels of superoxide dismutase, catalase, and glutathione [21, 130]. Consequently, a transient adaptive increase in the percentage of the nuclear factor erythroid 2-related factor 2-positive neurons was also observed. Nuclear factor erythroid 2-related factor 2 expression usually culminates in the activation of a strong antioxidant response, with the transcription of detoxifying enzymes and increased levels of antioxidants [21]. As a consequence of oxidative stress, CP also increases nuclear factor-kappa B expression in the hippocampus and frontal cortex with a consequent augmentation in the proinflammatory cytokines TNF-α, IL-6, and IL-1β and a diminution in anti-inflammatory IL-10 levels [21]. Induction of oxidative stress in the CNS together with increases in inflammation-associated molecules after CP treatment are also observed [22], as well as higher numbers of activated microglia in the hippocampus [25]. As discussed above, this combination of oxidative stress and neuroinflammation can cause brain damage and cognitive dysfunction.
Another possible mechanism behind CP-related cognitive impairment is inhibition of neurogenesis. Cyclophosphamide alkylates DNA and prevents genome duplication in dividing cells, compromising the formation of new neurons. Cyclophosphamide has the potential to inhibit adult hippocampal neurogenesis, reducing the amounts of proliferating and differentiating neurons, at least in a transient manner [25, 27, 28]. In a specific study [80], CP did not affect mice neurogenesis either in the short term (3 weeks after treatment) or the long term (16 weeks after treatment). However, animals received only one injection of CP (150 mg/kg) and neurogenesis was evaluated after at least 3 weeks of recovery. In other studies in which reduced neurogenesis was detected, the analyses were mostly carried out 1 or 2 days after CP administration [25, 27, 28]. In one of them, neurogenesis returned to the control levels after 2–10 days of recovery [27], and in another study [25], neurogenesis was still significantly reduced 3–4 weeks after treatment, but animals received four injections of CP (total of 200 mg/kg). Therefore, when evaluating the effects of CP in neurogenesis using animal models, it may be relevant to use more than one administration of the drug, as multiple doses are used in the clinic.
Cyclophosphamide can also induce abnormal morphological changes in neurons, either alone [22] or in combination with other chemotherapeutics [151]. A higher percentage of degenerated cells was observed in the hippocampus and frontal cortex of mice after treatment [21]. Abnormalities in dendrites of granule cells of the rat hippocampus, including less branching, shorter length, and thinner and torturous dendritic shafts with intermittent appearances of varicosities, were further detected after CP treatment [32]. Moreover, CP treatment altered dendritic spines, critical for learning and memory. Once the morphology of spines is critical in receiving excitatory projections, abnormalities such as those are expected to result in lower excitability and insufficient integration of newborn granule cells, which could contribute to the development of cognitive decline [32]. Last, deficits in cognitive function after chemotherapy are associated with a reduced metabolic activity (with lower glucose consumption) in certain brain areas [152]. Positron emission tomography analysis in rats confirmed that CP reduces glucose metabolism both in the medial prefrontal cortex and hippocampus [82].
2.3 Taxanes
Paclitaxel (PTX) and docetaxel (DTX), the main representative drugs of the taxane class, cause cytotoxicity by disruption of microtubule function, being therefore called anti-microtubule agents. They stabilize guanosine diphosphate-tubulin in the microtubules, thereby inhibiting their depolymerization and the process of cell division as a consequence [153].
Taxanes are particularly sensitive to the action of P-glycoprotein exporter at the BBB, an efflux pump that exhibits high efficiency in limiting the passage of these substances to the CNS [154, 155]. However, small amounts of the drug have been detected in the CNS in different studies [95, 156,157,158]. Radiolabeled PTX, for example, was detectable in the brain tissues of mice after intravenous administration [156]; and radiolabeled DTX was detected in the brain of patients with advanced solid tumors in quantities corresponding to less than 1% of the total administered dose [157]. In another study [95], plasma and brain concentrations of DTX were quantified by mass spectrometry in mice without tumors. Docetaxel brain concentrations reached a peak of 28.3 ± 3.2 ng/g 1 h after administration followed by a rapid decline within 24 hours, and levels below the limit of detection after 48 hours post-injection [95].
Despite their limited passage through the BBB, both PTX and DTX have been related to cognitive impairment, suggesting that even these small amounts of taxanes that reach the CNS may be sufficient to cause serious damage. Docetaxel caused object recognition impairment in rats shortly after treatment (~ 1–3 weeks), although it did not appear to affect spatial reference memory [97]. Similar patterns of cognitive impairment were observed in a later study using a mouse model [95]. In contrast, opposite results were described in another study after DTX administration to rats, in which long-term impairment of spatial memory was found, but no effect on object recognition was observed [88]. Regarding PTX, studies using different drug doses and schedules of administration found significant cognitive impairment related to spatial memory both in mice [66] and rat models [23].
The specific capacity of taxanes to inhibit microtubules can distinctly affect neuronal function. Microtubules are important for the development and maintenance of neurons, providing structural support, participating in neurite growth, and mediating axonal transport in the neuron. As such, interference with microtubule function can culminate in defective neuronal development [159] and affect hippocampal neurogenesis [28]. In this line, treatment with PTX [66] was shown to induce the disruption of vesicular zinc stores in hippocampal mossy fiber terminals, reducing zinc levels, likely owing to a deficient axonal transport. Zinc is an important factor for neurogenesis. As a consequence, the authors observed that PTX significantly impaired neuronal differentiation in the subgranular zone of the hippocampus, together with the induction of cognitive impairment [66].
However, studies in the literature have been proposing microtubule-independent mechanisms through which taxanes can lead to CRCI. One of these mechanisms is inflammation. Taxanes can induce proinflammatory factors, increasing the production of TNF-α and IL-1β in patients and animal models [23, 160], and can cause neuronal apoptosis in the hippocampus of rats [23]. Although blocking inflammation before PTX treatment may not protect from cognitive impairment [161], some studies showed that specific inhibition of TNF-α can revert PTX-induced impairment of spatial learning and memory [23]. Additionally, intermittent treatment with DTX was already shown to elevate the number of glial fibrillary, acidic protein-positive activated astrocytes in the hippocampus and also to provoke a transient increase in autophagy levels in the brain [95]. These patterns appear to be consistent with the transient increases in brain levels of DTX shortly after each new injection of the drug in mice [95]. Docetaxel was also linked to a redox imbalance in neuroblastoma cells [162].
Another potential microtubule-independent mechanism is the dysregulation of calcium homeostasis. Calcium signaling is a crucial factor for several neuronal functions, including the control of neurotransmitter release in the synaptic cleft and the maintenance of spines and dendrites [163]. Recent evidence has suggested PTX-induced intracellular calcium signaling alterations as a potential cause of CRCI [158, 164]. Mitochondrial dysfunction, through opening of the mitochondrial permeability transition pore and loss of mitochondrial Ca2+, is likely involved [165, 166].
Interestingly, treatment with taxanes is associated with both peripheral and central neurotoxicity. Peripheral neuropathy, for example, is observed upon treatment with taxanes both in animals and humans [167, 168]. One of the possible mechanisms leading to peripheral neuropathy is the dysfunction of microtubules in dorsal root ganglia, axons, and Schwann cells [167, 169], but other studies also suggest calcium signaling dysregulation as a potential cause [170,171,172]. By altering microtubule dynamics and causing mitochondrial dysfunction, taxanes damage the peripheral nerves, which then triggers both peripheral and central inflammation [173], and the latter can be a potential cause to CRCI, as already mentioned.
Importantly, the solvents used to solubilize taxanes in aqueous medium can be an additional source of neurotoxicity. Although polyethoxylated castor oil (Cremophor EL), for example, has a low volume of distribution to the tissues, it has been shown in in vitro studies to lead to axonal degeneration and demyelination [174].
2.4 Other Classes
2.4.1 Antimetabolites
Antimetabolites used to treat BC mainly include 5-fluorouracil (5FU), methotrexate (MTX), and capecitabine, which are small molecules that interfere with cellular metabolism. Acting as false substrates for enzymes involved in the DNA or RNA synthesis, these molecules impair nucleic acid synthesis and progression through the cell cycle [175].
5-Fluorouracil, a fluorinated analog of uracil, inhibits thymidylate synthase, blocking the synthesis of thymidine. 5-Fluorouracil penetrates the brain by passive diffusion, readily crossing the BBB [176]. In this way, 5FU concentration in the cerebrospinal fluid can reach 11–50% of the serum concentration [177]. Potential neurotoxic effects associated with this drug have been investigated, including CRCI.
Systemic treatment with 5FU alone caused a syndrome of delayed myelin destruction in the CNS of mice, lasting for 6 months after treatment [33]. A significant reduction in proliferation and an increase in apoptosis in neurogenic regions after treatment was also observed. The degeneration caused by 5FU seemed to have no correlation with chronic inflammation or vascular damage, representing yet another mechanism of CNS degenerative damage [33]. Reduction in neural cell proliferation in the dentate gyrus of hippocampus in mice [28] and decreased levels of BDNF and doublecortin, markers of immature progenitor cells, in rats after 5FU treatment were also reported [117].
Regarding the evaluation of cognitive symptoms in preclinical models, subtle impairment in object recognition [106], slight deficits in spatial memory [117], or more significant effects on cognition [111] have been reported. However, in other studies, 5FU affected neither memory function nor object recognition [49]. Differences in animal species, drug doses, treatment frequency, and the cognitive performance tests used may account for the discrepancies found among studies, although these significant variations may indicate that the deficits caused by 5FU are subtle [177].
Methotrexate, a folate antagonist (inhibiting dihydrofolate reductase), is also shown to have an impact on the CNS. Regimens containing MTX and 5FU together caused an increase in IL-1α levels in the rat brain [178]. In addition, MTX alone was found to cause chronic microglial activation and astrocyte reactivity, which further leads to a dysregulation in the oligodendrocyte lineage cells in mouse models [41]. This dysregulation significantly affects myelin production and impairs cognitive function. The myelination process impairment is likely related to BDNF signaling disruption in oligodendrocyte cells by MTX [40]. The white matter damage seems to be a common characteristic of antimetabolite agents.
Capecitabine, as an oral antineoplastic prodrug of 5FU, can potentially cross the BBB [179] and cause similar damage to the CNS. However, in a clinical trial that compared the isolated prodrug with the regimens CMF and AC (consisting of DOX plus CP) [100], measurements of self-reported cognitive function performed before and after treatment showed that average cognitive function scores for all treatments remained within “normal ability”, and did not differ among groups.
2.4.2 Platinum-Based Compounds
Platinum-based compounds cause cytotoxicity because of a direct interaction with DNA bases (especially purines), causing DNA crosslinking (monoadducts, inter-strand or intra-strand crosslinks) [180]. This results in inhibition of DNA repair and/or DNA synthesis in cancer cells. Carboplatin, cisplatin, and oxaliplatin are the most popular drugs in this group. Cisplatin crosses the BBB, especially through uptake mediated by the copper transporter 1 protein. Direct damage to mitochondrial DNA has been implicated as one of the main mechanisms behind cisplatin-induced and possibly other platinum compound-induced cognitive impairment. Cisplatin-induced mitochondrial DNA damage results in mitochondrial dysfunction and structural abnormalities that can increase oxidative stress levels and lead to apoptosis. As neurons, especially dendritic and presynaptic regions, are rich in mitochondria, they are also extremely vulnerable to cisplatin [36]. Recently, it has been suggested that a treatment regimen including carboplatin led to a more significant shortening of telomeres in blood cells of patients with BC than other regimens, although it is not certain whether this effect is associated with significant cognitive deficits [181].
3 Clinical Manifestations of CRCI in Patients Treated with Different Chemotherapeutic Regimens
Several chemotherapy regimens are considered appropriate for the treatment of patients with early-stage BC [182]. In general, multiple drug regimens containing an anthracycline (DOX or EPI) and a taxane are selected for the treatment of patients with high-risk disease, while shorter and less complex regimens may be used for patients with lymph node-negative or certain lymph node-positive BC with more favorable disease biology. There are also non-anthracycline-containing regimens (Table 1).
The chemotherapy regimen selection is made considering multiple patient and disease factors. Treatment choice depends on BC stage and on receptor status [195]: hormone receptor-positive, HER2-positive, or triple-negative BC. It also depends on the estimated risk of recurrence and relative risk reduction effect of chemotherapy, balanced by drug toxicity profile, patient comorbidities, and patient preferences. The main findings regarding CRCI manifestations in patients after chemotherapy for BC are discussed below.
3.1 Anthracycline-Based Regimens
In a recently published meta-analysis, DOX-containing regimens were shown to significantly impair the cognition of patients with BC compared with healthy controls [196]. In a study evaluating patients with early-stage BC (median age 52.4 years), standard-dose AC adjuvant chemotherapy induced significant deterioration in delayed memory (affecting 19% of the patients, as assessed up to 30 days after treatment), although not altering immediate memory and verbal learning process [64]. Moreover, AC chemotherapy was also shown to negatively impact semantic memory in patients with early-stage BC [69]. Other longitudinal studies evaluating both AC and AC-T regimens observed that 1 week after completion of chemotherapy, 52% of women experienced a decline in a variety of cognitive domains, including total cognitive score, attention, delayed memory, motor function, and visuospatial skills [109, 118]. In that cohort, 23% were classified as having cognitive impairments at baseline. At the 6-month follow-up, 20% of patients still had a significant decrease from baseline scores for two or more tests, 7% had some improvement, and the remainder were stable [109]. In a study evaluating the effects of anthracycline-based chemotherapies including AC, AC-T, FEC (5FU plus EPI plus CP), and FEC-T (FEC followed by a taxane) in 418 newly diagnosed patients with BC with no cognitive impairment at baseline, approximately 8% of patients presented with incident cognitive impairment during the first year of follow-up. In particular, patients receiving AC (regardless of the use of taxanes) had a significantly higher risk of developing cognitive impairment than those not treated with chemotherapy [70].
Association between cognitive impairment and changes in white matter integrity, evaluated by magnetic resonance imaging (MRI), has been observed in several studies of patients with BC treated with anthracycline-based regimens. For instance, patients treated with AC or AC-T and antiestrogen therapy had a slower processing speed combined with lower fractional anisotropy, a measure of white matter integrity in MRI, in the genu of corpus callosum (responsible for communication between brain hemispheres), when compared with healthy controls [120]. Furthermore, patients receiving AC, AC-T, or FEC had a decline in white matter integrity in the superior longitudinal fasciculus and corticospinal tract detected after 6 months of treatment, when compared with patients who did not receive systemic chemotherapy [60]. In another study, a comparison between the effects of anthracycline-based and non-anthracycline-based regimens on cognitive status and functional brain connectivity, performed 2 years after treatment, demonstrated that the anthracycline-based group had significantly lower verbal memory performance (including immediate and delayed recall), as well as lower left precuneus connectivity compared with the non-anthracycline or no systemic chemotherapy groups, suggesting decreased efficiency of information processing [81]. Other reports showed that adjuvant therapy with EPI and DTX [51], or EPI plus CP plus DTX (followed by tamoxifen) [197], was also capable of affecting the integrity of white matter, and this lower integrity was correlated with poorer performances in neuropsychological scales [51, 197]. Finally, MRI analyses revealed that patients treated with both anthracycline or non-anthracycline regimens demonstrated significantly increased brain perfusion 1 month post-treatment relative to baseline, particularly in the right precentral gyrus [92]. This perfusion increase was negatively correlated with baseline overall neuropsychological performance, and may reflect a compensation mechanism for chemotherapy-induced cellular, vascular, or tissue damage.
Neuropsychological tests did not reveal differences in cognitive function between patients treated with FEC chemotherapy and healthy controls 6 months after treatment [112], even though treated patients still were up to three times more likely than controls to rate themselves as cognitively impaired. However, in another longitudinal study, both FEC and FEC-T regimens had significant negative effects on cognitive performance [50, 68]. In a short-term assessment (after completion chemotherapy), a broader range of cognitive abilities was affected in the taxane-added group. Nevertheless, cognitive dysfunction in attention and executive functions was still found in both groups after approximately 75 weeks from baseline [68]. Importantly, authors observed the importance of correcting for practice effects on repeated assessments with cognitive tests when analyzing neuropsychological test results to detect decrements in cognition [50].
Comparably to FEC or FEC-T, treatment with FAC or FAC-T (5FU plus DOX plus CP with or without taxanes) have been shown to significantly impair cognitive performance. When evaluated during or shortly after chemotherapy, 65% of patients with BC demonstrated cognitive decline (compared with 21% at baseline) [9]. At a long-term evaluation, approximately 1 year after chemotherapy, 61% demonstrated cognitive decline. In this study, 71% showed continuous decline, while 29% presented new delayed cognitive decline. Deficits in cognition were most common in the domains of learning and memory, executive function, and processing speed; improvements in late intervals were rare [9]. Cognitive impairment upon FAC treatment was also self-reported by patients with BC [198].
3.2 Non-Anthracycline-Containing Regimens
A series of cross-sectional studies evaluating patients with BC (age range 50–80 years) who received CMF chemotherapy (CP plus MTX plus 5FU) investigated its effects on cognitive performance, inflammation levels, and white matter integrity up to 20 years after the end of treatment. The results of these studies have shown that patients treated with chemotherapy still presented with lower cognitive scores compared with healthy controls, mainly in tests of immediate and delayed verbal memory, processing speed, executive functioning, and psychomotor speed [103]. A significantly long-term, worse fine motor functioning was detected in treated patients, and age was generally associated with poorer performances [84]. A lower general cognitive factor was associated with higher levels of blood inflammatory markers in BC survivors [53]. No significant difference was observed in white matter integrity. However, within BC survivors, time since treatment was inversely associated with lower global and focal white matter integrity [94].
In another cross-sectional study, impairment in cognitive function was found in 28% of patients with BC treated with CMF chemotherapy compared with 12% of the patients in the non-chemotherapy control group up to approximately 2 years after treatment [127]. This cognitive impairment was unaffected by anxiety, depression, fatigue, and time since treatment.
In a longitudinal study evaluating the effects of CMF, 33% of the treated patients were classified as cognitively impaired about 1 year after treatment, compared with 10% of the subjects in the control group (healthy women) [119]. Four years after the first evaluation, a neurophysiological analysis (electroencephalogram) was carried out. CMF-treated patients who were cognitively impaired in the first assessment had compatible electroencephalogram abnormalities and made more errors in an information-processing task compared with unimpaired patients who received CMF. These results indicate that the neurocognitive problems found 1 year after treatment may persist as neurophysiological abnormalities until 5 or more years after treatment [119]. However, CMF-related deficits in attention and concentration, particularly involving executive functioning, may return to normal levels with time after treatment [101]. In a longitudinal analysis of patients with BC treated with either CMF or EC/CMF (EPI plus CP and then CMF), it was observed that, on completion of chemotherapy, a significant impairment occurred in short-term verbal memory and verbal learning, which improved 6 months later [121]. In all other domains, the cognitive function either remained stable or even improved.
CMF chemotherapy was compared to the AC-T regimen in a cross-sectional study of patients with advanced or metastatic BC approximately 3.3 years after treatment [114]. There were no significant differences between the groups in stress-related variables. However, functional MRI revealed that women who received CMF demonstrated lower prefrontal cortex activation during memory encoding compared with healthy controls and with women treated with AC-T, suggesting that they may have more difficulty attending to stimuli and/or engaging in organizational or mnemonic strategies for memorization [114]. In general, women with BC treated with chemotherapy had significantly lower prefrontal cortex activation during the memory encoding condition, showing significantly greater activation than controls during the recall condition in multiple and diffuse brain regions. This diffuse activation probably reflects a significant increase in neural effort to recall a stimulus that was not properly encoded [114]. In another longitudinal study, CMF was compared to AC/FAC treatments in patients with early-stage BC [104]. Results revealed that the levels of inflammatory cytokines IL-6, IL-8, and monocyte chemoattractant protein-1 increased in the AC/FAC group and decreased in the CMF group after treatment, indicating that AC/FAC chemotherapy is more cytokine inducing than CMF. Objective neuropsychological tests were not performed, although a self-reported cognitive evaluation indicated that heavy headedness, difficulty thinking, and difficulty with concentration were all higher in the AC/FAC group; muddled thoughts were higher in the CMF group, and forgetfulness was the same in both groups [104].
3.3 Other Analyses
There may be a dose-dependent effect of chemotherapy on CRCI [99, 199]. In the past, many women with high-risk early-stage BC were treated with high-dose chemotherapy and bone marrow/stem cell support. A group of studies sought to evaluate the effects in CRCI of high-dose chemotherapy with CP plus thiotepa plus carboplatin (HD CTC), compared with standard-dose FEC treatment, CMF, or no chemotherapy. Neuropsychological tests, after correction for the practicing effects of a repeated assessment, indicated a higher deterioration in cognitive performance over time (~ 12 months after chemotherapy) in patients who received HD CTC when compared with healthy controls (25% vs 6.7% of patients), and such a difference was not observed for standard-dose FEC or no chemotherapy [123]. In a cross-sectional evaluation 4 years after treatment, analysis of brain electrophysiology showed that patients treated with HD CTC had abnormal electroencephalograms (reduction in P3 amplitude) compared with patients not treated with systemic chemotherapy [124]. Cognitive dysfunction in these patients, however, seemed to be transient. As indicated in another follow-up study, 4 years after treatment both subjective and objective cognitive performances improved in patients treated with HD CTC or FEC when compared with assessments made 2 years after chemotherapy [126]. Nevertheless, it is important to mention that for the HD CTC group, 45% of the patients classified as cognitively impaired on the first assessment (2 years after chemotherapy) could not participate on the second assessment (4 years after chemotherapy) because of relapse or death; for the FEC group, this percentage was 33%. For patients treated with CMF, no objective cognitive deficits were observed 4 years after chemotherapy when compared to the control, although patient complaints in both assessments were significantly higher [126]. Finally, a different study compared the effects of standard dose EC-T chemotherapy (EPI plus CP followed by PTX) versus high-dose E-T-CMF (EIP followed by PTX followed by CMF) as neoadjuvant chemotherapy in patients with early-stage BC. Toward the completion of chemotherapy, approximately a quarter of patients showed a decline in cognitive function (27%), whereas another quarter demonstrated improvement (28%), and the remaining patients had stable cognitive performance. There were no effects associated with the treatment arm and no consistent pattern of affected cognitive domains [122].
Treatment with the TC combination (DTX plus CP) was investigated for the induction of changes in cognition and brain MRI in older patients with early-stage BC (age > 60 years) [58]. No significant differences were observed in brain volumes between patients receiving chemotherapy and a healthy control group; however, there was a treatment-specific reduction in the temporal lobe volume of patients receiving TC compared with healthy controls and with patients treated with non-TC chemotherapy. A reduction in verbal reading recognition scores was also detected in TC-treated patients compared with controls [58]. TC chemotherapy was also observed to elevate the blood cortisol levels, which were correlated with poor performances in short-term memory tests during chemotherapy [98].
3.4 Endocrine Therapy
In many cases, in addition to chemotherapy, patients also receive endocrine therapy. Endocrine therapy is typically initiated following completion of chemotherapy and is usually administered for years. Although an evaluation of the association between endocrine therapy and CRCI is not in the scope of this review, these treatments have also been implicated in cognitive impairment [116, 200,201,202]. Recent clinical studies suggest that the effects of endocrine therapy alone on cognitive impairment may equate those of chemoendocrine therapy in long-term assessments [201].
4 Possible Therapeutic Interventions for CRCI
An increasing number of interventional strategies have been proposed considering the CRCI mechanisms. Nevertheless, there is still no standard treatment for patients, and the clinical approach consists mainly of symptom management [163, 203]. The repurpose of drugs is one of the most investigated strategies, together with non-pharmacological approaches including cognitive training and rehabilitation, physical activity, and dietary approaches.
With regard to drug repurpose, several clinical trials have been conducted using psychostimulants (modafinil and methylphenidate [46, 204,205,206,207,208,209]) antidepressants [210] (fluoxetine [46]), acetylcholinesterase inhibitors (donepezil [211, 212]), and anti-inflammatory and antioxidant drugs [213] (sodium 2-mercaptoethane sulfonate [36, 90, 110], flavonoids and polyphenols [57, 59, 214, 215]), among others [216,217,218].
Among the CNS-acting drugs tested against CRCI, two out of three main studies that have investigated the effects of modafinil in CRCI showed improvements in some aspects of cognitive function [205, 206]. For methylphenidate, a stimulant that enhances dopamine and norepinephrine availability, unfortunately, two initial studies failed to improve cognitive function of cancer survivors [207, 208]. However, a third and more recent randomized trial indicated benefits of the drug for verbal learning, memory, visual perception, analysis, and scanning speed [209]. Antidepressant drugs such as fluoxetine, likely owing to their capacity to increase neurogenesis in the adult hippocampus [210], were shown to improve memory function in neurodegenerative disorders by increasing the levels of BDNF and promoting hippocampal neurogenesis [46]. Additionally, the acetylcholinesterase inhibitor donepezil, currently used to treat Alzheimer’s disease, was also reported as able to mitigate oxidative stress and inflammation [211], and was shown to improve verbal memory in BC survivors who underwent adjuvant chemotherapy [212].
As already discussed, chemotherapy-induced inflammation can further generate oxidative stress. Sodium 2-mercaptoethane sulfonate, an antioxidant, neutralizes free radicals and oxidative products generated by chemotherapy [36]. Two preliminary clinical studies also revealed that sodium 2-mercaptoethane sulfonate co-administration decreased plasma levels of TNF-α [90, 110] and its receptors [90]. In addition, flavonoids and polyphenols, acting as free radical scavengers, also have the potential to reduce oxidative stress [57, 59, 214]. However, despite several studies investigating these agents, most natural plant-derived compounds did not reach clinical trials so far, and their efficacy in patients with cancer remains uncertain. As an example, a Ginkgo biloba extract administration showed no differences in subjective or objective measures of cognitive function compared with a placebo [215].
Finally, chemotherapy-related anemia could predispose patients to fatigue and cognitive dysfunction [216, 219]. Nevertheless, epoetin-alpha, a cytokine that stimulates red blood cell production, failed to demonstrate a clear and durable benefit in clinical studies against CRCI in women after DOX-based adjuvant or neoadjuvant chemotherapy for BC [216, 217].
Among the non-pharmacological interventions, diets containing aliments with anti-inflammatory properties are one of the lifestyle approaches investigated to prevent or treat CRCI. Omega-3 enriched diets, for example, are known for having protective properties against neuroinflammation [42, 52], likely through the anti-inflammatory activity of the fatty acids eicosapentaenoic acid and docosahexaenoic acid [52]. Thus far, however, dietary approaches were mainly tested in pre-clinical models with modest results regarding improvements of cognitive performance [220].
Other interventions to CRCI that are non-pharmacological include cognitive training and rehabilitation. Cognitive training normally involves guided practice on standardized tasks designed to reflect specific cognitive functions (e.g., memory or attention), and may be offered by individual or group sessions with therapist support [221]. In contrast, cognitive rehabilitation refers to an individualized approach to help people with cognitive impairments. Instead of enhancing performance on particular cognitive tasks, the emphasis is on improving functioning in the everyday context [221]. Both cognitive rehabilitation and training have been long used to help patients with dementia at different stages, and are now being widely tested for CRCI [222]. Thus far, however, there are no practice standards established, for example, regarding the number of sessions or duration of these cognition-focused programs for CRCI.
The Memory and Attention Adaptation Training intervention, consisting of education about chemotherapy-associated memory problems and training in memory and attention compensatory strategies applied to daily life, was one of the first approaches tested in patients with BC. Memory and Attention Adaptation Training was shown to improve self-reporting of cognitive function, quality of life, and standard neuropsychological test performance in one trial [223], and improved verbal memory and the spiritual well-being subscale of a quality-of-life measure in another trial, although without changing self-reporting of daily cognitive complaints [224]. A similar cognitive rehabilitation program with psychoeducation and cognitive exercises improved cognition compared with baseline, especially in the domains of memory and processing speed [225]. In a subsequent randomized trial, the same cognitive rehabilitation protocol showed better performance on different memory and neurocognitive tests [226]. Interestingly, cognitive rehabilitation with patient education and the practice of compensatory strategies was also suggested as beneficial during chemotherapy [227].
The preliminary efficacy of two specific cognitive trainings on improving memory (intervention 1) or processing speed (intervention 2) showed domain-specific positive effects, with memory training improving memory performance and speed of processing training improving processing speed, and both interventions improved perceived cognitive functioning and quality of life [228]. In another trial, an individual online cognitive training for executive function in long-term BC survivors led to significant improvements in cognitive flexibility, verbal fluency, and processing speed, with marginally significant improvements in verbal memory [229]. Contrary to the studies discussed above, a randomized trial comparing two different cognition-focused interventions to a no-intervention group (control) within patients with BC after adjuvant chemotherapy did not observe significant therapeutic benefits [230]. Both interventions showed improvements in performance for most of the neuropsychological parameters. Nevertheless, benefits were also observed for the control group, indicating no treatment effect [230]. Neurofeedback, a treatment based on training the patients to be able to control the upregulation and downregulation of brain activity by providing feedback [231], significantly improved self-reported outcomes of cognition during intervention in BC survivors [231]. In the follow-up period, participants no longer differed from normative populations in three of the four measures evaluated [231].
Finally, physical activity has been associated with improvements in cognitive performance and quality of life in patients with cancer treated with chemotherapy [232]. A comparison of cardiorespiratory fitness, self-reported physical activity, and cognitive function revealed a significant correlation between exercise behavior and better visual memory aspects [233]. In addition, EXCAP (Exercise for Breast Cancer Patients), a phase III randomized trial, showed that a 6-week exercise program during chemotherapy resulted in enhancement of total and self-perceived cognitive functions scores together with a reduction in the levels of proinflammatory markers [234]. In addition to reducing inflammation, exercise improves cognitive function, likely owing to its ability to protect neuronal integrity and increase both hippocampal neurogenesis and the levels of growth factors that promote cognitive function [235]. Interestingly, a correlation between physical activity and white matter integrity has been determined [236]. The treated patients with cancer were more engaged in physical activity, showed less lesions in white matter (assessed by MRI), and an improved cognitive performance [236]. Association of physical exercise with improvements in neurogenesis was demonstrated in different pre-clinical studies, as in rats treated with DOX [26] or MTX plus 5FU [237], for example.
5 Discussion
Chemotherapy-related cognitive impairment is a well-recognized potential adverse effect of chemotherapy, and many important reviews about its incidence, potential risk factors, and main clinical characteristics have been previously published [238,239,240,241]. In the present review, special attention was given to understand how the most common chemotherapeutic agents used to treat BC were potentially linked to cognitive impairments, both in terms of molecular mechanisms and clinical manifestations. Chemotherapy-related cognitive impairment incidence and clinical manifestation patterns may vary according to the specific drug, dose, and length of use. It may occur even with drugs that are not expected to cross the BBB. Therefore, indirect mechanisms, such as those mediated by generated metabolites or toxic species (especially inflammatory cytokines and ROS) play an important role in triggering cognitive deficits. As could be observed by the data discussed here, there is no single mechanism leading to CRCI, which is likely caused by a blend of processes.
Considering the original articles included in the literature survey performed for this review, approximately 85% of all clinical and preclinical studies reported cognitive performance impairment after treatment with chemotherapeutic agents (Fig. 2). The most common agents evaluated in the field of BC, either alone or in drug combination regimens, were DOX and CP. Among clinical studies evaluating AC effects on patients’ cognition, 77% were indicative of impairment. Regimens adding a taxane following AC (AC-T) were associated with cognitive impairment in 86% of the reviewed original studies. CMF was shown to cause cognitive impairment in 85% of the studies. Not all studies presented data on the incidence or prevalence of CRCI among patients, as several works provide scores of cognitive performance instead; in those who reported, however, CRCI affected up to 75% of patients [7]. Baseline cognitive impairment (before chemotherapy) was reported in approximately 20–30% of patients with cancer, with some studies showing no baseline impairment [68] and one study suggesting almost 50% of patients with previous impairments in two or more neuropsychological tests [121]. Furthermore, studies have shown that before treatment, patients with BC have more intra-individual variability in cognitive performance, which may reflect disruptions in the allocation of attention or cognitive control in different tasks when compared with healthy controls [242].

Percentage of preclinical and clinical studies, from the literature survey of chemotherapy-related cognitive impairment in breast cancer performed in this review. reporting cognitive impairment. The determination of cognitive impairment versus no impairment in this analysis was solely based on the results reported by the authors of each paper and does not account for publication biases
Most studies in BC with a longitudinal design did not follow patients for more than 6 months after the last chemotherapy cycle, and assessments of cognitive performance occurred at a maximum of around 4 years after chemotherapy, limiting long-term evaluation of patients. Some longitudinal studies indicated that impairments observed shortly after the end of chemotherapy recovered after 6 months [109, 121]. Others revealed that impairments persisted after more than 1 year post-treatment [9, 68, 119]. It is interesting to observe that the patterns of time to develop CRCI and the persistence of impairment appear to be different between patients with BC and those with other solid malignancies such as colorectal or head and neck cancer, for which treatments affect cognitive function in a more delayed manner [243, 244], progressing over 2 years after treatment [244]. The longest time after treatment of BC for which cognitive impairment was observed was around 20 years, reported in a series of cross-sectional studies in which patients had been treated with CMF [53, 84, 94, 103].
A better understanding of CRCI etiologic factors and clinical manifestations in patients with BC is crucial to improve diagnosis, to facilitate the understanding of epidemiology, severity, and duration, and to permit the development of specific management strategies, either to prevent BC or remediate it. As also discussed in this review, several pharmacological and non-pharmacological interventions are under investigation. However, so far there is still no standard of care to prevent or treat CRCI [163, 203], and the clinical approach consists mainly of symptom management [204].
6 Conclusions
Chemotherapy-related cognitive impairment has the potential to become a devastating event for individual cancer survivors. Symptoms may affect patients professionally, socially, and ultimately interfere with their autonomy. Discussions as the one presented in this work, added to the growing scientific knowledge on the theme, are important to help increase the awareness of CRCI, lead to the development of more effective diagnostics and treatment, as well as improve communication with patients. As people live longer and better after BC, non-resolving toxicities become progressively more relevant, and all these aspects mentioned are of ultimate importance to provide the best care to them.
References
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71:7–33. https://doi.org/10.3322/caac.21654.
Siembida EJ, Smith AW, Potosky AL, et al. Examination of individual and multiple comorbid conditions and health-related quality of life in older cancer survivors. Qual Life Res. 2021;30:1119–29.
Gibson EM, Monje M. Emerging mechanistic underpinnings and therapeutic targets for chemotherapy-related cognitive impairment. Curr Opin Oncol. 2019;31:531–9. https://doi.org/10.1097/CCO.0000000000000578.
Wefel JS, Schagen SB. Chemotherapy-related cognitive dysfunction. Curr Neurol Neurosci Rep. 2012;12:267–75. https://doi.org/10.1007/s11910-012-0264-9.
Huehnchen P, van Kampen A, Boehmerle W, Endres M. Cognitive impairment after cytotoxic chemotherapy. Neuro Oncol Pract. 2020;7:11–21. https://doi.org/10.1093/nop/npz052.
Harbeck N, Penault-Llorca F, Cortes J, et al. Breast cancer. Nat Rev Dis Primer. 2019;5:1–31. https://doi.org/10.1038/s41572-019-0111-2.
Wieneke MH, Dienst ER. Neuropsychological assessment of cognitive functioning following chemotherapy for breast cancer. Psychooncology. 1995;4:61–6. https://doi.org/10.1002/pon.2960040108.
Dijkshoorn ABC, van Stralen HE, Sloots M, et al. Prevalence of cognitive impairment and change in patients with breast cancer: a systematic review of longitudinal studies. Psychooncology. 2021;30:635–48. https://doi.org/10.1002/pon.5623.
Wefel JS, Saleeba AK, Buzdar AU, Meyers CA. Acute and late onset cognitive dysfunction associated with chemotherapy in women with breast cancer. Cancer. 2010;116:3348–56. https://doi.org/10.1002/cncr.25098.
Ahles TA, Saykin AJ, Furstenberg CT, et al. Neuropsychologic impact of standard-dose systemic chemotherapy in long-term survivors of breast cancer and lymphoma. J Clin Oncol. 2002;20:485–93. https://doi.org/10.1200/JCO.2002.20.2.485.
Scherwath A, Mehnert A, Schleimer B, et al. Neuropsychological function in high-risk breast cancer survivors after stem-cell supported high-dose therapy versus standard-dose chemotherapy: evaluation of long-term treatment effects. Ann Oncol. 2006;17:415–23. https://doi.org/10.1093/annonc/mdj108.
Wefel JS, Vardy J, Ahles T, Schagen SB. International Cognition and Cancer Task Force recommendations to harmonise studies of cognitive function in patients with cancer. Lancet Oncol. 2011;12:703–8. https://doi.org/10.1016/S1470-2045(10)70294-1.
Bernstein LJ, McCreath GA, Komeylian Z, Rich JB. Cognitive impairment in breast cancer survivors treated with chemotherapy depends on control group type and cognitive domains assessed: a multilevel meta-analysis. Neurosci Biobehav Rev. 2017;83:417–28. https://doi.org/10.1016/j.neubiorev.2017.10.028.
Ahles TA, Saykin AJ, McDonald BC, et al. Longitudinal assessment of cognitive changes associated with adjuvant treatment for breast cancer: impact of age and cognitive reserve. J Clin Oncol. 2010;28(29):4434–40. https://doi.org/10.1200/JCO.2009.27.0827.
Ganz PA, Van Dyk K. Cognitive impairment in patients with breast cancer: understanding the impact of chemotherapy and endocrine therapy. J Clin Oncol. 2020;38:1871–4. https://doi.org/10.1200/JCO.20.00336.
Tangpong J, Cole MP, Sultana R, et al. Adriamycin-induced, TNF-alpha-mediated central nervous system toxicity. Neurobiol Dis. 2006;23:127–39. https://doi.org/10.1016/j.nbd.2006.02.013.
Tangpong J, Miriyala S, Noel T, et al. Doxorubicin-induved central nervous system toxicity and protection by xanthone derivative of Garcinia mangostana. Neuroscience. 2011;175:292–9. https://doi.org/10.1016/j.neuroscience.2010.11.007.
Ramalingayya GV, Cheruku SP, Nayak PG, et al. Rutin protects against neuronal damage in vitro and ameliorates doxorubicin-induced memory deficits in vivo in Wistar rats. Drug Des Devel Ther. 2017;11:1011–26. https://doi.org/10.2147/DDDT.S103511.
Allen BD, Apodaca LA, Syage AR, et al. Attenuation of neuroinflammation reverses adriamycin-induced cognitive impairments. Acta Neuropathol Commun. 2019;7:186. https://doi.org/10.1186/s40478-019-0838-8.
El-Agamy SE, Abdel-Aziz AK, Wahdan S, et al. Astaxanthin ameliorates doxorubicin-induced cognitive impairment (chemobrain) in experimental rat model: impact on oxidative, inflammatory, and apoptotic machineries. Mol Neurobiol. 2018;55:5727–40. https://doi.org/10.1007/s12035-017-0797-7.
Iqubal A, Sharma S, Najmi AK, et al. Nerolidol ameliorates cyclophosphamide-induced oxidative stress, neuroinflammation and cognitive dysfunction: plausible role of Nrf2 and NF- κB. Life Sci. 2019;236: 116867. https://doi.org/10.1016/j.lfs.2019.116867.
Gaman AM, Uzoni A, Popa-Wagner A, et al. The role of oxidative stress in etiopathogenesis of chemotherapy induced cognitive impairment (CICI): “chemobrain.” Aging Dis. 2016;7:307–17. https://doi.org/10.14336/AD.2015.1022.
Li Z, Zhao S, Zhang H-L, et al. Proinflammatory factors mediate paclitaxel-induced impairment of learning and memory. Mediat Inflamm. 2018. https://doi.org/10.1155/2018/3941840.
Brown T, Sykes D, Allen AR. Implications of breast cancer chemotherapy-induced inflammation on the gut, liver, and central nervous system. Biomedicines. 2021;9:189. https://doi.org/10.3390/biomedicines9020189.
Christie L-A, Acharya MM, Parihar VK, et al. Impaired cognitive function and hippocampal neurogenesis following cancer chemotherapy. Clin Cancer Res. 2012;18:1954–65. https://doi.org/10.1158/1078-0432.CCR-11-2000.
Park H-S, Kim C-J, Kwak H-B, et al. Physical exercise prevents cognitive impairment by enhancing hippocampal neuroplasticity and mitochondrial function in doxorubicin-induced chemobrain. Neuropharmacology. 2018;133:451–61. https://doi.org/10.1016/j.neuropharm.2018.02.013.
Yang M, Kim J-S, Song M-S, et al. Cyclophosphamide impairs hippocampus-dependent learning and memory in adult mice: possible involvement of hippocampal neurogenesis in chemotherapy-induced memory deficits. Neurobiol Learn Mem. 2010;93:487–94. https://doi.org/10.1016/j.nlm.2010.01.006.
Janelsins MC, Roscoe JA, Berg MJ, et al. IGF-1 partially restores chemotherapy-induced reductions in neural cell proliferation in adult C57BL/6 mice. Cancer Invest. 2010;28:544–53. https://doi.org/10.3109/07357900903405942.
Kwatra M, Jangra A, Mishra M, et al. Naringin and sertraline ameliorate doxorubicin-induced behavioral deficits through modulation of serotonin level and mitochondrial complexes protection pathway in rat hippocampus. Neurochem Res. 2016;41:2352–66. https://doi.org/10.1007/s11064-016-1949-2.
Antkiewicz-Michaluk L, Krzemieniecki K, Romanska I, et al. Acute treatment with doxorubicin induced neurochemical impairment of the function of dopamine system in rat brain structures. Pharmacol Rep. 2016;68:627–30. https://doi.org/10.1016/j.pharep.2016.01.009.
Manchon JFM, Dabaghian Y, Uzor N-E, et al. Levetiracetam mitigates doxorubicin-induced DNA and synaptic damage in neurons. Sci Rep. 2016;6:25705. https://doi.org/10.1038/srep25705.
Wu L, Guo D, Liu Q, et al. Abnormal development of dendrites in adult-born rat hippocampal granule cells induced by cyclophosphamide. Front Cell Neurosci. 2017. https://doi.org/10.3389/fncel.2017.00171.
Han R, Yang YM, Dietrich J, et al. Systemic 5-fluorouracil treatment causes a syndrome of delayed myelin destruction in the central nervous system. J Biol. 2008;7:12. https://doi.org/10.1186/jbiol69.
Demby TC, Rodriguez O, McCarthy CW, et al. A mouse model of chemotherapy-related cognitive impairments integrating the risk factors of aging and APOE4 genotype. Behav Brain Res. 2020;384: 112534. https://doi.org/10.1016/j.bbr.2020.112534.
McElroy T, Brown T, Kiffer F, et al. Assessing the effects of redox modifier MnTnBuOE-2-PyP 5+ on cognition and hippocampal physiology following doxorubicin, cyclophosphamide, and paclitaxel treatment. Int J Mol Sci. 2020. https://doi.org/10.3390/ijms21051867.
Ongnok B, Chattipakorn N, Chattipakorn SC. Doxorubicin and cisplatin induced cognitive impairment: the possible mechanisms and interventions. Exp Neurol. 2020;324: 113118. https://doi.org/10.1016/j.expneurol.2019.113118.
Tong Y, Wang K, Sheng S, Cui J. Polydatin ameliorates chemotherapy-induced cognitive impairment (chemobrain) by inhibiting oxidative stress, inflammatory response, and apoptosis in rats. Biosci Biotechnol Biochem. 2020;84:1201–10. https://doi.org/10.1080/09168451.2020.1722057.
Anderson JE, Trujillo M, McElroy T, et al. Early effects of cyclophosphamide, methotrexate, and 5-fluorouracil on neuronal morphology and hippocampal-dependent behavior in a murine model. Toxicol Sci. 2020;173:156–70. https://doi.org/10.1093/toxsci/kfz213.
Lange M, Joly F, Vardy J, et al. Cancer-related cognitive impairment: an update on state of the art, detection, and management strategies in cancer survivors. Ann Oncol. 2019;30:1925–40. https://doi.org/10.1093/annonc/mdz410.
Geraghty AC, Gibson EM, Ghanem RA, et al. Loss of adaptive myelination contributes to methotrexate chemotherapy-related cognitive impairment. Neuron. 2019;103:250-65.e8. https://doi.org/10.1016/j.neuron.2019.04.032.
Gibson EM, Nagaraja S, Ocampo A, et al. Methotrexate chemotherapy induces persistent tri-glial dysregulation that underlies chemotherapy-related cognitive impairment. Cell. 2019;176:43-55.e13. https://doi.org/10.1016/j.cell.2018.10.049.
Bennouna D, Solano M, Orchard TS, et al. The effects of doxorubicin-based chemotherapy and omega-3 supplementation on mouse brain lipids. Metabolites. 2019. https://doi.org/10.3390/metabo9100208.
Bagnall-Moreau C, Chaudhry S, Salas-Ramirez K, et al. Chemotherapy-induced cognitive impairment is associated with increased inflammation and oxidative damage in the hippocampus. Mol Neurobiol. 2019;56:7159–72. https://doi.org/10.1007/s12035-019-1589-z.
Philpot RM, Ficken M, Johns BE, et al. Spatial memory deficits in mice induced by chemotherapeutic agents are prevented by acetylcholinesterase inhibitors. Cancer Chemother Pharmacol. 2019;84:579–89. https://doi.org/10.1007/s00280-019-03881-8.
Shi D-D, Huang Y-H, Lai CSW, et al. Ginsenoside Rg1 prevents chemotherapyiInduced cognitive impairment: associations with microglia-mediated cytokines. Neuroinflamm Neuroplast Mol Neurobiol. 2019;56:5626–42. https://doi.org/10.1007/s12035-019-1474-9.
El-Agamy SE, Abdel-Aziz AK, Esmat A, Azab SS. Chemotherapy and cognition: comprehensive review on doxorubicin-induced chemobrain. Cancer Chemother Pharmacol. 2019;84:1–14. https://doi.org/10.1007/s00280-019-03827-0.
Gourishetti K. Medhya rasayana restores memory function against doxorubicin-induced cognitive decline: possibly by its neuroprotective effect. Indian J Pharm Educ Res. 2019;53:s104–11. https://doi.org/10.5530/ijper.53.2s.54.
Speidell AP, Demby T, Lee Y, et al. Development of a human APOE knock-in mouse model for study of cognitive function after cancer chemotherapy. Neurotox Res. 2019;35:291–303. https://doi.org/10.1007/s12640-018-9954-7.
Alhowail AH. Preserved memory function of rats following fluorouracil treatment. J Pharm Res Int. 2019. https://doi.org/10.9734/jpri/2019/v30i330269.
Cerulla N, Arcusa À, Navarro J-B, et al. Cognitive impairment following chemotherapy for breast cancer: the impact of practice effect on results. J Clin Exp Neuropsychol. 2019;41:290–9. https://doi.org/10.1080/13803395.2018.1546381.
Li T-Y, Chen VC-H, Yeh D-C, et al. Investigation of chemotherapy-induced brain structural alterations in breast cancer patients with generalized q-sampling MRI and graph theoretical analysis. BMC Cancer. 2018. doi: https://doi.org/10.1186/s12885-018-5113-z.
Orchard TS, Gaudier-Diaz MM, Phuwamongkolwiwat-Chu P, et al. Low sucrose, omega-3 enriched diet has region-specific effects on neuroinflammation and synaptic function markers in a mouse model of doxorubicin-based chemotherapy. Nutrients. 2018. https://doi.org/10.3390/nu10122004.
van der Willik KD, Koppelmans V, Hauptmann M, et al. Inflammation markers and cognitive performance in breast cancer survivors 20 years after completion of chemotherapy: a cohort study. Breast Cancer Res. 2018;20:135. https://doi.org/10.1186/s13058-018-1062-3.
Kang S, Lee S, Kim J, et al. Chronic treatment with combined chemotherapeutic agents affects hippocampal micromorphometry and function in mice, independently of neuroinflammation. Exp Neurobiol. 2018;27:419–36. https://doi.org/10.5607/en.2018.27.5.419.
Ng T, Phey XY, Yeo HL, et al. Impact of adjuvant anthracycline-based and taxane-based chemotherapy on plasma VEGF levels and cognitive function in breast cancer patients: a longitudinal study. Clin Breast Cancer. 2018;18:e927–37. https://doi.org/10.1016/j.clbc.2018.03.016.
Almeida D, Pinho R, Correia V, et al. Mitoxantrone is more toxic than doxorubicin in SH-SY5Y human cells: a “chemobrain” in vitro study. Pharm Basel Switz. 2018. https://doi.org/10.3390/ph11020041.
Shi D-D, Dong CM, Ho LC, et al. Resveratrol, a natural polyphenol, prevents chemotherapy-induced cognitive impairment: involvement of cytokine modulation and neuroprotection. Neurobiol Dis. 2018;114:164–73. https://doi.org/10.1016/j.nbd.2018.03.006.
Chen BT, Sethi SK, Jin T, et al. Assessing brain volume changes in older women with breast cancer receiving adjuvant chemotherapy: a brain magnetic resonance imaging pilot study. Breast Cancer Res. 2018;20:38. https://doi.org/10.1186/s13058-018-0965-3.
Ramalingayya GV, Nayak PG, Shenoy RR, et al. Naringin ameliorates doxorubicin-induced neurotoxicity in vitro and cognitive dysfunction in vivo. Pharmacogn Mag. 2018;14:S197-207. https://doi.org/10.4103/pm.pm_364_17.
Menning S, de Ruiter MB, Veltman DJ, et al. Changes in brain white matter integrity after systemic treatment for breast cancer: a prospective longitudinal study. Brain Imaging Behav. 2018;12:324–34. https://doi.org/10.1007/s11682-017-9695-x.
Flanigan TJ, Anderson JE, Elayan I, et al. Effects of cyclophosphamide and/or doxorubicin in a murine model of postchemotherapy cognitive impairment. Toxicol Sci. 2018;162:462–74. https://doi.org/10.1093/toxsci/kfx267.
Barry RL, Byun NE, Tantawy MN, et al. In vivo neuroimaging and behavioral correlates in a rat model of chemotherapy-induced cognitive dysfunction. Brain Imaging Behav. 2018;12:87–95. https://doi.org/10.1007/s11682-017-9674-2.
Moruno-Manchon JF, Uzor N-E, Kesler SR, et al. Peroxisomes contribute to oxidative stress in neurons during doxorubicin-based chemotherapy. Mol Cell Neurosci. 2018;86:65–71. https://doi.org/10.1016/j.mcn.2017.11.014.
Andryszak P, Wiłkość M, Żurawski B, Izdebski P. Verbal memory in breast cancer patients treated with chemotherapy with doxorubicin and cyclophosphamide. Eur J Cancer Care (Engl). 2018. https://doi.org/10.1111/ecc.12749.
Thomas TC, Beitchman JA, Pomerleau F, et al. Acute treatment with doxorubicin affects glutamate neurotransmission in the mouse frontal cortex and hippocampus. Brain Res. 2017;1672:10–7. https://doi.org/10.1016/j.brainres.2017.07.003.
Lee BE, Choi BY, Hong DK, et al. The cancer chemotherapeutic agent paclitaxel (Taxol) reduces hippocampal neurogenesis via down-regulation of vesicular zinc. Sci Rep. 2017;7:11667. https://doi.org/10.1038/s41598-017-12054-7.
Lakshminarasimhan H, Coughlin BL, Darr AS, Byrne JH. Characterization and reversal of doxorubicin-mediated biphasic activation of ERK and persistent excitability in sensory neurons of Aplysia californica. Sci Rep. 2017;7:4533. https://doi.org/10.1038/s41598-017-04634-4.
Cerulla N, Arcusa À, Navarro J-B, et al. Role of taxanes in chemotherapy-related cognitive impairment: a prospective longitudinal study. Breast Cancer Res Treat. 2017;164:179–87. https://doi.org/10.1007/s10549-017-4240-6.
Andryszak P, Wiłkość M, Żurawski B, Izdebski P. Verbal fluency in breast cancer patients treated with chemotherapy. Breast Cancer Tokyo Jpn. 2017;24:376–83. https://doi.org/10.1007/s12282-016-0713-4.
Ramalho M, Fontes F, Ruano L, et al. Cognitive impairment in the first year after breast cancer diagnosis: a prospective cohort study. Breast Edinb Scotl. 2017;32:173–8. https://doi.org/10.1016/j.breast.2017.01.018.
Orchard TS, Gaudier-Diaz MM, Weinhold KR, Courtney DA. Clearing the fog: a review of the effects of dietary omega-3 fatty acids and added sugars on chemotherapy-induced cognitive deficits. Breast Cancer Res Treat. 2017;61:391–8. https://doi.org/10.1007/s10549-016-4073-8.
Ramalingayya GV, Sonawane V, Cheruku SP, et al. Insulin protects against brain oxidative stress with an apparent effect on episodic memory in doxorubicin-induced cognitive dysfunction in Wistar rats. J Environ Pathol Toxicol Oncol. 2017;36:121–30. https://doi.org/10.1615/JEnvironPatholToxicolOncol.2017017087.
Rendeiro C, Sheriff A, Bhattacharya TK, et al. Long-lasting impairments in adult neurogenesis, spatial learning and memory from a standard chemotherapy regimen used to treat breast cancer. Behav Brain Res. 2016;315:10–22. https://doi.org/10.1016/j.bbr.2016.07.043.
Moruno Manchon JF, Uzor N-E, Kesler SR, et al. TFEB ameliorates the impairment of the autophagy-lysosome pathway in neurons induced by doxorubicin. Aging. 2016;8:3507–19. https://doi.org/10.18632/aging.101144.
Lange M, Heutte N, Rigal O, et al. Decline in cognitive function in older adults with early-stage breast cancer after adjuvant treatment. Oncologist. 2016;21:1337–48. https://doi.org/10.1634/theoncologist.2016-0014.
Iarkov A, Appunn D, Echeverria V. Post-treatment with cotinine improved memory and decreased depressive-like behavior after chemotherapy in rats. Cancer Chemother Pharmacol. 2016;78:1033–9. https://doi.org/10.1007/s00280-016-3161-0.
Himmel LE, Lustberg MB, DeVries AC, et al. Minocycline, a putative neuroprotectant, co-administered with doxorubicin-cyclophosphamide chemotherapy in a xenograft model of triple-negative breast cancer. Exp Toxicol Pathol. 2016;68:505–15. https://doi.org/10.1016/j.etp.2016.08.001.
Janelsins MC, Heckler CE, Thompson BD, et al. A clinically relevant dose of cyclophosphamide chemotherapy impairs memory performance on the delayed spatial alternation task that is sustained over time as mice age. Neurotoxicology. 2016;56:287–93. https://doi.org/10.1016/j.neuro.2016.06.013.
Philpot RM, Ficken M, Wecker L. Doxorubicin and cyclophosphamide lead to long-lasting impairment of spatial memory in female, but not male mice. Behav Brain Res. 2016;307:165–75. https://doi.org/10.1016/j.bbr.2016.04.017.
Seigers R, Loos M, Van Tellingen O, et al. Neurobiological changes by cytotoxic agents in mice. Behav Brain Res. 2016;299:19–26. https://doi.org/10.1016/j.bbr.2015.10.057.
Kesler SR, Blayney DW. Neurotoxic effects of anthracycline- vs nonanthracycline-based chemotherapy on cognition in breast cancer survivors. JAMA Oncol. 2016;2:185–92. https://doi.org/10.1001/jamaoncol.2015.4333.
Lim I, Joung H-Y, Yu AR, et al. PET evidence of the effect of donepezil on cognitive performance in an animal model of chemobrain. BioMed Res Int. 2016. https://doi.org/10.1155/2016/6945415.
Marques-Aleixo I, Santos-Alves E, Balça MM, et al. Physical exercise mitigates doxorubicin-induced brain cortex and cerebellum mitochondrial alterations and cellular quality control signaling. Mitochondrion. 2016;26:43–57. https://doi.org/10.1016/j.mito.2015.12.002.
Hoogendam YY, Schagen SB, Ikram MA, et al. Late effects of adjuvant chemotherapy for breast cancer on fine motor function. Psychooncology. 2015;24:1799–807. https://doi.org/10.1002/pon.3796.
Petrovic M, Simillion C, Kruzliak P, et al. Doxorubicin affects expression of proteins of neuronal pathways in MCF-7 breast cancer cells. Cancer Genom Proteomics. 2015;12:347–58.
Salas-Ramirez KY, Bagnall C, Frias L, et al. Doxorubicin and cyclophosphamide induce cognitive dysfunction and activate the ERK and AKT signaling pathways. Behav Brain Res. 2015;292:133–41. https://doi.org/10.1016/j.bbr.2015.06.028.
Kitamura Y, Hattori S, Yoneda S, et al. Doxorubicin and cyclophosphamide treatment produces anxiety-like behavior and spatial cognition impairment in rats: possible involvement of hippocampal neurogenesis via brain-derived neurotrophic factor and cyclin D1 regulation. Behav Brain Res. 2015;292:184–93. https://doi.org/10.1016/j.bbr.2015.06.007.
Callaghan CK, O’Mara SM. Long-term cognitive dysfunction in the rat following docetaxel treatment is ameliorated by the phosphodiesterase-4 inhibitor, rolipram. Behav Brain Res. 2015;290:84–9. https://doi.org/10.1016/j.bbr.2015.04.044.
Aboalela N, Lyon D, Elswick RK, et al. Perceived stress levels, chemotherapy, radiation treatment and tumor characteristics are associated with a persistent increased frequency of somatic chromosomal instability in women diagnosed with breast cancer: a one year longitudinal study. PLoS ONE. 2015;10: e0133380. https://doi.org/10.1371/journal.pone.0133380.
Hayslip J, Dressler EV, Weiss H, et al. Plasma TNF-α and soluble TNF receptor levels after doxorubicin with or without co-administration of mesna: a randomized, cross-over clinical study. PLoS ONE. 2015. https://doi.org/10.1371/journal.pone.0124988.
Ng T, Chan M, Khor CC, et al. The genetic variants underlying breast cancer treatment-induced chronic and late toxicities: a systematic review. Cancer Treat Rev. 2014;40:1199–214. https://doi.org/10.1016/j.ctrv.2014.10.001.
Nudelman KNH, Wang Y, McDonald BC, et al. Altered cerebral blood flow one month after systemic chemotherapy for breast cancer: a prospective study using pulsed arterial spin labeling MRI perfusion. PLoS ONE. 2014;9: e96713. https://doi.org/10.1371/journal.pone.0096713.
Ahles TA, Li Y, McDonald BC, et al. Longitudinal assessment of cognitive changes associated with adjuvant treatment for breast cancer: the impact of APOE and smoking. Psychooncology. 2014;23:1382–90. https://doi.org/10.1002/pon.3545.
Koppelmans V, de Groot M, de Ruiter MB, et al. Global and focal white matter integrity in breast cancer survivors 20 years after adjuvant chemotherapy. Hum Brain Mapp. 2014;35:889–99. https://doi.org/10.1002/hbm.22221.
Fardell JE, Zhang J, De Souza R, et al. The impact of sustained and intermittent docetaxel chemotherapy regimens on cognition and neural morphology in healthy mice. Psychopharmacology. 2014;231:841–52. https://doi.org/10.1007/s00213-013-3301-8.
Briones TL, Woods J. Dysregulation in myelination mediated by persistent neuroinflammation: possible mechanisms in chemotherapy-related cognitive impairment. Brain Behav Immun. 2014;35:23–32. https://doi.org/10.1016/j.bbi.2013.07.175.
Fardell JE, Vardy J, Johnston IN. The short and long term effects of docetaxel chemotherapy on rodent object recognition and spatial reference memory. Life Sci. 2013;93:596–604. https://doi.org/10.1016/j.lfs.2013.05.006.
Loo WT, Yip MC, Chow LW, et al. A pilot study: application of hemoglobin and cortisol levels, and a memory test to evaluate the quality of life of breast cancer patients on chemotherapy. Int J Biol Markers. 2013;28:348–56. https://doi.org/10.5301/JBM.5000053.
Collins B, MacKenzie J, Tasca GA, et al. Cognitive effects of chemotherapy in breast cancer patients: a dose-response study. Psychooncology. 2013;22:1517–27. https://doi.org/10.1002/pon.3163.
Freedman RA, Pitcher B, Keating NL, et al. Cognitive function in older women with breast cancer treated with standard chemotherapy and capecitabine on Cancer and Leukemia Group B 49907. Breast Cancer Res Treat. 2013;139:607–16. https://doi.org/10.1007/s10549-013-2562-6.
Mandilaras V, Wan-Chow-Wah D, Monette J, et al. The impact of cancer therapy on cognition in the elderly. Front Pharmacol. 2013. https://doi.org/10.3389/fphar.2013.00048.
Cheung YT, Chui WK, Chan A. Neuro-cognitive impairment in breast cancer patients: pharmacological considerations. Crit Rev Oncol Hematol. 2012;83:99–111. https://doi.org/10.1016/j.critrevonc.2011.09.001.
Koppelmans V, Breteler MMB, Boogerd W, et al. Neuropsychological performance in survivors of breast cancer more than 20 years after adjuvant chemotherapy. J Clin Oncol. 2012;30:1080–6. https://doi.org/10.1200/JCO.2011.37.0189.
Janelsins MC, Mustian KM, Palesh OG, et al. Differential expression of cytokines in breast cancer patients receiving different chemotherapies: implications for cognitive impairment research. Support Care Cancer. 2012;20:831–9. https://doi.org/10.1007/s00520-011-1158-0.
Fremouw T, Fessler CL, Ferguson RJ, Burguete Y. Preserved learning and memory in mice following chemotherapy: 5-fluorouracil and doxorubicin single agent treatment, doxorubicin-cyclophosphamide combination treatment. Behav Brain Res. 2012;226:154–62. https://doi.org/10.1016/j.bbr.2011.09.013.
Fardell JE, Vardy J, Shah JD, Johnston IN. Cognitive impairments caused by oxaliplatin and 5-fluorouracil chemotherapy are ameliorated by physical activity. Psychopharmacology. 2012;220:183–93. https://doi.org/10.1007/s00213-011-2466-2.
Briones TL, Woods J. Chemotherapy-induced cognitive impairment is associated with decreases in cell proliferation and histone modifications. BMC Neurosci. 2011;12:124. https://doi.org/10.1186/1471-2202-12-124.
Long JM, Lee GD, Kelley-Bell B, et al. Preserved learning and memory following 5-fluorouracil and cyclophosphamide treatment in rats. Pharmacol Biochem Behav. 2011;100:205–11. https://doi.org/10.1016/j.pbb.2011.08.012.
Jansen CE, Cooper BA, Dodd MJ, Miaskowski CA. A prospective longitudinal study of chemotherapy-induced cognitive changes in breast cancer patients. Support Care Cancer. 2011;19:1647–56. https://doi.org/10.1007/s00520-010-0997-4.
Aluise CD, Miriyala S, Noel T, et al. 2-Mercaptoethane sulfonate prevents doxorubicin-induced plasma protein oxidation and TNF-α release: implications for the reactive oxygen species-mediated mechanisms of chemobrain. Free Radic Biol Med. 2011;50:1630–8. https://doi.org/10.1016/j.freeradbiomed.2011.03.009.
ElBeltagy M, Mustafa S, Umka J, et al. Fluoxetine improves the memory deficits caused by the chemotherapy agent 5-fluorouracil. Behav Brain Res. 2010;208:112–7. https://doi.org/10.1016/j.bbr.2009.11.017.
Debess J, Riis JØ, Engebjerg MC, Ewertz M. Cognitive function after adjuvant treatment for early breast cancer: a population-based longitudinal study. Breast Cancer Res Treat. 2010;121:91–100. https://doi.org/10.1007/s10549-010-0756-8.
Liedke PER, Reolon GK, Kilpp B, et al. Systemic administration of doxorubicin impairs aversively motivated memory in rats. Pharmacol Biochem Behav. 2009;94:239–43. https://doi.org/10.1016/j.pbb.2009.09.001.
Kesler SR, Bennett FC, Mahaffey ML, Spiegel D. Regional brain activation during verbal declarative memory in metastatic breast cancer. Clin Cancer Res. 2009;15:6665–73. https://doi.org/10.1158/1078-0432.CCR-09-1227.
Boyette-Davis JA, Fuchs PN. Differential effects of paclitaxel treatment on cognitive functioning and mechanical sensitivity. Neurosci Lett. 2009;453:170–4. https://doi.org/10.1016/j.neulet.2009.02.031.
Schilder CM, Eggens PC, Seynaeve C, et al. Neuropsychological functioning in postmenopausal breast cancer patients treated with tamoxifen or exemestane after AC-chemotherapy: cross-sectional findings from the neuropsychological TEAM-side study. Acta Oncol Stockh Swed. 2009;48:76–85. https://doi.org/10.1080/02841860802314738.
Mustafa S, Walker A, Bennett G, Wigmore PM. 5-Fluorouracil chemotherapy affects spatial working memory and newborn neurons in the adult rat hippocampus. Eur J Neurosci. 2008;28:323–30. https://doi.org/10.1111/j.1460-9568.2008.06325.x.
Jansen CE, Dodd MJ, Miaskowski CA, et al. Preliminary results of a longitudinal study of changes in cognitive function in breast cancer patients undergoing chemotherapy with doxorubicin and cyclophosphamide. Psychooncology. 2008;17:1189–95. https://doi.org/10.1002/pon.1342.
Kreukels BPC, van Dam FS, Ridderinkhof KR, et al. Persistent neurocognitive problems after adjuvant chemotherapy for breast cancer. Clin Breast Cancer. 2008;8:80–7. https://doi.org/10.3816/CBC.2008.n.006.
Abraham J, Haut MW, Moran MT, et al. Adjuvant chemotherapy for breast cancer: effects on cerebral white matter seen in diffusion tensor imaging. Clin Breast Cancer. 2008;8:88–91. https://doi.org/10.3816/CBC.2008.n.007.
Ruzich M, Ryan B, Owen C, et al. Prospective evaluation of cognitive function in patients with early breast cancer receiving adjuvant chemotherapy. Asia Pac J Clin Oncol. 2007;3:125–33. https://doi.org/10.1111/j.1743-7563.2007.00109.x.
Hermelink K, Untch M, Lux MP, et al. Cognitive function during neoadjuvant chemotherapy for breast cancer: results of a prospective, multicenter, longitudinal study. Cancer. 2007;109:1905–13. https://doi.org/10.1002/cncr.22610.
Schagen SB, Muller MJ, Boogerd W, et al. Change in cognitive function after chemotherapy: a prospective longitudinal study in breast cancer patients. J Natl Cancer Inst. 2006;98:1742–5. https://doi.org/10.1093/jnci/djj470.
Kreukels BPC, Schagen SB, Ridderinkhof KR, et al. Effects of high-dose and conventional-dose adjuvant chemotherapy on long-term cognitive sequelae in patients with breast cancer: an electrophysiologic study. Clin Breast Cancer. 2006;7:67–78. https://doi.org/10.3816/CBC.2006.n.015.
Lee GD, Longo DL, Wang Y, et al. Transient improvement in cognitive function and synaptic plasticity in rats following cancer chemotherapy. Clin Cancer Res. 2006;12:198–205. https://doi.org/10.1158/1078-0432.CCR-05-1286.
Schagen SB, Muller MJ, Boogerd W, et al. Late effects of adjuvant chemotherapy on cognitive function: a follow-up study in breast cancer patients. Ann Oncol. 2002;13:1387–97. https://doi.org/10.1093/annonc/mdf241.
Schagen SB, van Dam FS, Muller MJ, et al.) Cognitive deficits after postoperative adjuvant chemotherapy for breast carcinoma. Cancer. 1999;85:640–50. doi: https://doi.org/10.1002/(sici)1097-0142(19990201)85:3<640::aid-cncr14>3.0.co;2-g.
Minotti G, Menna P, Salvatorelli E, et al. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185–229. https://doi.org/10.1124/pr.56.2.6.
Bigotte L, Arvidson B, Olsson Y. Cytofluorescence localization of adriamycin in the nervous system. I. Distribution of the drug in the central nervous system of normal adult mice after intravenous injection. Acta Neuropathol 1982;57:121–9. https://doi.org/10.1007/BF00685379.
Kitamura Y, Ushio S, Sumiyoshi Y, et al. N-acetylcysteine attenuates the anxiety-like behavior and spatial cognition impairment induced by doxorubicin and cyclophosphamide combination treatment in rats. Pharmacology. 2021;106:286–93. https://doi.org/10.1159/000512117.
Khadrawy YA, Hosny EN, Mohammed HS. Protective effect of nanocurcumin against neurotoxicity induced by doxorubicin in rat’s brain. Neurotoxicology. 2021;85:1–9. https://doi.org/10.1016/j.neuro.2021.04.003.
Ibrahim SS, Elseoud OGA, Mohamedy MH, et al. Nose-to-brain delivery of chrysin transfersomal and composite vesicles in doxorubicin-induced cognitive impairment in rats: Insights on formulation, oxidative stress and TLR4/NF-kB/NLRP3 pathways. Neuropharmacology. 2021. https://doi.org/10.1016/j.neuropharm.2021.108738.
de la Hoz-Camacho R, Rivera-Lazarín AL, Vázquez-Guillen JM, et al. Cyclophosphamide and epirubicin induce high apoptosis in microglia cells while epirubicin provokes DNA damage and microglial activation at sub-lethal concentrations. EXCLI J. 2022;21:197–212. https://doi.org/10.17179/excli2021-4160.
Jung H, Kim SY, Canbakis Cecen FS, et al. Dysfunction of mitochondrial Ca2+ regulatory machineries in brain aging and neurodegenerative diseases. Front Cell Dev Biol 2020;8:599792. https://doi.org/10.3389/fcell.2020.599792
Saris N-EL, Carafoli E. A historical review of cellular calcium handling, with emphasis on mitochondria. Biochem (Mosc). 2005;70:187–94. doi: https://doi.org/10.1007/s10541-005-0100-9.
Morgan MJ, Liu Z. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21:103–15. https://doi.org/10.1038/cr.2010.178.
Gloire G, Legrand-Poels S, Piette J. NF-κB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol. 2006;72:1493–505. https://doi.org/10.1016/j.bcp.2006.04.011.
Trachootham D, Lu W, Ogasawara MA, et al. Redox regulation of cell survival. Antioxid Redox Signal. 2008;10:1343–74. https://doi.org/10.1089/ars.2007.1957.
Goossens V, Grooten J, De Vos K, Fiers W. Direct evidence for tumor necrosis factor-induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity. Proc Natl Acad Sci USA. 1995;92:8115–9.
Baune B, Camara M-L, Eyre H, et al. Tumour necrosis factor-alpha mediated mechanisms of cognitive dysfunction. Transl Neurosci. 2012;3:263–77. https://doi.org/10.2478/s13380-012-0027-8.
Gutierrez EG, Banks WA, Kastin AJ. Murine tumor necrosis factor alpha is transported from blood to brain in the mouse. J Neuroimmunol. 1993;47:169–76. https://doi.org/10.1016/0165-5728(93)90027-v.
Pan W, Banks WA, Kastin AJ. Permeability of the blood–brain and blood–spinal cord barriers to interferons. J Neuroimmunol. 1997;76:105–11. https://doi.org/10.1016/S0165-5728(97)00034-9.
Keeney JTR, Swomley AM, Förster S, et al. Apolipoprotein A-I: insights from redox proteomics for its role in neurodegeneration. Proteomics Clin Appl. 2013;7:109–22. https://doi.org/10.1002/prca.201200087.
Hyka N, Dayer J-M, Modoux C, et al. Apolipoprotein A-I inhibits the production of interleukin-1β and tumor necrosis factor-α by blocking contact-mediated activation of monocytes by T lymphocytes. Blood. 2001;97:2381–9. https://doi.org/10.1182/blood.V97.8.2381.
Du J, Zhang A, Li J, et al. Doxorubicin-induced cognitive impairment: the mechanistic insights. Front Oncol. 2021;11: 673340. https://doi.org/10.3389/fonc.2021.673340.
Zou JY, Crews FT. TNF alpha potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: neuroprotection by NF kappa B inhibition. Brain Res. 2005;1034:11–24. https://doi.org/10.1016/j.brainres.2004.11.014.
Ongnok B, Khuanjing T, Chunchai T, et al. Donepezil protects against doxorubicin-induced chemobrain in rats via attenuation of inflammation and oxidative stress without interfering with doxorubicin efficacy. Neurother J Am Soc Exp Neurother. 2021;18:2107–25. https://doi.org/10.1007/s13311-021-01092-9.
Fu D, Calvo JA, Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer. 2012;12:104–20. https://doi.org/10.1038/nrc3185.
Dorr RT, Lagel K. Effect of sulfhydryl compounds and glutathione depletion on rat heart myocyte toxicity induced by 4-hydroperoxycyclophosphamide and acrolein in vitro. Chem Biol Interact. 1994;93:117–28. https://doi.org/10.1016/0009-2797(94)90091-4.
Kawanishi M, Matsuda T, Nakayama A, et al. Molecular analysis of mutations induced by acrolein in human fibroblast cells using supF shuttle vector plasmids. Mutat Res. 1998;417:65–73. https://doi.org/10.1016/s1383-5718(98)00093-x.
Brown T, McElroy T, Simmons P, et al. Cognitive impairment resulting from treatment with docetaxel, doxorubicin, and cyclophosphamide. Brain Res. 2021;1760: 147397. https://doi.org/10.1016/j.brainres.2021.147397.
Baudino B, D’agata F, Caroppo P, et al. The chemotherapy long-term effect on cognitive functions and brain metabolism in lymphoma patients. Q J Nucl Med Mol Imaging. 2012;56:559–68.
Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–65. https://doi.org/10.1038/nrc1317.
Kemper EM, van Zandbergen AE, Cleypool C, et al. Increased penetration of paclitaxel into the brain by inhibition of P-glycoprotein. Clin Cancer Res. 2003;9:2849–55.
Fellner S, Bauer B, Miller DS, et al. Transport of paclitaxel (Taxol) across the blood-brain barrier in vitro and in vivo. J Clin Invest. 2002;110:1309–18. https://doi.org/10.1172/JCI15451.
Gangloff A, Hsueh W-A, Kesner AL, et al. Estimation of paclitaxel biodistribution and uptake in human-derived xenografts in vivo with 18F-fluoropaclitaxel. J Nucl Med. 2005;46:1866–71.
van der Veldt AAM, Hendrikse NH, Smit EF, et al. Biodistribution and radiation dosimetry of 11C-labelled docetaxel in cancer patients. Eur J Nucl Med Mol Imaging. 2010;37:1950–8. https://doi.org/10.1007/s00259-010-1489-y.
Huehnchen P, Boehmerle W, Springer A, et al. A novel preventive therapy for paclitaxel-induced cognitive deficits: preclinical evidence from C57BL/6 mice. Transl Psychiatry. 2017;7: e1185. https://doi.org/10.1038/tp.2017.149.
Scripture CD, Figg WD, Sparreboom A. Peripheral neuropathy induced by paclitaxel: recent insights and future perspectives. Curr Neuropharmacol. 2006;4:165–72.
Zhao J, Zuo H, Ding K, et al. Changes in plasma IL-1β, TNF-α and IL-4 levels are involved in chemotherapy-related cognitive impairment in early-stage breast cancer patients. Am J Transl Res. 2020;12:3046–56.
Chang A, Chung N-C, Lawther AJ, et al. The Anti-Inflammatory Drug Aspirin Does Not Protect Against Chemotherapy-Induced Memory Impairment by Paclitaxel in Mice. Front Oncol 2020;10:564965. https://doi.org/10.3389/fonc.2020.564965.
Micheli L, Collodel G, Moretti E, et al. Redox imbalance induced by docetaxel in the neuroblastoma SH-SY5Y cells: a study of docetaxel-induced neuronal damage. Redox Rep Commun Free Radic Res. 2021;26:18–28. https://doi.org/10.1080/13510002.2021.1884802.
Nguyen LD, Ehrlich BE. Cellular mechanisms and treatments for chemobrain: insight from aging and neurodegenerative diseases. EMBO Mol Med. 2020. https://doi.org/10.15252/emmm.202012075.
Nguyen LD, Fischer TT, Ehrlich BE. Pharmacological rescue of cognitive function in a mouse model of chemobrain. Mol Neurodegener. 2021;16:41. https://doi.org/10.1186/s13024-021-00463-2.
Varbiro G, Veres B, Gallyas F, Sumegi B. Direct effect of Taxol on free radical formation and mitochondrial permeability transition. Free Radic Biol Med. 2001;31:548–58. https://doi.org/10.1016/S0891-5849(01)00616-5.
Kidd JF, Pilkington MF, Schell MJ, et al. Paclitaxel affects cytosolic calcium signals by opening the mitochondrial permeability transition pore. J Biol Chem. 2022;277:6504–10. https://doi.org/10.1074/jbc.M106802200.
Carlson K, Ocean AJ. Peripheral neuropathy with microtubule-targeting agents: occurrence and management approach. Clin Breast Cancer. 2011;11:73–81. https://doi.org/10.1016/j.clbc.2011.03.006.
Argyriou AA, Karteri S, Bruna J, et al. Serum neurofilament light chain levels as biomarker of paclitaxel-induced cognitive impairment in patients with breast cancer: a prospective study. Support Care Cancer. 2022;30:1807–14. https://doi.org/10.1007/s00520-021-06509-x.
Swain SM, Arezzo JC. Neuropathy associated with microtubule inhibitors: diagnosis, incidence, and management. Clin Adv Hematol Oncol. 2008;6:455–67.
Boehmerle W, Splittgerber U, Lazarus MB, et al. Paclitaxel induces calcium oscillations via an inositol 1,4,5-trisphosphate receptor and neuronal calcium sensor 1-dependent mechanism. Proc Natl Acad Sci U S A. 2006;103:18356–61. https://doi.org/10.1073/pnas.0607240103.
Boehmerle W, Zhang K, Sivula M, et al. Chronic exposure to paclitaxel diminishes phosphoinositide signaling by calpain-mediated neuronal calcium sensor-1 degradation. Proc Natl Acad Sci. 2007;104:11103–8. https://doi.org/10.1073/pnas.0701546104.
Mo M, Erdelyi I, Szigeti-Buck K, et al. Prevention of paclitaxel-induced peripheral neuropathy by lithium pretreatment. FASEB J. 2012;26:4696–709. https://doi.org/10.1096/fj.12-214643.
da Costa R, Passos GF, Quintão NLM, et al. Taxane-induced neurotoxicity: pathophysiology and therapeutic perspectives. Br J Pharmacol. 2020;177:3127–46. https://doi.org/10.1111/bph.15086.
Gelderblom H, Verweij J, Nooter K, Sparreboom A. Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer. 2001;37:1590–8. https://doi.org/10.1016/s0959-8049(01)00171-x.
Kaye SB. New antimetabolites in cancer chemotherapy and their clinical impact. Br J Cancer. 1998;78:1–7. https://doi.org/10.1038/bjc.1998.747.
Bourke RS, West CR, Chheda G, Tower DB. Kinetics of entry and distribution of 5-fluorouracil in cerebrospinal fluid and brain following intravenous injection in a primate. Cancer Res. 1973;33:1735–46.
Raffa RB, Tallarida RJ. Chemo fog: cancer chemotherapy-related cognitive impairment. New York (NY), Austin (TX): Springer Science+Business Media; Landes Bioscience; 2010.
Alhowail AH, Almogbel YS, Abdellatif AAH, et al. CMF and MET treatment induce cognitive impairment through upregulation of IL-1α in rat brain. Eur Rev Med Pharmacol Sci. 2021;25:4385–93. https://doi.org/10.26355/eurrev_202106_26148.
Morikawa A, Peereboom DM, Smith QR, et al. Clinical evidence for drug penetration of capecitabine and lapatinib uptake in resected brain metastases from women with metastatic breast cancer. J Clin Oncol. 2013;1:514. https://doi.org/10.1200/jco.2013.31.15_suppl.514.
Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4:307–20. https://doi.org/10.1038/nrd1691.
Alhareeri AA, Archer KJ, Fu H, et al. Telomere lengths in women treated for breast cancer show associations with chemotherapy, pain symptoms, and cognitive domain measures: a longitudinal study. Breast Cancer Res. 2020;22:137. https://doi.org/10.1186/s13058-020-01368-6.
Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365:1687–717. https://doi.org/10.1016/S0140-6736(05)66544-0.
Fisher B, Brown AM, Dimitrov NV, et al. Two months of doxorubicin-cyclophosphamide with and without interval reinduction therapy compared with 6 months of cyclophosphamide, methotrexate, and fluorouracil in positive-node breast cancer patients with tamoxifen-nonresponsive tumors: results from the National Surgical Adjuvant Breast and Bowel Project B-15. J Clin Oncol. 1990;8:1483–96. https://doi.org/10.1200/JCO.1990.8.9.1483.
Blum JL, Flynn PJ, Yothers G, et al. Anthracyclines in early breast cancer: the ABC Trials-USOR 06–090, NSABP B-46-I/USOR 07132, and NSABP B-49 (NRG Oncology). J Clin Oncol. 2017;35:2647–55. https://doi.org/10.1200/JCO.2016.71.4147.
Jones S, Holmes FA, O’Shaughnessy J, et al. Docetaxel with cyclophosphamide is associated with an overall survival benefit compared with dxorubicin and cyclophosphamide: 7-year follow-up of US Oncology Research Trial 9735. J Clin Oncol. 2009;27:1177–83. https://doi.org/10.1200/JCO.2008.18.4028.
Peethambaram PP, Hoskin TL, Heins CN, et al. Abstract PD7-05: how 21-gene recurrence score assay is being used to individualize adjuvant chemotherapy recommendations in ER+/HER2 -node positive breast cance: a national cancer data base study. Cancer Res. 2017;77:PD7-PD7-05. doi: https://doi.org/10.1158/1538-7445.SABCS16-PD7-05.
Citron ML, Berry DA, Cirrincione C, et al. Randomized trial of dose-dense versus conventionally scheduled and sequential versus concurrent combination chemotherapy as postoperative adjuvant treatment of node-positive primary breast cancer: first report of Intergroup Trial C9741/Cancer and Leukemia Group B Trial 9741. J Clin Oncol. 2003;21:1431–9. https://doi.org/10.1200/JCO.2003.09.081.
Sparano JA, Wang M, Martino S, et al. Weekly paclitaxel in the adjuvant treatment of breast cancer. N Engl J Med. 2008;358:1663–71. https://doi.org/10.1056/NEJMoa0707056.
Martin M, Pienkowski T, Mackey J, et al. Adjuvant docetaxel for node-positive breast cancer. 2009. https://doi.org/10.1056/NEJMoa043681. Accessed 19 Mar 2021.
Perez EA, Romond EH, Suman VJ, et al. Four-year follow-up of trastuzumab plus adjuvant chemotherapy for operable human epidermal growth factor receptor 2-positive breast cancer: joint analysis of data from NCCTG N9831 and NSABP B-31. J Clin Oncol. 2011;29:3366–73. https://doi.org/10.1200/JCO.2011.35.0868.
von Minckwitz G, Procter M, de Azambuja E, et al. Adjuvant pertuzumab and trastuzumab in early HER2-positive breast cancer. N Engl J Med. 2017;377:122–31. https://doi.org/10.1056/NEJMoa1703643.
Slamon D, Eiermann W, Robert N, et al. Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J Med. 2011;365:1273–83. https://doi.org/10.1056/NEJMoa0910383.
Tolaney SM, Barry WT, Dang CT, et al. Adjuvant paclitaxel and trastuzumab for node-negative, HER2-positive breast cancer. N Engl J Med. 2015;372:134–41. https://doi.org/10.1056/NEJMoa1406281.
Gianni L, Pienkowski T, Im Y-H, et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): a randomised multicentre, open-label, phase 2 trial. Lancet Oncol 2012;13:25-32. https://doi.org/10.1016/S1470-2045(11)70336-9.
National Comprehensive Cancer Network (NCCN). NCCN clinical practice guidelines in oncology. Breast cancer. Version 3. 2021.
Eide S, Feng Z-P. Doxorubicin chemotherapy-induced “chemo-brain”: meta-analysis. Eur J Pharmacol. 2020;881: 173078. https://doi.org/10.1016/j.ejphar.2020.173078.
Zhang H, Li P, Liu T, et al. Focal white matter microstructural alteration after anthracycline-based systemic treatment in long-term breast cancer survivors: a structural magnetic resonance imaging study. Brain Imaging Behav. 2021. https://doi.org/10.1007/s11682-021-00551-3.
Keetile NM, Osuch E, Lentoor AG. Chemotherapy-related subjective cognitive impairment in breast cancer patients in semi-rural South Africa. Health SA. 2021;26:1605. https://doi.org/10.4102/hsag.v26i0.1605.
van Dam FS, Schagen SB, Muller MJ, et al. Impairment of cognitive function in women receiving adjuvant treatment for high-risk breast cancer: high-dose versus standard-dose chemotherapy. J Natl Cancer Inst. 1998;90:210–8. https://doi.org/10.1093/jnci/90.3.210.
Buwalda B, Schagen SB. Is basic research providing answers if adjuvant anti-estrogen treatment of breast cancer can induce cognitive impairment? Life Sci. 2013;93:581–8. https://doi.org/10.1016/j.lfs.2012.12.012.
Wagner LI, Gray RJ, Sparano JA, et al. Patient-reported cognitive impairment among women with early breast cancer randomly assigned to endocrine therapy alone versus chemoendocrine therapy: results from TAILORx. J Clin Oncol. 2020;38:1875–86. https://doi.org/10.1200/JCO.19.01866.
Wu LM, Amidi A. Cognitive impairment following hormone therapy: current opinion of research in breast and prostate cancer patients. Curr Opin Support Palliat Care. 2017;11:38–45. https://doi.org/10.1097/SPC.0000000000000251.
Karschnia P, Parsons MW, Dietrich J. Pharmacologic management of cognitive impairment induced by cancer therapy. Lancet Oncol. 2019;20:e92-102. https://doi.org/10.1016/S1470-2045(18)30938-0.
Wefel JS, Kesler SR, Noll KR, Schagen SB. Clinical characteristics, pathophysiology, and management of noncentral nervous system cancer-related cognitive impairment in adults. CA Cancer J Clin. 2015;65:123–38. https://doi.org/10.3322/caac.21258.
Kohli S, Fisher SG, Tra Y, et al. The effect of modafinil on cognitive function in breast cancer survivors. Cancer. 2009;115:2605–16. https://doi.org/10.1002/cncr.24287.
Lundorff L, Jønsson B, Sjøgren P. Modafinil for attentional and psychomotor dysfunction in advanced cancer: a double-blind, randomised, cross-over trial. Palliat Med. 2009;23:731–8. https://doi.org/10.1177/0269216309106872.
Mar Fan HG, Clemons M, Xu W, et al. A randomised, placebo-controlled, double-blind trial of the effects of d-methylphenidate on fatigue and cognitive dysfunction in women undergoing adjuvant chemotherapy for breast cancer. Support Care Cancer. 2008;16:577–83. https://doi.org/10.1007/s00520-007-0341-9.
Lower EE, Fleishman S, Cooper A, et al. Efficacy of dexmethylphenidate for the treatment of fatigue after cancer chemotherapy: a randomized clinical trial. J Pain Symptom Manage. 2009;38:650–62. https://doi.org/10.1016/j.jpainsymman.2009.03.011.
Escalante CP, Meyers C, Reuben JM, et al. A randomized, double-blind, 2-period, placebo-controlled crossover trial of a sustained-release methylphenidate in the treatment of fatigue in cancer patients. Cancer J. 2014;20:8–14. https://doi.org/10.1097/PPO.0000000000000018.
Santarelli L, Saxe M, Gross C, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 2003;301:805–9. https://doi.org/10.1126/science.1083328.
Jacobson SA, Sabbagh MN. Donepezil: potential neuroprotective and disease-modifying effects. Expert Opin Drug Metab Toxicol. 2008;4:1363–9. https://doi.org/10.1517/17425255.4.10.1363.
Lawrence JA, Griffin L, Balcueva EP, et al. A study of donepezil in female breast cancer survivors with self-reported cognitive dysfunction 1 to 5 years following adjuvant chemotherapy. J Cancer Surviv. 2016;10:176–84. https://doi.org/10.1007/s11764-015-0463-x.
Cauli O. Oxidative stress and cognitive alterations induced by cancer chemotherapy drugs: a scoping review. Antioxidants (Basel). 2021;10:1116. https://doi.org/10.3390/antiox10071116.
Chtourou Y, Gargouri B, Kebieche M, Fetoui H. Naringin abrogates cisplatin-induced cognitive deficits and cholinergic dysfunction through the down-regulation of AChE expression and iNOS signaling pathways in hippocampus of aged rats. J Mol Neurosci. 2015;56:349–62. https://doi.org/10.1007/s12031-015-0547-0.
Barton DL, Burger K, Novotny PJ, et al. The use of Ginkgo biloba for the prevention of chemotherapy-related cognitive dysfunction in women receiving adjuvant treatment for breast cancer, N00C9. Support Care Cancer. 2013;21:1185–92. https://doi.org/10.1007/s00520-012-1647-9.
O’Shaughnessy JA, Vukelja SJ, Holmes FA, et al. Feasibility of quantifying the effects of epoetin alfa therapy on cognitive function in women with breast cancer undergoing adjuvant or neoadjuvant chemotherapy. Clin Breast Cancer. 2005;5:439–46. https://doi.org/10.3816/cbc.2005.n.002.
Fan HGM, Park A, Xu W, et al. The influence of erythropoietin on cognitive function in women following chemotherapy for breast cancer. Psychooncology. 2009;18:156–61. https://doi.org/10.1002/pon.1372.
Alhowail A, Chigurupati S. Research advances on how metformin improves memory impairment in “chemobrain.” Neural Regen Res. 2021;17:15–9. https://doi.org/10.4103/1673-5374.314284.
Jacobsen PB, Garland LL, Booth-Jones M, et al. Relationship of hemoglobin levels to fatigue and cognitive functioning among cancer patients receiving chemotherapy. J Pain Symptom Manage. 2004;28:7–18. https://doi.org/10.1016/j.jpainsymman.2003.11.002.
Wu Y-Q, Dang R-L, Tang M-M, et al. Long chain omega-3 polyunsaturated fatty acid supplementation alleviates doxorubicin-induced depressive-like behaviors andnNeurotoxicity in rats: involvement of oxidative stress and neuroinflammation. Nutrients. 2016;8:243. https://doi.org/10.3390/nu8040243.
Clare L, Woods RT, Moniz Cook ED, et al. Cognitive rehabilitation and cognitive training for early-stage Alzheimer’s disease and vascular dementia. Cochrane Database Syst Rev. 2003;(4):CD003260. https://doi.org/10.1002/14651858.CD003260.
Di Iulio F, Cravello L, Shofany J, et al. Neuropsychological disorders in non-central nervous system cancer: a review of objective cognitive impairment, depression, and related rehabilitation options. Neurol Sci. 2019;40:1759–74. https://doi.org/10.1007/s10072-019-03898-0.
Ferguson RJ, Ahles TA, Saykin AJ, et al. Cognitive-behavioral management of chemotherapy-related cognitive change. Psychooncology. 2007;16:772–7. https://doi.org/10.1002/pon.1133.
Ferguson RJ, McDonald BC, Rocque MA, et al. Development of CBT for chemotherapy-related cognitive change: results of a waitlist control trial. Psychooncology. 2012;21:176–86. https://doi.org/10.1002/pon.1878.
Ercoli LM, Castellon SA, Hunter AM, et al. Assessment of the feasibility of a rehabilitation intervention program for breast cancer survivors with cognitive complaints. Brain Imaging Behav. 2013;7:543–53. https://doi.org/10.1007/s11682-013-9237-0.
Ercoli LM, Petersen L, Hunter AM, et al. Cognitive rehabilitation group intervention for breast cancer survivors: results of a randomized clinical trial. Psychooncology. 2015;24:1360–7. https://doi.org/10.1002/pon.3769.
Park J-H, Jung YS, Kim KS, Bae SH. Effects of compensatory cognitive training intervention for breast cancer patients undergoing chemotherapy: a pilot study. Support Care Cancer. 2017;25:1887–96. https://doi.org/10.1007/s00520-017-3589-8.
Von Ah D, Carpenter JS, Saykin A, et al. Advanced cognitive training for breast cancer survivors: a randomized controlled trial. Breast Cancer Res Treat. 2012;135:799–809. https://doi.org/10.1007/s10549-012-2210-6.
Kesler S, Hadi Hosseini SM, Heckler C, et al. Cognitive training for improving executive function in chemotherapy-treated breast cancer survivors. Clin Breast Cancer. 2013;13:299–306. https://doi.org/10.1016/j.clbc.2013.02.004.
Poppelreuter M, Weis J, Bartsch HH. Effects of specific neuropsychological training programs for breast cancer patients after adjuvant chemotherapy. J Psychosoc Oncol. 2009;27:274–96. https://doi.org/10.1080/07347330902776044.
Alvarez J, Meyer FL, Granoff DL, Lundy A. The effect of EEG biofeedback on reducing postcancer cognitive impairment. Integr Cancer Ther. 2013;12:475–87. https://doi.org/10.1177/1534735413477192.
Fitzpatrick TR, Edgar L, Holcroft C. Assessing the relationship between physical fitness activities, cognitive health, and quality of life among older cancer survivors. J Psychosoc Oncol. 2012;30:556–72. https://doi.org/10.1080/07347332.2012.703768.
Crowgey T, Peters KB, Hornsby WE, et al. Relationship between exercise behavior, cardiorespiratory fitness, and cognitive function in early breast cancer patients treated with doxorubicin-containing chemotherapy: a pilot study. Appl Physiol Nutr Metab. 2014;39:724–9. https://doi.org/10.1139/apnm-2013-0380.
Mustian KM, Janelsins MC, Peppone LJ, et al. EXCAP exercise effects on cognitive impairment and inflammation: a URCC NCORP RCT in 479 cancer patients. J Clin Oncol. 2015;33:9504. https://doi.org/10.1200/jco.2015.33.15_suppl.9504.
Wong-Goodrich SJE, Pfau ML, Flores CT, et al. Voluntary running prevents progressive memory decline and increases adult hippocampal neurogenesis and growth factor expression after whole-brain irradiation. Cancer Res. 2010;70:9329–38. https://doi.org/10.1158/0008-5472.CAN-10-1854.
Cooke GE, Wetter NC, Banducci SE, et al. Moderate physical activity mediates the association between white matter lesion volume and memory recall in breast cancer Ssrvivors. PLoS ONE. 2016;11: e0149552. https://doi.org/10.1371/journal.pone.0149552.
Winocur G, Wojtowicz JM, Huang J, Tannock IF. Physical exercise prevents suppression of hippocampal neurogenesis and reduces cognitive impairment in chemotherapy-treated rats. Psychopharmacology. 2014;231:2311–20. https://doi.org/10.1007/s00213-013-3394-0.
Edelstein K, Bernstein LJ. Cognitive dysfunction after chemotherapy for breast cancer. J Int Neuropsychol Soc. 2014;20:351–6. https://doi.org/10.1017/S1355617714000149.
Ahles TA, Saykin A. Cognitive effects of standard-dose chemotherapy in patients with cancer. Cancer Invest. 2001;19:812–20. https://doi.org/10.1081/cnv-100107743.
Ahles TA, Root JC, Ryan EL. Cancer- and cancer treatment-associated cognitive change: an update on the state of the science. J Clin Oncol. 2012;30:3675–86. https://doi.org/10.1200/JCO.2012.43.0116.
Vardy J. Tannock I Cognitive function after chemotherapy in adults with solid tumours. Crit Rev Oncol Hematol. 2007;63:183–202. https://doi.org/10.1016/j.critrevonc.2007.06.001.
Yao C, Rich JB, Tirona K, Bernstein LJ. Intraindividual variability in reaction time before and after neoadjuvant chemotherapy in women diagnosed with breast cancer. Psychooncology. 2017;26:2261–8. https://doi.org/10.1002/pon.4351.
Vardy JL, Dhillon HM, Pond GR, et al. Cognitive function in patients with colorectal cancer who do and do not receive chemotherapy: a prospective, longitudinal, controlled study. J Clin Oncol. 2015;33:4085–92. https://doi.org/10.1200/JCO.2015.63.0905.
Zer A, Pond GR, Razak ARA, et al. Association of neurocognitive deficits with radiotherapy or chemoradiotherapy for patients with head and neck Cancer. JAMA Otolaryngol Neck Surg. 2018;144:71–9. https://doi.org/10.1001/jamaoto.2017.2235.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
Giovana R. Onzi received a research fellowship from the National Council for Scientific and Technological Development (CNPq #150654/2020-0). Adriana R. Pohlmann received a research grant from the National Council for Scientific and Technological Development (CNPq # 305343/2019-0). Silvia S. Guterres received a research grant and fellowship from FAPERGS (#18.2551.0000513-4).
Conflict of interest
Giovana R. Onzi works for Astrazeneca Brazil as a full-time employee. This manuscript was elaborated while the author was still working as a postdoctoral researcher at Federal University of Rio Grande do Sul. Paula R. Pohlmann has received consulting fees or honorarium from Perthera, Immunonet, Sirtex, CARIS, OncoPLex Diagnostics, Pfizer, Heron, Puma, BOLT, and Abbvie; fees for participation in review activities such as data monitoring boards from SEAGEN; payment for lectures including service on speakers bureaus from Dava Oncology, United Medical lecture, ION Oral Oncolytics Lecture, and Genentech/Roche; and holds stock/stock options in Immunonet. Daniela D. Rosa has received consulting fees or honorarium from Roche, Novartis, AstraZeneca, and Lilly; fees for participation in review activities such as data monitoring boards from Daiichi-Sankyo, Lilly, and Teva; and payment for lectures including service on speakers bureaus from Novartis, Pfizer, and AstraZeneca. Nathalia D’Agustini, Solange C. Garcia, Silvia S. Guterres, and Adriana R. Pohlmann have no conflicts of interest to declare.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Author contributions
All authors participated in the conceptualization of the manuscript. Giovana R. Onzi and Nathalia D’Agustini wrote the manuscript. Giovana R. Onzi and Adriana R. Pohlmann performed the data curation and analysis. All authors reviewed the final manuscript. Silvia S. Guterres, Daniela D. Rosa, and Adriana R. Pohlmann supervised the work and were responsible for funding acquisition.
Supplementary Information
Below is the link to the electronic supplementary material.
40264_2022_1182_MOESM2_ESM.xlsx
Supplementary file2 Electronic Supplementary Material 2 Full list of documents obtained in the literature survey performed on CRCI in breast cancer (XLSX 39 KB)
Rights and permissions
About this article

Cite this article
Onzi, G.R., D’Agustini, N., Garcia, S.C. et al. Chemobrain in Breast Cancer: Mechanisms, Clinical Manifestations, and Potential Interventions. Drug Saf 45, 601–621 (2022). https://doi.org/10.1007/s40264-022-01182-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-022-01182-3