
Abstract
Background
Since the approval and availability of the first biosimilar in 2015 in the United States (US), evidence regarding the post-marketing safety of cancer supportive care biosimilars remains limited.
Objective
The aim was to explore the adverse event (AE) reporting patterns and detect disproportionate reporting signals for cancer supportive care biosimilars in the US compared to their originator biologics.
Methods
The US Food and Drug Administration Adverse Event Reporting System database (January 1, 2004–March 31, 2020) was used to identify AE reports for filgrastim, pegfilgrastim, and epoetin alpha by type of product (originator biologics vs. biosimilars) and report characteristics. Plots of AE reports against years were used to reveal the reporting patterns. Disproportionality analyses using reporting odds ratios (RORs) were conducted to detect differences in serious and specific AEs between studied drugs and all other drugs. Breslow–Day tests were used to determine homogeneity between the originator biologic–biosimilar pair RORs for the same AE.
Results
Total numbers of AEs for all studied biosimilars increased after marketing. More AE reports were from female patients for all of the studied drugs. More AEs for originator biologics and filgrastim biosimilar were reported by health professionals, while the highest proportion of reports came from consumers for pegfilgrastim and epoetin alpha biosimilars (29% and 44.1%, respectively). Signals of disproportionate reporting in serious AEs were detected for a pegfilgrastim biosimilar (Fulphila®) compared to its originator biologic.
Conclusion
Our findings support the similarity in the signals of disproportionate reporting between cancer supportive care originator biologics and biosimilars, except for Fulphila®.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
An increase in the proportion of adverse event (AE) reports for cancer supportive care biosimilar products has been observed since their market launch. |
Heterogeneity in reporting patterns existed between studied originator biologic and corresponding biosimilar products. |
Although no signals of disproportionate reporting were detected between cancer supportive care biosimilar and originator biologic products for filgrastim and epoetin alpha, the signal of disproportionate reporting for serious AEs with the pegfilgrastim biosimilar Fulphila® was different compared to that of its corresponding biologic product, Neulasta®. |
1 Introduction
Chemotherapy-induced neutropenia (CIN) is one of the most frequently reported adverse events (AEs). It is observed in 0.8% of patients receiving chemotherapy and 42–72% of patients receiving myelosuppressive chemotherapy [1]. This complication puts cancer patients at significant risk for morbidity and mortality and leads to substantial medical costs for hospitalization. In 2012, CIN accounted for $2.3 billion for adult hospitalization and $439 million for children in the United States (US) [2]. In addition, chemotherapy-induced anemia (CIA) is a common and serious condition presenting in 67–83% of patients undergoing myelosuppressive chemotherapy [3, 4]. CIA results in reduced functional activity and quality of life, causing fatigue, dyspnea, and nausea [5, 6].
Granulocyte colony-stimulating factors (G-CSFs) and erythropoiesis-stimulating agents (ESAs) are important supportive care biologics in oncology for treatment and prevention of CIN and CIA, respectively. Biologics tend to be at the forefront of medical advances today, representing one of the fastest growing sectors of cancer treatment. However, their high prices place a huge burden on patients and healthcare systems globally [7]. According to a cost-effectiveness study in 2017, the cost of Neupogen® (filgrastim, a short-acting G-CSF), used to prevent CIN for a single chemotherapy cycle, including medication cost and cost of administration, ranged from $324 (1-day regimen) to $4559 (14-day regimen) per patient [8]. In the US, the Biologics Price Competition and Innovation Act of 2009 created an abbreviated licensure pathway for biologic products that are demonstrated to be biosimilar or interchangeable with a Food and Drug Administration (FDA)-approved biologic product [9]. A biosimilar is a biologic that is highly similar to the reference biologic, with no clinically significant differences in purity, safety, and efficacy [10]. An interchangeable biologic product is a biosimilar that may be substituted for a reference biologic by a pharmacist, subject to local state policies. Different from European countries, in the US, the FDA requires demonstration of interchangeability supported by clinical pharmacokinetic/pharmacodynamic, safety, and immunogenicity studies coupled with post-marketing data [11]. To date, there is no interchangeable biosimilar approved in the US [12].
Biosimilars are developed to reduce healthcare costs and increase access to medications. In Europe, 62% of the ESA market is dominated by biosimilars [13]. A simulation study based on the number of patients receiving chemotherapy in European G5 countries found that replacing epoetin alpha originators by its biosimilars resulted in €110 million of savings [14]. The first biosimilar in the US, Zarxio® (filgrastim-sndz), was approved in March 6, 2015 [15]. A recent comprehensive systematic review and meta-analysis demonstrated no significant difference in efficacy and safety profiles between cancer supportive biologics (including filgrastim, pegfilgrastim, and epoetin alpha) and their corresponding biosimilars [16]. However, most of these studies were conducted in Europe, where the biosimilars were approved and marketed long before. Research to evaluate the post-marketing safety profiles of biosimilars in the US is very limited. To address this important knowledge gap in post-marketing surveillance of biosimilars marketed in the US, this study described AE reporting patterns and generated signals of disproportionate reporting of cancer supportive care biosimilars compared to their biologics using the FDA Adverse Event Reporting System (FAERS) database.
2 Methods
We conducted retrospective analyses of AE reports from the public version of the FAERS database for three supportive cancer care medications (filgrastim, pegfilgrastim, and epoetin alpha) approved and marketed in the US. The analyses included comparisons of AE reports between biologic originators and corresponding biosimilars.
2.1 Data Source
The FAERS database is a public, spontaneous reporting system receiving AE reports from both US and non-US regions [17]. The reports are submitted voluntarily from healthcare providers and consumers and mandatorily from pharmaceutical companies due to FDA regulations [17]. This database aims to support the FDA’s post-marketing surveillance program for drug and therapeutic biologic products [17]. The data files are published quarterly, containing seven files: DEMOyyQq.TXT (patient demographic and administrative information), DRUGyyQq.TXT (drug/biologic information), REACyyQq.TXT (Medical Dictionary for Regulatory Activities [MedDRA®] terms coded for AEs), OUTCyyQq.TXT (patient outcomes and seriousness), RPSRyyQq.TXT (reporting sources), THERyyQq.TXT (drug therapy start date and end date), and INDIyyQq.TXT (MedDRA® terms coded for the indications of reported drugs). We retrieved the FAERS data files from January 1, 2004 to March 31, 2020. Duplicate reports were removed from the analyses, using the FDA's recommendation of adopting the most recent case number for the same report [18]. This study was granted exemption from human subjects research by the Auburn University Institutional Review Board.
2.2 Originator Biologic and Biosimilar Product Identification
Information on the US FDA-approved filgrastim, pegfilgrastim, and epoetin alpha originator biologics and biosimilars were collected using the National Drug Code (NDC) Directory and Purple Book Database [19, 20]. Each biosimilar’s and corresponding originator biologic’s proprietary and nonproprietary names, manufacturers, labelers, and approval and first marketed dates in the US were extracted (Table 1).
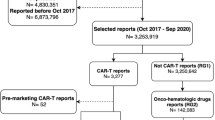
From the FAERS DRUGyyQq.TXT file, we identified AE reports that had originator biologics and biosimilar products in our list of study drugs (Table 1). Records were retained irrespective of their role in the mentioned AEs, such as primary suspect or secondary suspect drugs, or concomitant use or interacting. Identification of relevant reports for a drug included text string searches containing the proprietary names as well as their abbreviations and potential misspellings. Searches for drug names also involved their nonproprietary name using FDA recommended nonproprietary naming guidance [21]. This naming convention requires a biologic product to have a nonproprietary name consisting of a core name along with a four-letter suffix (e.g., filgrastim-sndz for Zarxio®). Next, we retained those reports with correct proprietary/nonproprietary name–manufacturer combination (e.g., ‘Neupogen®-Amgen’; ‘Zarxio®/filgrastim-sndz–Sandoz’; ‘Nivestym®/filgrastim-aafi–Pfizer’) to avoid originator biologic versus biosimilar product misclassification [22]. We excluded reports without any information or unmatched information of manufacturers in the manufacturer name column of the DEMOyyQq.TXT file. However, we included reports with missing manufacturer names or manufacturer names incorrectly recoded as ‘FDA’ while the correct manufacturer name was included in the drug name column (about 10% of filgrastim reports and less than 1% of reports for pegfilgrastim and epoetin alpha). Next, AE reports from countries other than the US were removed (about 22.25%) due to this study’s focus on the US market. In addition, reports with event dates before the marketed dates (Table 1) for each product were excluded to avoid misclassification. For example, all reports for Zarxio® were excluded when the event date mentioned in the DEMOyyQq.TXT file showed a date prior to the first marketed date of Zarxio® (September 3, 2015). A flowchart of the AE report selection process is depicted in Fig. 1. Detailed inclusion and exclusion criteria regarding the identification of studied biologics and biosimilars reports in the FAERS are provided in electronic supplementary material Table 1.

Flowchart of the FAERS AE reports selection process for originator biologics and biosimilars. AE adverse event, ESM electronic supplementary material, FAERS Food and Drug Administration Adverse Event Reporting System, PT Preferred Term
2.3 Adverse Event (AE) Identification
The AEs and medication errors in FAERS are coded using the standard MedDRA® terminology [23]. Serious AEs have been pre-defined by the FDA and categorized as death, disability, congenital anomaly, life-threatening events that required hospitalization or intervention to prevent permanent impairment or damage, and other serious events [24]. These serious AEs were identified from the outcome column in the OUTCyyQq.TXT file for each of the products. AEs that did not fall under this definition were categorized as non-serious AEs. In addition, we identified the following specific AEs for studied products based off of systematic reviews on randomized controlled trials (RCTs) and cohort studies as well as product label information: filgrastim (bone pain), pegfilgrastim (bone pain), and epoetin alpha (cardiovascular events, nausea/vomiting) [16, 25,26,27,28,29,30,31,32,33]. To identify these specific AEs, the MedDRA® version 23.0 was used [34]. If our AEs of interest fell into a broader level term of the high level term (HLT), then all Preferred Terms (PTs) included in the level were used to define the AEs. For example, ‘nausea and vomiting symptoms’ is an HLT in MedDRA®, under which there are 20 PTs. In order to identify nausea and vomiting reports for a product, we searched for all the 20 PTs listed in MedDRA® from the REACyyQq.TXT files of the FAERS database. A similar method was used to identify reports related to bone pain and cardiovascular AEs. A list of the PTs used to define these specific AEs is provided in electronic supplementary material Table 2.
2.4 Data Analysis
First, we identified the total number of FAERS AEs reported during the study period for studied originator biologic and biosimilar products. Total number of AE reports for biosimilars and corresponding biologics were then plotted against year (Figs. 2, 3, 4). AE reports were further categorized by patients’ sex (male, female, and not specified/missing), age (0–17 years, 18–64 years, 65–85 years, 85 + years, and missing), and type of reporter (physician, pharmacist, other health professional, lawyer, consumer, and missing). Chi-squared tests were used to compare the proportions of AE reports across these report characteristics for each originator biologic-biosimilar pair.

Reporting pattern for filgrastim originator biologics vs. biosimilars (January 1, 2004–March 31, 2020); first biosimilar (Zarxio®) marketed date for filgrastim is September 3, 2015

Reporting pattern for pegfilgrastim originator biologics vs. biosimilars (January 1, 2004–March 31, 2020); first biosimilar (Fulphila®) marketed date for pegfilgrastim is July 9, 2018

Reporting pattern for epoetin alpha originator biologics vs. biosimilars (January 1, 2004–March 31, 2020); first biosimilar (Retacrit®) marketed date for epoetin alpha is June 18, 2018
Signal‐detection algorithms (SDAs) such as reporting odds ratio (ROR) and proportional reporting ratio (PRR) have been applied to spontaneously reported data (such as the FAERS) in order to monitor, prioritize, and identify new product safety signals [35]. The ROR estimates the odds of reporting the event of interest in those exposed to each target drug of interest divided by the odds of reporting the event of interest in those not exposed to the drug of interest [36]. Disproportionality analyses were carried out based on total reports from the entire study duration using RORs with related 95% confidence intervals (CIs) to compare reporting rates of serious and specific AEs between an originator biologic/biosimilar (index drugs) and other drugs (reference drugs) in the FAERS database. For example, the ROR for serious AEs with filgrastim biologics was calculated as the odds of serious AEs reported with filgrastim biologics compared to the odds of the same serious AEs reported with all other drugs in the FAERS during the same observation period [36]. A potential signal in the form of a significant disproportionality was defined as the lower bound of the 95% CI exceeding 1 [37]. This SDA was performed for each studied originator biologic and biosimilar product that had a minimum of three combinations in the presence of the studied drug and AE [38]. If either of the 95% CIs of the RORs for an originator biologic–biosimilar pair did not include 1 (could be larger or smaller than 1), a further Breslow–Day statistic was used to identify the homogeneity of the biologic’s and corresponding biosimilar drug’s RORs for the same type of AE [22]. A p value less than 0.05 from the Breslow–Day test indicated a significant difference in RORs for a specific event between an originator biologic and the corresponding biosimilar. Since the biologics had been approved and marketed several years before the marketed dates of corresponding biosimilars, sensitivity analyses were conducted to analyze RORs of AE reports for all studied products starting from the marketed date of the first marketed biosimilar instead of the entire study duration used in the main analysis. A second sensitivity analysis was conducted to limit the identification of AE reports to only primary or secondary suspect drug exposure, and excluded concomitant use or interacting drugs. SAS (version 9.4; SAS Institute, Cary, NC, USA) was used to conduct all statistical analyses at p < 0.05.
3 Results
3.1 Reporting Patterns of Studied Biologics and Biosimilars in FAERS
Figures 2, 3, and 4 describe the patterns of FAERS AE reports for filgrastim, pegfilgrastim, and epoetin alpha biologics and biosimilars, respectively. The number of reports for filgrastim increased significantly starting in 2015 (57.1% of total reports for filgrastim). Filgrastim biosimilar (Zarxio® and Nivestym®) reports appeared after the products were marketed in 2015 and 2018, respectively, increasing gradually over time (Fig. 2). Total reports for pegfilgrastim increased dramatically in 2016 and reached a peak in 2018 (Fig. 3). Pegfilgrastim biosimilar reports (Udenyca® and Fulphila®) accounted for 0.13%, 0.92%, and 1.16% of total pegfilgrastim reports in 2018, 2019, and 2020, respectively, since their availability in 2018. Epoetin alpha biosimilar reports accounted for 0.16%, 13.16%, and 46.94% of total epoetin alpha reports in 2018, 2019, and 2020, respectively, after the first marketed date of Retacrit® in 2018 (Fig. 4).
3.2 Characteristics of AE Reports
Table 2 summarizes the characteristics of the FAERS AE reports for filgrastim, pegfilgrastim, and epoetin alpha originator biologics and biosimilars. Overall, 47,283 AE reports for these products were identified in the US, of which a total of 481 reports (1.02%) belonged to the biosimilars. Among the 99,727 PTs reported in these 47,283 AE reports, 1174 PTs (1.17%) were identified from AE reports for biosimilars. The proportions of reports from females were higher than from males, and this pattern was consistent for all studied originator biologic and biosimilar products. However, more than half (56%) of the pegfilgrastim biosimilar reports had missing information on sex. The unadjusted associations of reports between patient sex and drug type (biologic vs. biosimilar) were found to be significant (p < 0.0001 and p = 0.001, respectively) for pegfilgrastim and epoetin alpha, but not for filgrastim (p = 0.421). At least half of the originator biologic and biosimilar reports had missing information with respect to patients’ age, except for epoetin alpha biosimilar reports (23.4% of missing information on age). No association between patient’s age and drug type was observed for filgrastim reports (p = 0.30), but the unadjusted associations were significant for pegfilgrastim and epoetin alpha originator biologics and biosimilars (p = 0.004 and < 0.0001, respectively). For all the originator biologics, physician reports constituted the highest proportion of total reports, while the reports for biosimilars had noticeable differences in reporter type. Most of the filgrastim biosimilar reports are from other health professionals (43%), whereas for both pegfilgrastim and epoetin alpha, the highest proportion of reports (29% and 44.1%, respectively) came from consumers. Significant associations (p < 0.0001) were found between reporter type and drug type for all three pairs of studied drugs.
3.3 RORs of Serious and Specific AEs for Studied Originator Biologic and Biosimilar Products
Tables 3, 4, and 5 exhibit the RORs of serious (vs. non-serious) and specific (vs. all other) AEs for the studied originator biologic and biosimilar products. In the main analyses, the ROR of serious (vs. non-serious) AEs for Neupogen® (ROR 1.15, 95% CI 1.10–1.19) was significantly different from that for the individual biosimilar Zarxio® (ROR 0.92, 95% CI 0.77–1.10) as well as that for the combination of both biosimilar products Zarxio® and Nivestym® (ROR 0.96, 95% CI 0.82–1.13), but it was not statistically different from that for the individual biosimilar Nivestym® (ROR 1.27, 95% CI 0.81–1.99). The RORs of bone pain were higher for all three filgrastim originator biologic and biosimilar products compared to all other drugs, and no difference was observed between Neupogen® with corresponding individual or combined biosimilar products (all p > 0.05 for Breslow–Day tests). The ROR of serious AEs for Neulasta® (ROR 0.113, 95% CI 0.111–0.115) was significantly different from that for the individual biosimilars Fulphila® (ROR 6.72, 95% CI 3.13–14.41) and Udenyca® (ROR 0.49, 95% CI 0.35–0.69), as well as that for the combination of both biosimilar products (ROR 1.11, 95% CI 0.84–1.46). Again, the RORs of bone pain were higher for both pegfilgrastim originator biologic and biosimilar products compared to all other drugs, while no significant difference was detected between Neulasta® with Udenyca® (p > 0.05 for Breslow–Day tests). Although the ROR of serious AEs for epoetin alpha originator biologics (ROR 0.88, 95% CI 0.86–0.90) was significantly different from that for the corresponding biosimilar (ROR 0.51, 95% CI 0.39–0.66), no difference was detected between the RORs of nausea/vomiting (ROR 0.72, 95% CI 0.65–0.79 vs. ROR 0.58, 95% CI 0.18–1.8) and cardiovascular events (ROR 1.2, 95% CI 1.08–1.34 vs. ROR 1.14, 95% CI 0.37–3.57). Sensitivity analyses were conducted by restricting all reports to the marketed date of the first biosimilar in each originator biologic–biosimilar comparison forward (#1), and by restricting reports to only primary or secondary suspect drug exposure (#2). Results from sensitivity analysis #1 showed that the ROR of serious (vs. non-serious) AEs for Neupogen® changed magnitude and was significantly different compared to that for individual biosimilars Zarxio® and Nivestym® as well as that for the combination of Zarxio® and Nivestym® (p < 0.05). For Neulasta®, the RORs of bone pain became significantly different compared to biosimilar Udenyca® (p < 0.05). The ROR of serious AEs for epoetin alpha biologics was no longer statistically different from that for the biosimilar Retacrit® (p > 0.05). Results from the rest of parallel sensitivity analysis #1 remained similar to findings from the main analyses. Results from sensitivity analysis #2 showed that the RORs of bone pain became different between Neulasta® and Udenyca® (p < 0.05, electronic supplementary material Table 4). Results from the rest of parallel sensitivity analysis #2 remained similar to findings from the main analyses.
4 Discussion
To the best of our knowledge, this study provided the most up-to-date post-marketing pharmacovigilance evidence in AE reporting of supportive care originator biologics and corresponding biosimilars marketed in the US. Given the limited post-marketing data for these newly approved biosimilar products in the US, especially for pegfilgrastim biosimilars and epoetin alpha, our study focused on the patterns of AE reporting and the detection of disproportionality signals by comparing the reports of serious (vs. non-serious) AEs and known specific AEs between these originator biologic–biosimilar pairs. Overall, we observed an increased proportion of AE reports for biosimilar products since their market launch. We found heterogeneity in reporting patterns between studied originator biologic and corresponding biosimilar products. Most importantly, we detected a potential signal of disproportionate reporting of AEs between originator biologic and corresponding biosimilar products. Specifically, the signal of disproportionate reporting for serious AEs with the pegfilgrastim biosimilar Fulphila® was different to that with its corresponding biologic product, Neulasta®.
The increased proportions of AE reports for biosimilar products were likely reflecting their market uptake. For example, since the first filgrastim biosimilar Zarxio® was marketed in the US in September 2015, it was reported that Zarxio® accounted for 47% of all filgrastim administrations among the commercially insured and 42% of those among Medicare Advantage beneficiaries by March 2018 [39]. Although the uptake of pegfilgrastim biosimilars has been increasing steadily since their launch, currently sharing about 29% (20.5% by Udenyca® alone) of the prefilled syringe form market, the main market share of the on-body injector Neulasta OnPro® has remained steady in the US [40, 41]. Therefore, we observed increasing but small proportions of AE reports with pegfilgrastim biosimilars. Finally, the proportion of AE reports for the epoetin alpha biosimilar Retacrit® has significantly increased following its marketing in June 2018, consistent with the reported market share of close to 20% [42]. The consistency between studied originator biologic and biosimilar products’ post-marketing AE reporting and their market share not only reflects the efficiency of the US FDA’s MedWatch medical product safety reporting program, but also ensures the validity of using the FAERS data for pharmaceutical products’ post-marketing surveillance and pharmacovigilance.
In addition, we found heterogeneity in reporting patterns between studied originator biologic and corresponding biosimilar products. Recent published literature on patterns of G-CSF and ESA use has demonstrated that females are more likely to receive prophylactic filgrastim, pegfilgrastim, and epoetin alpha [43,44,45,46], which is in line with our finding that a majority of AE reports for these products in the FAERS data were associated with female patients. Likewise, G-CSFs have been primarily prescribed for patients with neutropenia, and the incidence of neutropenia is 70% in women receiving chemotherapy for breast cancer [47, 48]. Although healthcare providers (e.g., physicians, nurses, pharmacists) were the main reporters for these AEs, we did observe the difference in reporter type between pegfilgrastim and epoetin alpha biologic and biosimilar reports. Specifically, more biosimilar AEs were reported from consumers compared with biologics AEs. A recent study reported that the completeness of FAERS reports from consumers was generally greater than that of reports from healthcare providers [49]. Although the Toki et al. study [49] only assessed two quarters of FAERS data in 2016, it is encouraging to observe consumers’ awareness of post-marketing AE reporting for pharmaceutical products. Consumers’ increased awareness of and involvement in post-marketing surveillance and pharmacovigilance will improve both the volume and completeness of AE reporting data, which, in turn, will facilitate clinical practice and health policy decision making.
In order to detect signals of disproportionate reporting of AEs for studied originator biologic and biosimilar products, we performed main and sensitivity analyses by including January 1, 2004–March 31, 2020 FAERS data, limiting this to reports after the marketing of the first biosimilar and to reports with only primary or secondary suspect drug exposure, respectively. We found no signals in serious AE reporting for pegfilgrastim and epoetin alpha originator biologics compared to all other drugs. The filgrastim biologic product Neupogen® had a signal of disproportionate reporting of serious AEs in the main analysis, but the signal disappeared in sensitivity analyses. The three corresponding biosimilar products performed differently. Specifically, no signal of disproportionate reporting in serious AEs for pegfilgrastim (Udenyca® only) and epoetin alpha biosimilars compared to all other drugs was detected. However, another pegfilgrastim biosimilar, Fulphila®, had higher serious AE reporting compared to all other drugs and its corresponding originator biologic product Neulasta®. Both filgrastim biosimilar products showed no signals in serious AE reporting in the main analysis, but they had higher serious AE reporting when limited to a post-biosimilar marketing period. Three takeaway messages can be generated based on our findings in both main and sensitivity analyses of signal detection in serious AE reporting: (1) epoetin alpha originator biologic and biosimilar products showed no signal of disproportionate reporting; (2) pegfilgrastim originator biologics and biosimilar Udenyca® showed no signal, but the biosimilar Fulphila® had a signal of disproportionate reporting in serious AEs compared to its originator biologic and all other drugs; and (3) because filgrastim originator biologic and biosimilar products showed inconsistent signals in the main and sensitivity analyses, we conservatively recommend to interpret it as no signals in serious AE reporting for these products compared to all other drugs. Two published post-marketing observational studies using administrative claims data found no difference in the incidence of serious AEs for Zarxio® compared to Neupogen® [44, 50]. The difference in signal of disproportionate reporting detection based on the observation period was also observed in a methodological article by Rahman et al., in which RORs for specific AEs for branded drugs were higher in the full study period compared to the post-generic approval period using the FAERS [22]. One explanation could be due to the length of marketing exposure for studied products. Originator biologic products were approved years before their corresponding biosimilar products; therefore, the longer marketing exposure period led to more AE reports and potentially higher reporting disproportionality. Taking this precaution, we determined to detect a signal that was statistically significant in both the main and sensitivity analyses. In addition, we did not observe the possible Weber effect on AE reporting for filgrastim biosimilar products, where tendency to report AEs is higher in the first 2 years after a drug is marketed [51]. The signal of disproportionate reporting in serious AEs for the biosimilar Fulphila® compared to all other drugs and the biologic Neulasta® identified in our study should be further evaluated using well-designed new user cohort studies or experimental studies to confirm the signal.
Regarding specific AEs, all individual filgrastim and pegfilgrastim originator biologics and biosimilars except for Fulphila® showed signals of disproportionate reporting in bone pain AE reporting, which is consistent with bone pain as a common adverse reaction in the drug labels for these products [25,26,27, 29,30,31]. We did not observe a difference in bone pain AE reporting between filgrastim originator biologics and biosimilars in our analysis. In a recent European study, a significant difference in bone pain time to onset (TTO) was observed between filgrastim originator biologic and biosimilar Zarxio® [52]. Another study reported a significantly higher ROR of bone pain for filgrastim biosimilar Nivestym® compared to its biologic Neupogen®, but no difference was found for Zarxio® [53]. An RCT conducted in Singapore showed no statistically significant difference in the proportion of patients reporting bone pain in the Nivestym® group compared to the Neupogen® group [54]. Since filgrastim biosimilar launched in markets outside of the US in 2008, the difference in signal detection between our study and European studies may be explained by different marketing exposure periods and differences in AE reporting to the US FDA FAERS and World Health Organization (WHO) Vigibase®, which was used in those studies. Pegfilgrastim biosimilars are new to both US and European markets since their introduction in 2018. Therefore, the post-marketing surveillance data are very limited in both regions. In the current study, we observed a difference in ROR of bone pain between pegfilgrastim originator biologic and its biosimilar Udenyca® in both sensitivity analyses, but not in the main analysis. Pegfilgrastim-related bone pain was found to be insignificantly different between originator biologics and biosimilars in previous phase III clinical trials in the US [55]. Caution is required to interpret any difference in bone pain AE reporting between Neulasta® and Udenyca® as the number of bone pain reports for Udenyca® was small and the exposure time period since its marketing launch is short. More evidence with longer observation is needed to determine the difference in risk of bone pain between these two products. Similarly, no difference in nausea and vomiting as well as cardiovascular AE reporting between epoetin alpha originator biologics and biosimilar products was observed in our study. The safety profile of epoetin alpha biosimilar has been studied in Europe, where its first biosimilar was marketed in 2007. In two observational studies using real-world data conducted in Italy, no difference was detected between epoetin alpha originator biologics and biosimilars in terms of major adverse cardiovascular events and cardiovascular/cerebrovascular conditions [56, 57]. However, in our analysis, a signal of disproportionate reporting of cardiovascular AE for epoetin alpha originator biologics was detected, which calls for attention for continuous post-marketing surveillance to monitor and assess safety of the product in the US market.
Our study has some limitations. First, the FAERS database includes spontaneous AE reports, which are prone to over and/or underreporting bias [58]. Second, the quality of the AE reports depends on the accuracy and completeness of the reports made by different sources. We did observe large proportions of missing data related to patient demographics and reporter type, which prevented us from assessing adjusted associations between these factors and AE reporting. Imprecise information for drug names, manufacturer names, and event dates exists in the FAERS. However, we used multiple stringent criteria for categorizing originator biologics and biosimilars to reduce potential misclassification. On the other hand, these stringent criteria led to the exclusion of a large proportion of the FAERS reports from our analyses. Third, small numbers of some specific AEs for newly marketed biosimilars resulted in RORs with wider CIs. Signals generated from a small number of reports need to be interpreted with caution; future data mining using the Bayesian Confidence Propagation Neural Network (BCPNN) could strengthen our analysis and reduce sensitivity to small fluctuations in the number of reports [59]. Fourth, our study focused on serious and known specific AEs included in the drug label and in systematic reviews of RCTs and observational studies. It is possible that we have missed detecting potential new or rare, as well as important, AE signals for the studied drugs. For example, a considerable barrier to biosimilar interchangeability is potential immunogenic AEs, and future pharmacovigilance studies should aim to resolve the ongoing debate [60]. Finally, findings from this retrospective analysis only generated the hypothesis about difference in AE reporting between biologic and corresponding biosimilar products. We could not establish the causal relationship between specific AEs and studied drugs because the role of other confounding factors (such as drug–drug and drug–food interactions, patient medical history, and concurrent medications) cannot be ruled out. Stronger study designs like a cohort study or an RCT need to be conducted to test our hypothesis.
5 Conclusions
Using the US FDA spontaneous reporting data, we did not observe any difference in signals of disproportionate reporting of serious AEs between filgrastim and epoetin alpha originator biologic and biosimilar products. However, the signal of disproportionate reporting for serious AEs with pegfilgrastim biosimilar (Fulphila®) was different compared to its corresponding originator biologic product Neulasta®. Although disproportionate reporting signals in bone pain for filgrastim and pegfilgrastim as well as in cardiovascular events for epoetin alpha were detected when compared to all other drugs in the FAERS database, no difference in signal of disproportionate reporting was observed between originator biologic and its corresponding biosimilar products. Our findings support the similarity in the signals of disproportionate reporting between US FDA-approved biosimilars and originator biologics. However, given the limitations of the spontaneous reporting data, future studies using larger data sources, longer observation period, and rigorous study designs to test these AE signals detected from our study are warranted.
References
Klastersky J, de Naurois J, Rolston K, Rapoport B, Maschmeyer G, Aapro M, et al. Management of febrile neutropaenia: ESMO clinical practice guidelines & #x2020. Ann Oncol. 2016;27:v111–8. https://doi.org/10.1093/annonc/mdw325.
Tai E Jr., Guy GP, Dunbar A, Richardson LC. Cost of cancer-related neutropenia or fever hospitalizations, United States, 2012. J Oncol Pract. 2017;13(6):e552–61. https://doi.org/10.1200/jop.2016.019588.
Ludwig H, Van Belle S, Barrett-Lee P, Birgegård G, Bokemeyer C, Gascón P, et al. The European Cancer Anaemia Survey (ECAS): a large, multinational, prospective survey defining the prevalence, incidence, and treatment of anaemia in cancer patients. Eur J Cancer. 2004;40(15):2293–306. https://doi.org/10.1016/j.ejca.2004.06.019.
Xu H, Xu L, Page JH, Cannavale K, Sattayapiwat O, Rodriguez R, et al. Incidence of anemia in patients diagnosed with solid tumors receiving chemotherapy, 2010–2013. Clin Epidemiol. 2016;8:61–71. https://doi.org/10.2147/CLEP.S89480.
Family L. Symptom burdens related to chemotherapy-induced anemia in stage IV cancer. J Community Support Oncol. 2018;16(6):e260–71. https://doi.org/10.12788/jcso.0432.
Österborg A, Brandberg Y. Relationship between changes in hemoglobin level and quality of life during chemotherapy in anemic cancer patients receiving epoetin alfa therapy. Cancer. 2003;97(12):3125–6. https://doi.org/10.1002/cncr.11430.
Patel KB, Arantes LH Jr, Tang WY, Fung S. The role of biosimilars in value-based oncology care. Cancer Manag Res. 2018;10:4591–602. https://doi.org/10.2147/CMAR.S164201.
McBride A, Campbell K, Bikkina M, MacDonald K, Abraham I, Balu S. Cost-efficiency analyses for the US of biosimilar filgrastim-sndz, reference filgrastim, pegfilgrastim, and pegfilgrastim with on-body injector in the prophylaxis of chemotherapy-induced (febrile) neutropenia. J Med Econ. 2017;20(10):1083–93. https://doi.org/10.1080/13696998.2017.1358173.
Biosimilars. US Food and Drug Administration. https://www.fda.gov/drugs/therapeutic-biologics-applications-bla/biosimilars. Accessed 12 Aug 2020.
Biosimilar and Interchangeable Products. US Food and Drug Administration. https://www.fda.gov/drugs/biosimilars/biosimilar-and-interchangeable-products. Accessed 12 Aug 2020.
Prescribing Biosimilar and Interchangeable Products. US Food and Drug Administration. https://www.fda.gov/drugs/biosimilars/prescribing-biosimilar-and-interchangeable-products. Accessed 12 Aug 2020.
Biosimilar and Interchangeable Biologics: More Treatment Choices. US Food and Drug Administration. https://www.fda.gov/consumers/consumer-updates/biosimilar-and-interchangeable-biologics-more-treatment-choices. Accessed 12 Aug 2020.
IMS Biosimilar report—The Impact of Biosimilar Competition in Europe. European Commission. 2017. https://ec.europa.eu/docsroom/documents/23102. Accessed 12 Aug 2020.
Abraham I, Han L, Sun D, MacDonald K, Aapro M. Cost savings from anemia management with biosimilar epoetin alfa and increased access to targeted antineoplastic treatment: a simulation for the EU G5 countries. Future Oncol. 2014;10(9):1599–609. https://doi.org/10.2217/fon.14.43.
Raedler LA. Zarxio (filgrastim-sndz): first biosimilar approved in the United States. Am Health Drug Benefits. 2016;9(Spec Feature):150–4.
Yang J, Yu S, Yang Z, Yan Y, Chen Y, Zeng H, et al. Efficacy and safety of supportive care biosimilars among cancer patients: a systematic review and meta-analysis. Biodrugs. 2019;33(4):373–89. https://doi.org/10.1007/s40259-019-00356-3.
Questions and Answers on FDA's Adverse Event Reporting System (FAERS). US Food and Drug Administration. 2018. https://www.fda.gov/drugs/surveillance/questions-and-answers-fdas-adverse-event-reporting-system-faers. Accessed 12 Aug 2020.
Sakaeda T, Tamon A, Kadoyama K, Okuno Y. Data mining of the public version of the FDA adverse event reporting system. Int J Med Sci. 2013;10(7):796–803. https://doi.org/10.7150/ijms.6048.
Purple Book: Database of Licensed Biological Products. US Food and Drug Administration. https://purplebooksearch.fda.gov/. Accessed 12 Aug 2020.
National Drug Code Directory. US Food and Drug Administration. https://www.fda.gov/drugs/drug-approvals-and-databases/national-drug-code-directory. Accessed 12 Aug 2020.
Nonproprietary Naming of Biological Products: Guidance for Industry. US Food and Drug Administration. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/nonproprietary-naming-biological-products-guidance-industry. Accessed 12 Aug 2020.
Rahman MM, Alatawi Y, Cheng N, Qian J, Peissig PL, Berg RL, et al. Methodological considerations for comparison of brand versus generic versus authorized generic adverse event reports in the US Food and Drug Administration Adverse Event Reporting System (FAERS). Clin Drug Investig. 2017;37(12):1143–52. https://doi.org/10.1007/s40261-017-0574-4.
FDA Adverse Event Reporting System. openFDA. 2020. https://open.fda.gov/data/faers/. Accessed 12 Aug 2020.
CFR—code of federal regulations title 21 Sec 314.80. Postmarketing Reporting of Adverse Drug Experiences. US Food and Drug Administration. 2015. www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm. Accessed 12 Aug 2020.
ZARXIO™ (filgrastim-sndz). US Food and Drug Administration. 2015. https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/125553lbl.pdf. Accessed 12 Aug 2020.
NEUPOGEN® (filgrastim) US Food and Drug Administration. 2015. https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/103353s5183lbl.pdf. Accessed 12 Aug 2020.
NEULASTA® (pegfilgrastim). US Food and Drug Administration. 2015. https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/125031s180lbl.pdf. Accessed 12 Aug 2020.
Epogen® (epoetin alfa). US Food and Drug Administration. 2017. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/103234s5363s5366lbl.pdf. Accessed 12 Aug 2020.
NIVESTYM™ (filgrastim-aafi) US Food and Drug Administration. 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761080s000lbl.pdf. Accessed 12 Aug 2020.
FULPHILA (pegfilgrastim-jmdb). US Food and Drug Administration. 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761075s000lbl.pdf. Accessed 12 Aug 2020.
UDENYCATM (pegfilgrastim-cbqv). US Food and Drug Administration. 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761039s000lbl.pdf. Accessed 12 Aug 2020.
RETACRIT™(epoetin alfa-epbx). US Food and Drug Administration. 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125545s000lbl.pdf. Accessed 12 Aug 2020.
Dale DC, Crawford J, Klippel Z, Reiner M, Osslund T, Fan E, et al. A systematic literature review of the efficacy, effectiveness, and safety of filgrastim. Support Care Cancer. 2018;26(1):7–20. https://doi.org/10.1007/s00520-017-3854-x.
Standardised MedDRA Queries. MedDRA-Medical dictionary for regulatory activities. https://www.meddra.org/standardised-meddra-queries. Accessed 12 Aug 2020.
Szarfman A, Machado SG, O’Neill RTJDS. Use of screening algorithms and computer systems to efficiently signal higher-than-expected combinations of drugs and events in the US FDA’s spontaneous reports database. Drug Saf. 2002;25(6):381–92. https://doi.org/10.2165/00002018-200225060-00001.
Rothman KJ, Lanes S, Sacks ST. The reporting odds ratio and its advantages over the proportional reporting ratio. Pharmacoepidemiol Drug Saf. 2004;13(8):519–23. https://doi.org/10.1002/pds.1001.
Egberts AC, Meyboom RH, van Puijenbroek EP. Use of measures of disproportionality in pharmacovigilance: three Dutch examples. Drug Saf. 2002;25(6):453–8. https://doi.org/10.2165/00002018-200225060-00010.
Shalviri G, Mohammad K, Majdzadeh R, Gholami K. Applying quantitative methods for detecting new drug safety signals in pharmacovigilance national database. Pharmacoepidemiol Drug Saf. 2007;16(10):1136–40. https://doi.org/10.1002/pds.1459.
Kozlowski S, Birger N, Brereton S, McKean SJ, Wernecke M, Christl L, et al. Uptake of the biologic filgrastim and its biosimilar product among the medicare population. JAMA. 2018;320(9):929–31. https://doi.org/10.1001/jama.2018.9014.
Mehr S. An interesting comparison: the latest data on US and EU biosimilar uptake. Biosimilars review and report. 2020. https://biosimilarsrr.com/2020/04/23/an-interesting-comparison-the-latest-data-on-us-and-eu-biosimilar-uptake/. Accessed 12 Aug 2020.
Bangia I. Biosimilar uptake varies by class of agent. AJMC—The Center for Biosimilars. 2020. https://www.centerforbiosimilars.com/news/biosimilar-uptake-varies-by-class-of-agent. Accessed 12 Aug 2020.
Cohen J. In U.S. Biosimilars run into more roadblocks. Forbes. 2019. https://www.forbes.com/sites/joshuacohen/2019/09/12/in-u-s-biosimilars-run-into-more-roadblocks/#de360f319e9a. Accessed 12 Aug 2020.
Barnes G, Pathak A, Schwartzberg L. G-CSF utilization rate and prescribing patterns in United States: associations between physician and patient factors and GCSF use. Cancer Med. 2014;3(6):1477–84. https://doi.org/10.1002/cam4.344.
Douglas AG, Schwab P, Lane D, Kennedy K, Slabaugh SL, Bowe A. A comparison of brand and biosimilar granulocyte-colony stimulating factors for prophylaxis of chemotherapy-induced febrile neutropenia. J Manag Care Spec Pharm. 2017;23(12):1221–6. https://doi.org/10.18553/jmcp.2017.23.12.1221.
Seetasith A, Holdford D, Shah A, Patterson J. On-label and off-label prescribing patterns of erythropoiesis-stimulating agents in inpatient hospital settings in the US during the period of major regulatory changes. Res Social Adm Pharm. 2017;13(4):778–88. https://doi.org/10.1016/j.sapharm.2016.07.005.
Weycker D, Doroff R, Hanau A, Bowers C, Belani R, Chandler D, et al. Use and effectiveness of pegfilgrastim prophylaxis in US clinical practice:a retrospective observational study. BMC Cancer. 2019;19(1):792. https://doi.org/10.1186/s12885-019-6010-9.
Culakova E, Thota R, Poniewierski MS, Kuderer NM, Wogu AF, Dale DC, et al. Patterns of chemotherapy-associated toxicity and supportive care in US oncology practice: a nationwide prospective cohort study. Cancer Med. 2014;3(2):434–44. https://doi.org/10.1002/cam4.200.
Chang J. Chemotherapy dose reduction and delay in clinical practice evaluating the risk to patient outcome in adjuvant chemotherapy for breast cancer. Eur J Cancer. 2000;36(Suppl 1):S11–4. https://doi.org/10.1016/s0959-8049(99)00259-2.
Toki T, Ono S. Assessment of factors associated with completeness of spontaneous adverse event reporting in the United States: a comparison between consumer reports and healthcare professional reports. J Clin Pharm Ther. 2020;45(3):462–9. https://doi.org/10.1111/jcpt.13086.
Chen X, Agiro A, Barron J, Debono D, Fisch M. Early adoption of biosimilar growth factors in supportive cancer care. JAMA Oncol. 2018;4(12):1779–81. https://doi.org/10.1001/jamaoncol.2018.5090.
Hoffman KB, Dimbil M, Erdman CB, Tatonetti NP, Overstreet BM. The Weber Effect and the United States Food and Drug Administration’s Adverse Event Reporting System (FAERS): analysis of sixty-two drugs approved from 2006 to 2010. Drug Saf. 2014;37(4):283–94. https://doi.org/10.1007/s40264-014-0150-2.
Kobayashi T, Kamada I, Komura J, Toyoshima S, Ishii-Watabe A. Comparative study of the number of report and time-to-onset of the reported adverse event between the biosimilars and the originator of filgrastim. Pharmacoepidemiol Drug Saf. 2017;26(8):917–24. https://doi.org/10.1002/pds.4218.
Rastogi S, Shukla S, Sharma AK, Sarwat M, Srivastava P, Katiyar T, et al. Towards a comprehensive safety understanding of granulocyte-colony stimulating factor biosimilars in treating chemotherapy associated febrile neutropenia: trends from decades of data. Toxicol Appl Pharmacol. 2020;395:114976. https://doi.org/10.1016/j.taap.2020.114976.
Chew C, Ng HY. Efficacy and safety of nivestim versus neupogen for mobilization of peripheral blood stem cells for autologous stem cell transplantation. Sci Rep. 2019;9(1):19938. https://doi.org/10.1038/s41598-019-56477-w.
Desai K, Misra P, Kher S, Shah N. Clinical confirmation to demonstrate similarity for a biosimilar pegfilgrastim: a 3-way randomized equivalence study for a proposed biosimilar pegfilgrastim versus US-licensed and EU-approved reference products in breast cancer patients receiving myelosuppressive chemotherapy. Exp Hematol Oncol. 2018;7(1):22. https://doi.org/10.1186/s40164-018-0114-9.
Trotta F, Belleudi V, Fusco D, Amato L, Mecozzi A, Mayer F, et al. Comparative effectiveness and safety of erythropoiesis-stimulating agents (biosimilars vs originators) in clinical practice: a population-based cohort study in Italy. BMJ Open. 2017;7(3):e011637. https://doi.org/10.1136/bmjopen-2016-011637.
Stoppa G, D’Amore C, Conforti A, Traversa G, Venegoni M, Taglialatela M, et al. Comparative safety of originator and biosimilar epoetin alfa drugs: an observational prospective multicenter study. Biodrugs. 2018;32(4):367–75. https://doi.org/10.1007/s40259-018-0293-2.
Ghosh P, Dewanji A. Effect of reporting bias in the analysis of spontaneous reporting data. Pharm Stat. 2015;14(1):20–5. https://doi.org/10.1002/pst.1657.
Bate A, Lindquist M, Orre R, Edwards I, Meyboom R. Data-mining analyses of pharmacovigilance signals in relation to relevant comparison drugs. Eur J Clin Pharmacol. 2002;58(7):483–90. https://doi.org/10.1007/s00228-002-0484-z.
Colbert RA, Cronstein BN. Biosimilars: the debate continues. Arthritis Rheum. 2011;63(10):2848–50. https://doi.org/10.1002/art.30505.
Acknowledgements
The authors would like to thank and acknowledge Drs. Surachat Ngorsuraches and Kimberly Garza, associate professors at Auburn University Harrison School of Pharmacy, for their feedback on research proposal development. We also thank Mr. Whitt Krehling, Auburn University, for his assistance in editing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No financial assistance was used to conduct the study described in the article and/or used to assist with the preparation of the article.
Conflict of Interest
There are no conflicts of interest to report for all authors.
Data Availability
The US FDA FAERS data supporting the results reported in the article can be accessed and downloaded from https://fis.fda.gov/extensions/FPD-QDE-FAERS/FPD-QDE-FAERS.html.
Ethics Approval
The study was granted exemption by the Auburn University Institutional Review Board, and the study was performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Informed Consent
Not applicable.
Author Contributions
KAT, CBT, and JQ had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: All authors. Acquisition, analysis, or interpretation of data: KAT, CBT, and JQ. Drafting of the paper: All authors. Critical revision of the paper for important intellectual content: All authors. Statistical analysis: KAT and CBT. Study supervision: JQ.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tanni, K.A., Truong, C.B., Almahasis, S. et al. Safety of Marketed Cancer Supportive Care Biosimilars in the US: A Disproportionality Analysis Using the Food and Drug Administration Adverse Event Reporting System (FAERS) Database. BioDrugs 35, 239–254 (2021). https://doi.org/10.1007/s40259-020-00466-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-020-00466-3