
Abstract
Background
The United States Food and Drug Administration’s (FDA) Adverse Event Reporting System (FAERS) consists of adverse event (AE) reports linked to approved drugs. The database is widely used to support post-marketing safety surveillance programs. Sometimes cited as a limitation to the usefulness of FAERS, however, is the ‘Weber effect,’ which is often summarized by stating that AE reporting peaks at the end of the second year after a regulatory authority approves a drug. Weber described this effect in 1984 based upon a single class of medications prescribed in the United Kingdom. Since that time, the FDA has made a concerted effort to improve both reporting and the database itself. Both volume and quality of AE reporting has dramatically improved since Weber’s report, with an estimated 800,000 yearly reports now being logged into FAERS.
Objective
The aim of this study was to determine if current FAERS reporting follows the trend described by Weber.
Methods
Sixty-two drugs approved by the FDA between 2006 and 2010 were included in this analysis. Publicly available FAERS data were used to assess the ‘primary suspect’ AE reporting pattern for up to a 4-year period following each drug’s approval date.
Results
A total of 334,984 AE reports were logged into FAERS for the 62 drugs analyzed here. While a few of the drugs demonstrated what could be considered ‘Weber effect’ curves, a majority of the drugs showed little evidence for the effect. In fact, the general AE reporting pattern observed in this study appears to consist simply of increasing case counts over the first three quarters after approval followed by relatively constant counts thereafter.
Conclusions
Our results suggest that most of the modern adverse event reporting into FAERS does not follow the pattern described by Weber. Factors that may have contributed to this finding include large increases in the volume of AE reports since the Weber effect was described, as well as a concerted effort by the FDA to increase awareness regarding the utility of post-marketing AE reporting.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Adverse event (AE) archives such as the US FDA’s Adverse Event Reporting database (FAERS) are sometimes assumed to suffer from the ‘Weber effect,’ often generalized as ‘after regulatory approval, AE reporting peaks by the end of year 2 and then rapidly diminishes with time.’ |
We did not find evidence for such a general trend in 62 drugs, therefore assertions that modern FAERS data are unreliable due to the ‘Weber effect’ appear unfounded. |
1 Introduction
By necessity, pre-approval clinical testing is conducted in relatively homogenous subjects and, accordingly, cannot delineate the complete adverse event (AE) profile of a drug. Limitations common to pre-approval testing include small test populations, brief drug exposure durations, and exclusion criteria that often eliminate testing of pregnant subjects, the elderly, the young, subjects with existing comorbidities, and patients who take multiple medications [1]. Therefore, unexpected actions often occur once a drug is approved and introduced into the broad and heterogeneous population of consumers. In fact, many serious and life-threatening adverse events are commonly encountered only after a drug obtains US Food and Drug Administration (FDA) approval [2–5]. Additionally, only half of all serious AEs are listed in the main source of AE information for many prescribers (the Physician’s Desk Reference) within 7 years after they win FDA approval [5]. In short, careful post-approval AE monitoring is vital to the ongoing drug evaluation process and patient safety.
The FDA’s Adverse Event Reporting System (FAERS) is a centralized, computerized, information database that is broadly used by the FDA and other pharmacovigilance experts for post-marketing drug safety surveillance [1, 6–21]. The FDA uses FAERS analyses to make post-marketing regulatory decisions such as the issuance of warnings, label changes, and/or market removal [22]. International government and related organizations (Australia’s Therapeutic Goods Administration, Canada’s Vigilance Adverse Reaction Online Database, Europe’s EudraVigilance, Japan’s Pharmaceuticals and Medical Devices Agency, The United Kingdom’s Yellow Card Scheme, and The World Health Organization’s VigiBase) also use spontaneous AE databases to identify post-approval drug safety concerns.
Two commonly assumed limitations of spontaneous AE databases is that they suffer from ‘stimulated reporting’ [23–27] and the ‘Weber effect’ [28]. With regard to the Weber effect, in 1984, JCP Weber published a paper in which he detailed adverse event reporting trends in the United Kingdom regarding oral drugs in the non-steroidal anti-inflammatory drug (NSAID) class [28]. Of note, the reporting trend he described occurred during a time period when the UK’s ‘Black Triangle’ reporting guidelines [29] directed prescribers to pay special attention to AEs during the first 2 years after a drug’s approval.
In many modern publications, the Weber effect is assumed to operate in various global AE databases and is often too simply summarized as ‘after regulatory approval of a drug, AE reporting increases over the first 2 years, peaks near the end of year 2, and then reliably, and rapidly, diminishes with further time on the market.’ An important point that Weber made about the pattern, however, is often overlooked by such a generalization. Weber stated “this decline is due to a reduction in the reporting of clinically mild or trivial reactions. The more serious ADR, such as haematemesis, perforation of peptic ulcers, blood dycrasias, etc. are reported from year to year in a quite constant manner” [28].
Since Weber’s original paper was published, multiple publications have either approximated or replicated the Weber effect [30, 31] while other publications that did not directly test for the effect cite it as an accepted principle [32–34]. Some of the same authors, however, who found a Weber effect with the NSAID class of drugs [31] reported only a partial Weber effect with the serotonin-reuptake class of medications [35]. Other authors discovered that some NSAIDs had a Weber-like pattern, while others did not [36]. A recent report that correlated usage data with reported AEs also did not observe Weber-like patterns [37]. An analysis of new drugs approved in 2006 also showed no evidence to support the contention that the modern generalization of the Weber effect accurately describes current AE reporting trends [38]. Most of the studies, however, that analyzed reporting trends have been conducted in a limited drug class or with a narrow time window. Additionally, the nature of AE reporting has dramatically changed since 1984, with improvements both in the quality of reports and the volume of data [39, 40]. In fact, an estimated 800,000 new case reports are currently logged into FAERS every year [41].
Accordingly, in order to determine general AE reporting patterns for recent FAERS data, we analyzed 62 drugs approved from 2006 to 2010.
2 Methods
2.1 Drugs Included
We included prescription drugs approved from 2006 to 2010 that had a total of at least 1,000 ‘primary suspect’ case reports in FAERS. (‘Primary suspect’ is a designation by the person who submitted a given case report, and is their estimate of which drug, if the subject was taking more than one, was likely responsible for the observed AE). Over-the-counter and recreational drugs, vaccines, and broad, undefined compounds listed in FAERS were not analyzed. Duplicate case reports were removed.
2.2 Inclusion Criteria for FAERS Case Reports
Case reports that were missing or contained malformed key identification fields (Individual Safety Report number [ISR], patient number, drug sequence identification, or MedDRA® AE term) were discarded. As long as the aforementioned key identification fields were contained in a given case report, allowable missing fields included age, gender, weight, outcome, and condition. Cases were discarded if the drug name was found to be indeterminate or if the name was determined to not represent an FDA-approved drug (e.g., dietary supplements, foods, etc). In an effort to exclude pre-approval AE case reports mistakenly logged into FAERS, the date of receipt for a given case report must have occurred after the drug’s FDA approval date.
2.3 Drug Name Mapping
Drug-name text mapping was accomplished as previously described by Hoffman et al. [42]. Drug names were normalized to RxNorm reference codes [43] using string searching and manual curation. National Drug File Reference Terminology [44] was used to provide ancillary information on class and mechanism of action.
2.4 Established Pharmacologic Classes (EPC)
EPC is a designation by the FDA that indicates the established pharmacologic class(es) of a drug or other therapeutic compound [44]. EPC data were obtained from the FDA National Drug Code file and mapped by researchers to the appropriate FAERS drug-name data by way of brand or generic compound name.
2.5 Case Counting
Quarterly primary suspect case reports submitted by ‘all reporters’ were counted from the first full quarter after a drug’s FDA approval through quarter 4 of 2012.
2.6 Normalization
For each drug, the highest case count total reached, in any of the 16 quarters analyzed, was normalized to 100. All remaining 15 quarters were normalized accordingly.
3 Results
Sixty-two drugs and 334,984 primary suspect reports were included in this analysis. Our results show that the Weber effect may no longer be a valid concern in modern FAERS reporting. AE reporting volume increased, as would be expected, over the first few quarters after a drug was introduced to the market. While no clear pattern could be ascribed to the reporting trends, most drugs reached a near maximum amount of reports by the third full quarter after approval. In general, after that point, case counts per quarter stayed relatively constant.
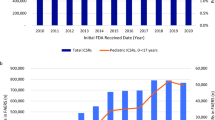
Tables 1, 2, 3, 4, and 5 list the drugs analyzed by year, with EPC, approval date, total number of primary suspect case reports, and the percentage of ‘healthcare professionals’ versus ‘other’ reporters. The brand name was noted for generic compounds marketed in the US under multiple commercial names.
Figures 1, 2, 3, 4, and 5 show primary suspect case reports submitted by all reporters from the first full quarter after a drug’s approval through quarter 4 of 2012. We also charted year-by-year results for healthcare professional reporters (data included in Electronic Supplementary Material as tables 1–5), but there were no significant differences between the reporter groups (see Figs. 6 and 7 for grand averages for both groups of reporters). The black line represents the average of all drugs for that year. In general, case counts increase until approximately 3–7 quarters after approval and then remain relatively constant.

Drugs approved in 2006

Drugs approved in 2007

Drugs approved in 2008

Drugs approved in 2009

Drugs approved in 2010

The average of all ‘all reporters’ for the 62 drugs

The average of all ‘healthcare professional reporters’ for the 62 drugs
Figures 6 and 7 show average primary case counts (± standard error) for all drugs, across all years, for up to 16 full quarters after approval. Figure 6 was derived by using the case counts logged into FAERS for all reporters, while Fig. 7 used healthcare professional reports only. As can be seen from Figs. 6 and 7, the average number of case reports filed for new drugs increases steadily until it reaches a plateau within quarter 3–7.
Table 6 (included in the Electronic Supplementary Material) and Fig. 8 show within-drug comparisons of normalized case counts compared with a hypothetical Weber-like decrease in case counts from quarter 8 after approval to quarter 16. For each drug, we modeled the quarterly report counts as a linear function of time and evaluated the fit above a constant (null) model using an ANOVA. Table 6 lists the p values for each drug regarding line fit and intercept to the hypothetical Weber-like decrease in reporting rates after quarter 8. For this analysis we used 42 of the 61 drugs in this study because they had case count data for quarters 8–16. Of the 20 drugs with significant estimates (p < 0.05) for the period coefficient, six drugs (carvedilol, certolizumab, eltrombopag, romiplostim, sitagliptin, and varenicline) had negative slopes (decreasing reporting) and thus fit the expected behavior of our experimental Weber-effect model (df = 7, F test).

Within-drug comparison of normalized case counts compared with a hypothetical linear Weber-like decrease in case counts from quarter 8 after approval to quarter 16
4 Discussion
In modern publications, the Weber effect [28] is typically generalized as an increase in AE reporting over the first 2 years after a drug’s approval, followed by a rapid decline in reporting rates. This generalization of what Weber described is often cited as a limitation to the interpretation and usefulness of post-marketing AE databases such as FAERS. We found little evidence of such a reporting trend regarding FAERS data for 62 drugs approved from 2006 to 2010. If a generalizable pattern to the data can be discerned, it is simply that case counts in FAERS tend to increase for approximately the first three quarters after a drug’s approval date and then stay relatively constant for at least the next 13 quarters thereafter. Our findings comport with both an internal FDA study that did not replicate the Weber effect [37] and a recent study that mapped 5-year AE reporting trends for drugs approved in 2006 [38].
We postulate that modern-day FAERS reporting may no longer exhibit the Weber effect due to increased focus on the importance and utility of post-approval AE reporting by both the FDA and key healthcare players over the last decades. With specific reference to potential AE reporting differences between when Weber published his findings and the results presented here, the following events may have contributed to improved AE reporting, and therefore a suppression of Weber-like reporting trends. In 1997, the FDA formed the FAERS (then referred to as AERS) database to facilitate the reporting of post-approval AEs. FAERS now has a total of over seven million AE case reports. In 2005, the FDA formed a permanent Drug Safety Oversight Board with the specific task of monitoring post-marketing AEs [45]. The FDA issued new guidance in 2006 for how AE data should be presented in drug labels [46]. In 2007, in order to enhance post-approval drug safety analysis even further, the FDA Amendments Act was implemented to improve FAERS and provide more resources (including regulatory enforcement) to the FDA [47, 48]. Also in 2007, the FDA began to require drug companies to enact Risk Evaluation and Mitigation Strategies (REMS) to better manage, track, and report AEs [49]. Finally, in 2008, Title IX of the FDA Amendments Act [50] was enacted, which required direct-to-consumer advertisements to contain, in prominent text, “you are encouraged to report negative side effects of prescription drugs to the FDA” with phone numbers and a website listed to facilitate such reporting.
4.1 Limitations
A number of constraints must be considered when analyzing these data. FAERS relies heavily upon voluntary reporting, and therefore, the frequency of actual AEs is very likely to be underestimated. We, however, see no obvious reason why overall underreporting would affect the type of trend analyses reported here. Even though we view primary suspect FAERS cases to be the most accurate for quantitative purposes, the designation is subjective and the influence of other drugs or factors cannot be ruled out from a given case report. Since we did not have usage data for these drugs, we could not analyze AE case counts as a ‘reporting rate.’ Our analysis was therefore limited to total case counts per drug. A related study, however, that normalized case counts with usage data did not find evidence of the Weber effect [37].
5 Conclusions
Our primary objective was to determine if modern FAERS case reports parallel the AE reporting trend first described by Weber for NSAIDs marketed in the UK in 1984. Based on the first 4 years of reporting after drug approval, we did not observe a Weber reporting pattern for drugs approved from 2006 to 2010 with over 1,000 primary suspect case reports. Instead, the general reporting pattern detected was a steep rise in case counts over the first three quarters after approval. After an initial plateau was reached by the third full quarter, relatively constant case count totals continued for the remaining 13 quarters. While a few of the drugs in the group of 62 analyzed here did show Weber-like trending, the vast majority did not. We do not know the exact reasons why our analysis did not replicate Weber’s findings, but we estimate that modern-day AE reporting is quite different than it was in 1984. Indeed, post-marketing AE reporting has dramatically increased in volume since Weber’s analysis. Additionally, the systems for capturing AE reports, as well as the value of analyzing such data, have greatly improved over the last decades. The Weber effect should not be assumed when analyzing modern-day FAERS reporting.
References
Ahmad SR. Adverse drug event monitoring at the Food and Drug Administration. J Gen Intern Med. 2003;18(1):57–60.
FDA. Follow-Up to the November 2009 Early Communication about an Ongoing Safety Review of Sibutramine, Marketed as Meridia. 2010. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/ucm198206.htm. Accessed Feb 2014.
Charatan F. Bayer decides to withdraw cholesterol lowering drug. BMJ (Clin Res Ed). 2001;323(7309):359.
FDA. Safety Information: Vioxx (rofecoxib). 2002. http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm154520.htm. Accessed Feb 2014.
Lasser KE, Allen PD, Woolhandler SJ, Himmelstein DU, Wolfe SM, Bor DH. Timing of new black box warnings and withdrawals for prescription medications. JAMA, J Am Med Assoc. 2002;287(17):2215–20.
Szarfman A, Tonning JM, Doraiswamy PM. Pharmacovigilance in the 21st century: new systematic tools for an old problem. Pharmacotherapy. 2004;24(9):1099–104.
Poluzzi E, Raschi E, Koci A, Moretti U, Spina E, Behr ER, et al. Antipsychotics and Torsadogenic risk: signals emerging from the US FDA Adverse Event Reporting System Database. Drug Saf Int J Med Toxicol Drug Exp. 2013;36(6):467–79. doi:10.1007/s40264-013-0032-z.
Hochberg AM, Hauben M. Time-to-signal comparison for drug safety data-mining algorithms vs. traditional signaling criteria. Clin Pharmacol Ther. 2009;85(6):600–6. doi:10.1038/clpt.2009.26.
Robertson HT, Allison DB. Drugs associated with more suicidal ideations are also associated with more suicide attempts. PLoS ONE. 2009;4(10):e7312. doi:10.1371/journal.pone.0007312.
Weaver J, Grenade LL, Kwon H, Avigan M. Finding, evaluating, and managing drug-related risks: approaches taken by the US Food and Drug Administration (FDA). Dermatol Ther. 2009;22(3):204–15. doi:10.1111/j.1529-8019.2009.01233.x.
Bailey S, Singh A, Azadian R, Huber P, Blum M. Prospective data mining of six products in the US FDA Adverse Event Reporting System: disposition of events identified and impact on product safety profiles. Drug Saf Int J Med Toxicol Drug Exp. 2010;33(2):139–46. doi:10.2165/11319000-000000000-00000.
Harpaz R, Chase HS, Friedman C. Mining multi-item drug adverse effect associations in spontaneous reporting systems. BMC Bioinform. 2010;11(Suppl 9):S7. doi:10.1186/1471-2105-11-S9-S7.
Moore TJ, Glenmullen J, Furberg CD. Prescription drugs associated with reports of violence towards others. PLoS ONE. 2010;5(12):e15337. doi:10.1371/journal.pone.0015337.
Wang HW, Hochberg AM, Pearson RK, Hauben M. An experimental investigation of masking in the US FDA adverse event reporting system database. Drug Saf Int J Med Toxicol Drug Exp. 2010;33(12):1117–33. doi:10.2165/11584390-000000000-00000.
Moore TJ, Furberg CD, Glenmullen J, Maltsberger JT, Singh S. Suicidal behavior and depression in smoking cessation treatments. PLoS ONE. 2011;6(11):e27016. doi:10.1371/journal.pone.0027016.
Sakaeda T, Kadoyama K, Okuno Y. Statin-associated muscular and renal adverse events: data mining of the public version of the FDA adverse event reporting system. PLoS ONE. 2011;6(12):e28124. doi:10.1371/journal.pone.0028124.
Takarabe M, Kotera M, Nishimura Y, Goto S, Yamanishi Y. Drug target prediction using adverse event report systems: a pharmacogenomic approach. Bioinformatics. 2012;28(18):i611–8. doi:10.1093/bioinformatics/bts413.
Tamura T, Sakaeda T, Kadoyama K, Okuno Y. Aspirin- and clopidogrel-associated bleeding complications: data mining of the public version of the FDA adverse event reporting system, AERS. Int J Med Sci. 2012;9(6):441–6. doi:10.7150/ijms.4549.
Chen HC, Tsong Y, Chen JJ. Data mining for signal detection of adverse event safety data. J Biopharm Stat. 2013;23(1):146–60. doi:10.1080/10543406.2013.735780.
Harpaz R, DuMouchel W, LePendu P, Bauer-Mehren A, Ryan P, Shah NH. Performance of pharmacovigilance signal-detection algorithms for the FDA adverse event reporting system. Clin Pharmacol Ther. 2013;93(6):539–46. doi:10.1038/clpt.2013.24.
Hoffman KB, Kraus C, Dimbil M, Golomb BA. A survey of the FDA’s AERS database regarding muscle and tendon adverse events linked to the statin drug class. PLoS ONE. 2012;7(8):e42866. doi:10.1371/journal.pone.0042866.
FDA U. FDA Adverse Event Reporting System (FAERS) (formerly AERS). 2012. http://www.fda.gov/drugs/guidancecomplianceregulatoryinformation/surveillance/adversedrugeffects/default.htm. Accessed Feb 2014.
Dusetzina SB, Higashi AS, Dorsey ER, Conti R, Huskamp HA, Zhu S, et al. Impact of FDA drug risk communications on health care utilization and health behaviors: a systematic review. Med Care. 2012;50(6):466–78. doi:10.1097/MLR.0b013e318245a160.
Piening S, Haaijer-Ruskamp FM, de Vries JT, van der Elst ME, de Graeff PA, Straus SM, et al. Impact of safety-related regulatory action on clinical practice: a systematic review. Drug Saf Int J Med Toxicol Drug Exp. 2012;35(5):373–85. doi:10.2165/11599100-000000000-00000.
Busch SH, Frank RG, Leslie DL, Martin A, Rosenheck RA, Martin EG, et al. Antidepressants and suicide risk: how did specific information in FDA safety warnings affect treatment patterns? Psychiatr Serv. 2010;61(1):11–6. doi:10.1176/appi.ps.61.1.11.
Dasgupta N, Mandl KD, Brownstein JS. Breaking the news or fueling the epidemic? Temporal association between news media report volume and opioid-related mortality. PLoS ONE. 2009;4(11):e7758. doi:10.1371/journal.pone.0007758.
Conti RM, Dusetzina SB, Herbert AC, Berndt ER, Huskamp HA, Keating NL. The impact of emerging safety and effectiveness evidence on the use of physician-administered drugs: the case of bevacizumab for breast cancer. Med Care. 2013;51(7):622–7. doi:10.1097/MLR.0b013e318290216f.
Weber J. Epidemiology of adverse reactions to nonsteroidal anti-inflammatory drugs. Adv Inflamm Res. 1984;6:1–7.
MHRA. Black Triangle Scheme—new medicines and vaccines subject to EU-wide additional monitoring. United Kingdom. 2014. http://www.mhra.gov.uk/Safetyinformation/Howwemonitorthesafetyofproducts/Medicines/BlackTriangleproducts/index.htm. Accessed Feb 2014.
Davenport MS, Dillman JR, Cohan RH, Hussain HK, Khalatbari S, McHugh JB, et al. Effect of abrupt substitution of gadobenate dimeglumine for gadopentetate dimeglumine on rate of allergic-like reactions. Radiology. 2013;266(3):773–82. doi:10.1148/radiol.12120253.
Hartnell NR, Wilson JP. Replication of the Weber effect using postmarketing adverse event reports voluntarily submitted to the United States Food and Drug Administration. Pharmacotherapy. 2004;24(6):743–9. doi:10.1592/phco.24.8.743.36068.
Berlin JA, Glasser SC, Ellenberg SS. Adverse event detection in drug development: recommendations and obligations beyond phase 3. Am J Public Health. 2008;98(8):1366–71. doi:10.2105/ajph.2007.124537.
Thomsen HS, Webb J. The Lalli and Weber effects and the incidence of acute non-renal adverse reactions to contrast media. Acta Radiol (Stockholm, Sweden: 1987). 2012;53(9):953–4. doi:10.1258/ar.2012.12a006.
Slade BA, Leidel L, Vellozzi C, Woo EJ, Hua W, Sutherland A, et al. Postlicensure safety surveillance for quadrivalent human papillomavirus recombinant vaccine. JAMA J Am Med Assoc. 2009;302(7):750–7. doi:10.1001/jama.2009.1201.
Hartnell NR, Wilson JP, Patel NC, Crismon ML. Adverse event reporting with selective serotonin-reuptake inhibitors. Ann Pharmacother. 2003;37(10):1387–91. doi:10.1345/aph.1C522.
Wallenstein EJ, Fife D. Temporal patterns of NSAID spontaneous adverse event reports: the Weber effect revisited. Drug Saf Int J Med Toxicol Drug Exp. 2001;24(3):233–7.
McAdams MA, Governale LA, Swartz L, Hammad TA, Dal Pan GJ. Identifying patterns of adverse event reporting for four members of the angiotensin II receptor blockers class of drugs: revisiting the Weber effect. Pharmacoepidemiol Drug Saf. 2008;17(9):882–9. doi:10.1002/pds.1633.
Chhabra P, Chen X, Weiss SR. Adverse event reporting patterns of newly approved drugs in the USA in 2006: an analysis of FDA Adverse Event Reporting System data. Drug Saf Int J Med Toxicol Drug Exp. 2013;36(11):1117–23. doi:10.1007/s40264-013-0115-x.
Staffa JA, Dal Pan GJ. Regulatory innovation in postmarketing risk assessment and management. Clin Pharmacol Ther. 2012;91(3):555–7. doi:10.1038/clpt.2011.289.
Weiss-Smith S, Deshpande G, Chung S, Gogolak V. The FDA drug safety surveillance program: adverse event reporting trends. Arch Intern Med. 2011;171(6):591–3. doi:10.1001/archinternmed.2011.89.
FDA U. Reports Received and Reports Entered into AERS by Year. 2013. http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Surveillance/AdverseDrugEffects/ucm070434.htm. Accessed Feb 2014.
Hoffman KB, Overstreet, BM, Doraiswamy, PM. A Drug Safety ePlatform for Physicians, Pharmacists and Consumers based on Post-Marketing Adverse Events. Drugs and Therapy Studies 2013;3(e4).
RxNorm. National Library of Medicine. http://www.nlm.nih.gov/research/umls/rxnorm/. Accessed Feb 2014.
FDA. Pharmacological Class: National Drug File Reference Terminology. 2013. http://www.fda.gov/ForIndustry/DataStandards/StructuredProductLabeling/ucm162549.htm. Accessed Feb 2014.
FDA. Milestones in Food and Drug Law History. 2005. http://www.fda.gov/aboutfda/whatwedo/history/milestones/ucm081229.htm. Accessed Feb 2014.
FDA. Guidance for Industry: Adverse Reactions Section of Labeling for Human Prescription Drug and Biological Products. 2006.
Schultz WB. Bolstering the FDA’s drug-safety authority. New Engl J Med. 2007;357(22):2217–9. doi:10.1056/NEJMp078212.
Traynor K. Law gives FDA new enforcement clout. Am J Health Syst Pharm Off J Am Soc Health Syst Pharm. 2007;64(22):2314–5. doi:10.2146/news070094.
FDA. Approved Risk Evaluation and Mitigation Strategies (REMS). 2007. http://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm111350.htm. Accessed Feb 2014.
FDA. FDAAA Title IX: enhanced authorities regarding postmarket safety of drugs. 2008.
Acknowledgments
The research leading to these results was funded entirely by AdverseEvents, Inc., a private corporation.
MedDRA®, the Medical Dictionary for Regulatory Activities terminology, is the international medical terminology developed under the auspices of the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH).
Keith B. Hoffman, Mo Dimbil, Colin B. Erdman, and Brian M. Overstreet have all declared employment- and stock-related conflicts of interests in their declaration forms related to AdverseEvents Inc. (AEI). Nicholas P. Tatonetti has declared a stock-related conflict of interest in his declaration forms related to AEI. Keith B. Hoffman, Mo Dimbil, Colin B. Erdman, Nicholas P. Tatonetti, and Brian M. Overstreet have no other conflicts of interest that are directly relevant to the content of this manuscript.
Keith B. Hoffman conceived of the study; analyzed and interpreted data; and drafted and approved the final submitted manuscript. Nicholas P. Tattonetti made suggestions for data interpretation and approved the final submitted manuscript. Mo Dimbil, Colin B. Erdman, and Brian M. Overstreet collected data and approved the final submitted manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Hoffman, K.B., Dimbil, M., Erdman, C.B. et al. The Weber Effect and the United States Food and Drug Administration’s Adverse Event Reporting System (FAERS): Analysis of Sixty-Two Drugs Approved from 2006 to 2010. Drug Saf 37, 283–294 (2014). https://doi.org/10.1007/s40264-014-0150-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-014-0150-2