
Abstract
Medicinally active compounds in the flavonoid class of phytochemicals are being studied for antiviral action against various DNA and RNA viruses. Quercetin is a flavonoid present in a wide range of foods, including fruits and vegetables. It is said to be efficient against a wide range of viruses. This research investigated the usefulness of Quercetin against Hepatitis C virus, Dengue type 2 virus, Ebola virus, and Influenza A using computational models. A molecular docking study using the online tool PockDrug was accomplished to identify the best binding sites between Quercetin and PubChem-based receptors. Network-pharmacological assay to opt to verify function-specific gene-compound interactions using STITCH, STRING, GSEA, Cytoscape plugin cytoHubba. Quercetin explored tremendous binding affinity against NS5A protein for HCV with a docking score of – 6.268 kcal/mol, NS5 for DENV-2 with a docking score of – 5.393 kcal/mol, VP35 protein for EBOV with a docking score of – 4.524 kcal/mol, and NP protein for IAV with a docking score of – 6.954 kcal/mol. In the network-pharmacology study, out of 39 hub genes, 38 genes have been found to interact with Quercetin and the top interconnected nodes in the protein–protein network were (based on the degree of interaction with other nodes) AKT1, EGFR, SRC, MMP9, MMP2, KDR, IGF1R, PTK2, ABCG2, and MET. Negative binding energies were noticed in Quercetin-receptor interaction. Results demonstrate that Quercetin could be a potential antiviral agent against these viral diseases with further study in in-vivo models.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Throughout history, virus-borne infectious diseases have caused millions of deaths and continue to be a hazard to public health. Scientists all over the world have made tremendous efforts to identify, treat, and prevent viral infections. However, discovering novel medicines may be difficult because of the complex life cycle of viruses and their rapid genetic alterations. Despite the availability of vaccines for many viral infectious diseases, some of the deadliest viruses, such as influenza A, Hepatitis C, Dengue type 2, and Ebola virus, continue to cause outbreaks worldwide and have resulted in significant morbidity and mortality in humans through epidemics or pandemics (Mourya et al. 2019). Highly contagious viruses are increasing the demand for vaccine research, but vaccine production is a long and complicated process. However, vaccines and antiviral medicines are often expensive, making them unavailable to most of the world's population. Both have a significant negative impact on the human body.
Natural compounds may be the best alternative for developing novel drugs. Many researchers have been working to synthesize new antiviral drugs over the last few years (Badshah et al. 2021). Natural substances have been more popular in recent decades because of their wide range of biological functions and direct usage as medications (Badshah et al. 2021; Ninfali et al. 2020). Quercetin is a unique flavonoid component that is well known for its antiviral properties. It has little or no side effects compared to synthetic medications, which could be an effective treatment option for different viral infections (Ninfali et al. 2020).
The name Quercetin comes from the Latin word ''Quercetum'' which means ‘Oak Forest’ (Batiha et al. 2020). The chemical formula of quercetin is 2-(3,4-dihydroxy phenyl)-3,5,7-trihydroxychromen-4-one and C15H10O7 (Batiha et al. 2020). Quercetin belongs to the flavonol class of flavonoids found widely among vegetables and fruits, including berries, lovage, capers, cilantro, dill, apples, and onions (Anand David et al. 2016). The color of quercetin is yellow and entirely soluble in lipids and alcohol, slightly soluble in hot water but insoluble in cold water (Batiha et al. 2020).
Quercetin (Fig. 1) is an active therapeutic agent, which shows potential antiviral activity and inhibits viruses by targeting viral infections at multiple stages. The main molecular mechanisms of quercetin's antiviral activities are the suppression of viral neuraminidase, proteases, and DNA/RNA polymerases and the alteration of many viral proteins. Quercetin has shown antiviral activity towards a wide range of viruses, including hepatitis C virus (HCV) (Khachatoorian et al. 2012), Mayaro virus (dos Santos et al. 2014), influenza A virus (IAV) (Kim et al. 2010), Chikungunya virus (CHIKV) (Lani et al. 2015), and Epstein-Barr virus (EBV) (Lee et al. 2015). Quercetin has been shown to inhibit HCV through binding and inactivating the viral NS3 protease (Bachmetov et al. 2012). Quercetin prevents DENV-2 replication by inhibiting viral RNA polymerase and blocking Ebola viral infection via VP24 interferon-inhibitory function (Fanunza et al. 2020). This manuscript, the mechanism of quercetin against four viruses such as influenza A, Hepatitis C, Dengue type 2, and Ebola virus will be discussed.

2D structure of quercetin (Ashwini et al. 2017)
Materials and methods
Antiviral drug design of Quercetin by computer-aided approach
Molecular docking study
Docking tools
The molecular docking was performed using Schrodinger suites-Maestro 2021 (Maestro 2021). Pockdrug online server was used to predict the best binding pocket and probable drug ability. Discovery studio (v 4.1) was used for the visualization.
Ligand preparation
The chemical structure of quercetin was extracted from the PubChem repository (https://pubchem.ncbi.nlm.nih.gov/). The ligand was prepared by using the LigPrep tool, which was embedded in Schrödinger suite-Maestro v 11.1, where the following parameters were used as follows: neutralized at pH 7.0 ± 2.0 using Epik 2.2 and the OPLS3 force field were used for minimization (Md. Amjad Hossen et al. 2021).
Protein preparation
To unravel quercetin activity against different viruses, four major proteins following NS5A (PDB ID: 4CL1) for HCV, NS5 (PDB ID: 3EVG) for DENV-2, VP35 (PDB ID: 4IBK) for EBOV, and NP-specific inhibitor (PDB ID: 6J1U) for IAV, were selected for molecular docking study based on literature review (Yang et al. 2021). The crystal structure of these proteins has been collected from the RCSB Protein Data Bank (PDB) an online database (https://www.rcsb.org/) and their active binding sites were selected based on drug probability score by using an online tool PockDrug (http://pockdrug.rpbs.univ-paris-diderot.fr/cgi-bin/index.py?page=home). Preprocessing, optimization and minimization processes were done by using Protein Preparation Wizard. This process is included in Schrodinger suit-maestro (v11.1). The structures were optimized at pH 7.0, water molecules fewer than 3 H-bonds to non-waters were removed. Restrained minimization was done where heavy atoms are converged to RMSD of 0.30 Å on the implemented OPLS3 force field. Then the receptor grids were generated after selecting the best binding sites by using an online tool PockDrug (Hossen et al. 2021).
Glide ligand molecular docking
To evaluate antiviral effects, prepared ligand and proteins were interacted by using Schrodinger suite-maestro (v11.1). Consequently, the spreadsheets and structures were exported for further research. Discovery Studio (v. 4.1) software was used to visualize receptor-ligand binding interactions in three dimensions (Md. Amjad Hossen et al. 2021).
Identifying the quercetin-interacting genes associated with functional enrichment in human
Quercetin–target protein network construction
We used STITCH 5 (http://stitch.embl.de/, ver. 5.0) to identify target proteins related to quercetin (Szklarczyk et al. 2016). It calculates a score for each pair of protein-chemical interactions.. We used the “single item by name” module of STITCH 5 tool to enter the term “quercetin” into the STITCH 5 tool. Then, we selected the organism as “Homo sapiens” and the “medium required interaction score” ⩾0.4 was set in the “settings” module. We used the “maximum number of interaction” as 500 by using “custom value” for getting the more interacted target genes with quercetin. We also utilized the Swiss Target Prediction tool for performing a combination of similarity measurements based on known 2D and 3D chemical structures to predict the corresponding potential bioactive targets (probability > 0.2, http://www.swisstargetprediction.ch/) (Gfeller et al. 2014). The online tool “calculate and draw custom Venn diagrams” (http://bioinformatics.psb.ugent.be/webtools/Venn/) was used to identify commonly interacted genes. We entered the total genes found in STITCH tool and the Swiss Target Prediction tool into the Venn diagrams tool (http://bioinformatics.psb.ugent.be/webtools/Venn/) to identify the commonly interacted genes with quercetin. Finally, we identified the common genes that are interacted with quercetin not only in STITCH tool but also in the Swiss Target Prediction tool, which indicated the interaction of quercetin with target genes in both tools.
Construction of protein–protein interaction (PPI) network and top-ranked gene identification of the predicted genes
We constructed a PPI network of the predicted 39 common genes by search tool for the retrieval of interacting genes (STRING) database (https://string-db.org/cgi/input.pl; STRING-DB v11.0) (Szklarczyk et al. 2019). We used the “multiple proteins” module of the STRING tool to enter the 39 commonly found genes into the STRING tool. Then, we selected the organism as “Homo sapiens” and the “medium required interaction score” ⩾0.4 was set in the “settings” module. We exported all interacted information of PPI network by using “exports” module. Then, we inputted the STRING-based information into the Cytoscape tool 3.8.2 (Shannon et al, 2003) to construct the PPI network. The cytoHubba (Chin et al. 2014) a user-friendly interface in Cytoscape tool to explore the nodes in the PPI networks. We installed the cytoHubba in the Cytoscape tool by using “app manager” module. Then calculated the degree of interaction in the original PPI network by using the “target network” module in the cytoHubba. Based on the degree of interaction, the rank of the target proteins in the PPI network was identified using the “Hubba nodes” module of cytoHubba. The obtained protein interaction data of 39 target proteins were imported into Cytoscape 3.8.2 software for visualizing the PPI network (Shannon et al, 2003).
Gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) pathway enrichment analyses of the target proteins
To identify the role of target proteins that interact with quercetin in gene functions and cellular signaling pathway, gene ontology and gene set enrichment analysis (GSEA) was employed (Subramanian et al. 2005). We inputted the commonly found 39 interacted target proteins into the GSEA by using the “Investigate Gene Sets” module. Then, the KEGG (Kanehisa et al. 2019) pathways significantly associated with the predicted genes were identified by selecting “KEGG: KEGG gene sets” in the GSEA. Moreover, we analyzed the gene ontology (GO) function enrichment of proteins (39 interacted target proteins with quercetin, Table 2) involved in the PPI network by selecting the “GO: Gene Ontology” module in the GSEA. The target proteins involved with the biological process (BP), cellular components (CC), molecular function (MF), and the KEGG pathways were also described. The FDR value < 0.05, calculated by the Benjamini–Hochberg method (Benjamini and Hochberg 1995) was considered statistically significant.
Results
Molecular docking analysis
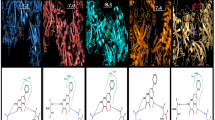
Quercetin demonstrated promising binding interaction against different viruses including HCV, DENV-2, EBOV, and IAV while the results have been shown in (Table 1) and (Fig. 2). Binding affinity and ligand efficiencies estimation of quercetin with different viruses including HCV, DENV-2, EBOV, and IAV via using MM-GBSA are summarized in Table 2. Firstly, the compound quercetin explored tremendous binding affinity against NS5A protein for HCV with a docking score of -6.268 kcal/mol (Fig. 2A). Besides, it also interacted with 5 conventional H-bond such as PRO 154 (2.78 Å), SER 157 (2.46 Å), GLN 158 (2.76 Å), CYS 111 (1.99 Å) along with single C-H bond PRO 160 (2.54 Å). At the same time, it interacted with two pi-alkyl including VAL 101 (4.20 Å), LYS 110 (5.05 Å), and one pi-pi stacked bond TYR 153 (5.55 Å) interactions. In addition, NS5 for DENV-2 was interacted (Fig. 2B) with quercetin which elucidated a docking score of -5.393 kcal/mol with a single water hydrogen bond HOH 546 (2.63 Å) and three conventional H-bond like LYS 110 (2.19 Å), as well as SO4904, showed two bands with their distance (1.62 and 1.89 Å). Conversely, the compound showed an unfavorable donor group (2.48 Å) but two pi anion bonds with ASP 146 (3.83 and 3.24 Å) and one conventional hydrogen bond GLY 83 (3.07 Å). Furthermore, quercetin was interacted with VP35 for EBOV remarkably (Fig. 2C) by four conventional H-bond following ALA 221 (2.45 Å), ARG 225 (2.82 Å), DMS 403 (3.06 Å), and VAL 294 (3.60 Å) whereas, docking score was 4.524 kcal/mol. The compound also demonstrated a single pi-alkyl bond ILE 295 (4.60 Å), two pi- cation bonds (4.64 and 4.97 Å) with LYS, and a pi-lone pair bond GLN 254 (3.00 Å). Moreover, the compound also showed a favorable binding score of -6.954 kcal/mol against NP for IAV while the compound was interacted with (Fig. 2D) by five H-bond such as ASP 51 (1.64 Å), SER 283 (1.68 and 2.61 Å), CYS 279 (2.68 Å) and THR 45 (1.83 Å) three pi alkyl bond following ALA 286 (4.41 and 4.16 Å), CYS 44 (4.86 Å)) and single amide pi stacked bond GLY 282 (4.22 Å). Hence, quercetin was unveiled as a potential agent based on the activity.

Illustration of Quercetin binding mode 3D (left) and 2D (right) against different viruses including HCV (A), DENV-2 (B), EBOV (C), and IAV (D) via molecular docking analysis
Quercetin significantly interacted with various genes
By using the STITCH tool, we found that the quercetin interacted with the 299 genes (Fig. 3 and Supplementary Table S1). Besides, we identified the 100 genes that interacted with quercetin with a minimum probability of 0.20 (Fig. 3 and Supplementary Table S2). After doing the Venn diagram, we found that 39 genes that are identified by both tools interact with quercetin (Fig. 3 and Supplementary Table S3). The interaction of quercetin with 39 genes is shown in Fig. 4 (supplementary Tables S3).

Identifying common 39 genes that are interacted with quercetin. Interaction of common 39 genes with quercetin

Interaction of common 39 genes with quercetin. The Cytoscape tool was utilized to illustrate the interactions
Quercetin-associated genes are involved in protein–protein interaction
We inputted all 39 common genes into the STRING to investigate their involvement in the PPI network. Interestingly, we found that 38 genes, except PYGL, are involved in the PPI network with PPI enrichment p-value < 1.0e-16, number of edges 153, average node degree 7.85, and average local clustering coefficient is 0.589. The STRING-based information was inputted into the Cytoscape tool for constructing the PPI network. The PPI network of 38 genes is displayed in Fig. 5. The redder node indicated the highest number of degrees of interaction in the network and yellow node indicated the smaller number of interactions with other genes. Then, we used the cytoHubba (Chin et al. 2014) a user-friendly interface in Cytoscape tool for exploring the highly interconnected nodes in the PPI network. We identified the top interconnected nodes in the PPI network from the original PPI network by using the cytoHubba. The top 10 interconnected nodes (based on the degree of interaction with other nodes) are AKT1, EGFR, SRC, MMP9, MMP2, KDR, IGF1R, PTK2, ABCG2, and MET (Table 3), indicating that the cellular signaling controlled by these genes may be regulated by the quercetin.

The protein–protein interaction (PPI) network of common 39 genes that are interacted with quercetin in STITCH and the Swiss Target Prediction tool
Functional enrichment analysis of quercetin associated genes
The functional analysis of gene ontology included the biological processes (Fig. 6, Supplementary Table S5), cellular components (Supplementary Table S6), and molecular functions (Supplementary Table S7). The significantly enriched top biological processes are a response to oxygen-containing compounds, cellular response to oxygen-containing compounds, regulation of cell death, cellular response to chemical stress, positive regulation of signaling, response to oxidative stress, apoptotic process, cell population proliferation, cellular response to reactive oxygen species, and response to nitrogen compound. The significant cellular components are mitochondrion, receptor complex, an extrinsic component of the cytoplasmic side of the plasma membrane, external side of the apical plasma membrane, an extrinsic component of membrane, plasma membrane region, an extrinsic component of the plasma membrane, apical plasma membrane, side of the membrane, and intrinsic component of the plasma membrane. Moreover, the top molecular functions are adenyl nucleotide binding, protein kinase activity, ribonucleotide binding, kinase activity, transferase activity transferring phosphorus-containing groups, identical protein binding, protein tyrosine kinase activity, oxidoreductase activity, transition metal ion binding, and transmembrane receptor protein tyrosine kinase activity.

The top ten biological processes (BP), cellular components (CC), and molecular functions (MF) are significantly associated with quercetin-targeted genes
Furthermore, KEGG pathways enrichment analysis identifies the 50 significant terms associated with quercetin interacted genes (Table 4). The pathways mainly involved cancer, metabolism, cellular signaling cascades, and immune regulations. The cancer-associated pathways are pathways in cancer, prostate cancer, melanoma, endometrial cancer, acute myeloid leukemia, glioma, small cell lung cancer, bladder cancer, non-small cell lung cancer, colorectal cancer, pancreatic cancer, renal cell carcinoma, and chronic myeloid leukemia. The metabolic pathways are arachidonic acid metabolism, tryptophan metabolism, tyrosine metabolism, and metabolism of xenobiotics by cytochrome P450. The cellular signaling and developmental pathways are focal adhesion, ErbB signaling pathway, VEGF signaling pathway, progesterone-mediated oocyte maturation, adherens junction, Endocytosis Insulin signaling pathway, ABC transporters, steroid hormone biosynthesis, gap junction, GnRH signaling pathway, oocyte meiosis, cell cycle, neurotrophin signaling pathway, axon guidance, Jak-STAT signaling pathway, regulation of actin cytoskeleton, mTOR signaling pathway, p53 signaling pathway, apoptosis, melanogenesis, tight junction, calcium signaling pathway. The enriched immune regulatory pathways are leukocyte transendothelial migration, chemokine signaling pathway, epithelial cell signaling in Helicobacter pylori infection B cell receptor signaling pathway, cytokine-cytokine receptor interaction, T cell receptor signaling pathway, Fc epsilon RI signaling pathway, hematopoietic cell lineage, Fc gamma R-mediated phagocytosis, and Toll-like receptor signaling pathway. These findings indicated that the quercetin-associated interactions of genes are regulating the key biological functions.
Discussion
The in vitro and in vivo findings are usually verified using the computational model in a drug discovery process. Molecular docking, a sort of computer study, is often used to predict ligand-target interactions and acquire a better understanding of natural product biological activity. It also outlines the likely mechanism(s) of action as well as the binding modalities within enzyme binding pockets (Rodenhuis-Zybert et al. 2010). Our anticipated compound Quercetin, a flavonoid, was found to be highly interacted with the receptor of four proteins such as NS5A (PDB ID: 4CL1) for HCV, NS5 (PDB ID: 3EVG) for DENV-2, VP35 (PDB ID: 4IBK) for EBOV, NP-specific inhibitor (PDB ID: 6J1U) for IAV while the bond-energy of quercetin with hepatitis C and influenza viruses are more negative than two others implying the higher binding affinity.
In the Big data era, molecular docking and network pharmacological studies are offering a great development opportunity in terms of algorithm development, theoretical analyses, and applications. However, researchers are concerned about the difficulty of combining massive clinical and experimental data with scientific verification to uncover the regulatory mechanisms of network pharmacology and conduct research more effectively. To avoid ambiguous usage of network pharmacology, data validation before the clinical application is an inevitable step (Hoofnagle, 2002). As a result, experimental validation is required before applying these findings to clinical practice. The PPI network is involved in numerous biological processes, and faulty PPI is at the root of many clinical disorders (Falck-Ytter et al. 2002). We discovered top 10 interconnected nodes (based on the degree of interaction with other nodes) are AKT1, EGFR, SRC, MMP9, MMP2, KDR, IGF1R, PTK2, ABCG2, and MET which are strongly linked to antivirals using the PPI of a targeted protein. The epidermal growth factor receptor (EGFR) is involved in the regulation of essential epithelial processes as well as transcription factor upregulation. IRF1 is first implicated in increased interferon-kappa production, which leads to STAT1 activation and further amplification of downstream interferon-induced genes, such as antiviral effectors and chemokines (Lulli et al. 2016). Matrix Metalloproteinase 9 exerts antiviral activity against respiratory syncytial virus. Persistent MMP9 signaling is associated with tissue remodeling (Dabo et al. 2015) and the progression of many lung diseases including chronic obstructive pulmonary disease (COPD), and asthma (Ventura et al. 2014), and airway infections (Lee et al. 2013).
In addition, we identified GO pathways enrichment analysis of target protein. The GO analysis indicates that the target proteins may bind with the plasma membrane, chromosome, chromatin, regulatory region nucleic acid, or/and cellular receptor of cells for mediating the process of metabolic, immunological, signaling, and/or other activities, to exert signaling and antiviral potential. Pathway analysis revealed some pathways which are involved in the attenuation of oxidative stress and cytokine production. Oxidative stress is found to upregulate the biological enzymes NADPH oxidase, xanthine oxidase, and dual oxidase which altogether defend the antioxidative enzymes (superoxide dismutase, glutathione peroxidase, catalase, etc.), and cause several metabolic and physiological disorders in viral infections while Quercetin reverses the phenomenon and enhances the healing process (Borchi et al. 2010). Moreover, antioxidative enzymes are controlling the PPI network of target proteins which are significantly associated with enriched pathways, suggesting that they are responsible for controlling these signaling networks and enriched pathways to control viral infections.
KEGG pathways are primarily involved in metabolism (cytochrome P450 xenobiotic metabolism, cytochrome metabolism of drugs, drug metabolism, other enzymes, biosynthesis of steroid hormones, ascorbate, and alternate metabolism, interconversion of pentose and glucuronate, porphyrin and chlorophyll metabolism, and starch and sucrose metabolism), immune regulation (Cytokine-cytokine receptor interaction, T cell receptor signaling pathway, natural killer cell-mediated cytotoxicity, adipocytokine signaling pathway, Toll-like receptor signaling pathway, allograft rejection, and systemic lupus erythematosus), neurological regulation (amyotrophic lateral sclerosis), cellular development (apoptosis and p53 signaling pathway), cellular signaling (NOD-like receptor signaling pathway, RIG-I-like receptor signaling pathway, and PPAR signaling pathway), molecular transportation (ABC transporters) and interconversion of pentose and glucuronate metabolism and PPAR signaling pathway (Shen et al. 2021). Our anticipated Quercetin molecule is found to be highly interacted with the JAK-STAT Pathway which is considered a novel target to tackle viral infections because the Janus kinase–signal transducer and activator of transcription (JAK-STAT) pathway is especially relevant due to its essential role in the regulation of local and systemic inflammation in response to viral infections, being, therefore, a putative therapeutic target. Therefore, the genes that interacted with Quercetin are responsible for controlling these signaling networks and enriched pathways (Ezeonwumelu et al. 2021). Based on the compounds–protein targets relationships, it is evident that Quercetin is acting on the multi-target genes and proteins which are substantial contributions of Quercetin to biological and physiological activities to manage viral infections.
Conclusion
Antiviral effects of Quercetin have been investigated in this research through computational model. Molecular docking study showed that Quercetin had a strong binding affinity against HCV and IAV among other studied viruses. Highest binding energy – 6.954 kcal/mol was noticed for IAV. In the network pharmacological study, 38 hub genes showed a top ten interconnected nodes were triggered by Quercetin leading to expedite its antiviral action. Results demonstrate that the Quercetin could be a potential antiviral agent through a comprehensive study in experimental antiviral models.
Data availability
Data will be available upon request.
References
Anand David AV, Arulmoli R, Parasuraman S (2016) Overviews of biological importance of quercetin: a bioactive flavonoid. Pharmacogn Rev 10(20):84–89. https://doi.org/10.4103/0973-7847.194044
Ashwini S, Varkey SP, Shantaram M (2017) In Silico Docking of Polyphenolic Compounds against Caspase 3-HeLa Cell Line Protein. Int J Drug Dev Res 9(3):28–32
Bachmetov L, Gal-Tanamy M, Shapira A, Vorobeychik M, Giterman-Galam T, Sathiyamoorthy P et al (2012) Suppression of hepatitis C virus by the flavonoid quercetin is mediated by inhibition of NS3 protease activity. J Viral Hepat 19(2):e81-88. https://doi.org/10.1111/j.1365-2893.2011.01507.x
Badshah SL, Faisal S, Muhammad A, Poulson BG, Emwas AH, Jaremko M (2021) Antiviral activities of flavonoids. Biomed Pharmacother 140:111596. https://doi.org/10.1016/j.biopha.2021.111596
Batiha GE, Beshbishy AM, Ikram M, Mulla ZS, El-Hack MEA, Taha AE, Elewa YHA (2020) The pharmacological activity, biochemical properties, and pharmacokinetics of the major natural polyphenolic flavonoid: Quercetin. Foods. https://doi.org/10.3390/foods9030374
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B (methodol) 57(1):289–300. https://doi.org/10.1111/j.2517-6161.1995.tb02031.x
Borchi E, Bargelli V, Stillitano F, Giordano C, Sebastiani M, Nassi PA, d'Amati G, Cerbai E, Nediani C (2010) Enhanced ROS production by NADPH oxidase is correlated to changes in antioxidant enzyme activity in human heart failure. Biochim Biophys Acta 1802(3):331–338. https://doi.org/10.1016/j.bbadis.2009.10.014
Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY (2014) cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol Suppl 4(Suppl 4):S11. https://doi.org/10.1186/1752-0509-8-S4-S11
Dabo AJ, Cummins N, Eden E, Geraghty P (2015) Matrix metalloproteinase 9 exerts antiviral activity against respiratory syncytial virus. PLoS ONE 10(8):e0135970. https://doi.org/10.1371/journal.pone.0135970
dos Santos A, dos Santos AE, Kuster RM, Yamamoto KA, Salles TS, Campos R, de Meneses MD, Soares MR, Ferreira D (2014) Quercetin and quercetin 3-O-glycosides from Bauhinia longifolia (Bong.) Steud. show anti-Mayaro virus activity. Parasit Vectors 28(7):130. https://doi.org/10.1186/1756-3305-7-130
Ezeonwumelu IJ, Garcia-Vidal E, Ballana E (2021) JAK-STAT pathway: a novel target to tackle viral infections. Viruses 13(12):2379. https://doi.org/10.3390/v13122379
Falck-Ytter Y, Kale H, Mullen KD, Sarbah SA, Sorescu L, McCullough AJ (2002) Surprisingly small effect of antiviral treatment in patients with hepatitis C. Ann Intern Med 136(4):288–292. https://doi.org/10.7326/0003-4819-136-4-200202190-00008
Fanunza E, Iampietro M, Distinto S, Corona A, Quartu M, Maccioni E et al (2020) Quercetin Blocks Ebola virus infection by counteracting the VP24 interferon-inhibitory function. Antimicrob Agents Chemother. https://doi.org/10.1128/AAC.00530-20
Gfeller D, Grosdidier A, Wirth M, Daina A, Michielin O, Zoete V (2014) SwissTargetPrediction: a web server for target prediction of bioactive small molecules. Nucleic Acids Res 42(Web Server issue):W32–W38. https://doi.org/10.1093/nar/gku293
Hoofnagle JH (2002) Course and outcome of hepatitis C. Hepatology 36(5 Suppl 1):S21–S29. https://doi.org/10.1053/jhep.2002.36227
Hossen MA, Ali Reza ASM, Amin MB, Nasrin MS, Khan TA, Rajib MHR et al (2021) Bioactive metabolites of Blumea lacera attenuate anxiety and depression in rodents and computer-aided model. Food Sci Nutr 9(7):3836–3851. https://doi.org/10.1002/fsn3.2362
Kanehisa M, Sato Y, Furumichi M, Morishima K, Tanabe M (2019) New approach for understanding genome variations in KEGG. Nucleic Acids Res 47(D1):D590–D595. https://doi.org/10.1093/nar/gky962
Khachatoorian R, Arumugaswami V, Raychaudhuri S, Yeh GK, Maloney EM, Wang J, Dasgupta A, French SW (2012) Divergent antiviral effects of bioflavonoids on the hepatitis C virus life cycle. Virology 433(2):346–355. https://doi.org/10.1016/j.virol.2012.08.029
Kim Y, Narayanan S, Chang KO (2010) Inhibition of influenza virus replication by plant derived isoquercetin. Antiviral Res 88(2):227–235. https://doi.org/10.1016/j.antiviral.2010.08.016
Lani R, Hassandarvish P, Chiam CW, Moghaddam E, Chu JJ, Rausalu K et al (2015) Antiviral activity of silymarin against chikungunya virus. Sci Rep 5:11421. https://doi.org/10.1038/srep11421
Lee YH, Lai CL, Hsieh SH, Shieh CC, Huang LM, Wu-Hsieh BA (2013) Influenza A virus induction of oxidative stress and MMP-9 is associated with severe lung pathology in a mouse model. Virus Res 178(2):411–422. https://doi.org/10.1016/j.virusres.2013.09.011
Lee M, Son M, Ryu E, Shin YS, Kim JG, Kang BW, Cho H, Kang H (2015) Quercetin-Induced Apoptosis Prevents EBV Infection. Oncotarget. 6(14):12603–12624. https://doi.org/10.18632/oncotarget.3687
Lulli D, Carbone ML, Pastore S (2016) Epidermal growth factor receptor inhibitors trigger a type I interferon response in human skin. Oncotarget 26;7(30):47777–47793. https://doi.org/10.18632/oncotarget.10013
Maestro (2021) Schrödinger Release 2021-3; Maestro, Schrödinger, LLC, New York, NY
Mourya DT, Yadav PD, Ullas PT, Bhardwaj SD, Sahay RR, Chadha MS, Shete AM, Jadhav S, Gupta N, Gangakhedkar RR, Khasnobis P, Singh SK (2019) Emerging/re-emerging viral diseases & new viruses on the Indian horizon. Indian J Med Res 149(4):447–467. https://doi.org/10.4103/ijmr.IJMR_1239_18
Ninfali P, Antonelli A, Magnani M, Scarpa ES (2020) Antiviral properties of flavonoids and delivery strategies. Nutrients 12(9):2534. https://doi.org/10.3390/nu12092534
Rodenhuis-Zybert IA, Wilschut J, Smit JM (2010) Dengue virus life cycle: viral and host factors modulating infectivity. Cell Mol Life Sci 67(16):2773–2786. https://doi.org/10.1007/s00018-010-0357-z
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13(11):2498–504.https://doi.org/10.1101/gr.1239303
Shen M, Xu M, Zhong F, Crist MC, Prior AB, Yang K, Allaire DM, Choueiry F, Zhu J, Shi H (2021) A multi-omics study revealing the metabolic effects of estrogen in liver cancer cells HepG2. Cells 10(2):455. https://doi.org/10.3390/cells10020455
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102(43):15545–15550. https://doi.org/10.1073/pnas.0506580102
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, Mering CV (2019) STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47(D1):D607–D613. https://doi.org/10.1093/nar/gky1131
Szklarczyk D, Santos A, Von Mering C, Jensen LJ, Bork P, Kuhn M (2016) STITCH 5: augmenting protein–chemical interaction networks with tissue and affinity data. Nucleic Acids Res 44(D1):D380–D384. https://doi.org/10.1093/nar/gkv1277
Ventura I, Vega A, Chacón P, Chamorro C, Aroca R, Gómez E, Bellido V, Puente Y, Blanca M, Monteseirín J (2014) Neutrophils from allergic asthmatic patients produce and release metalloproteinase-9 upon direct exposure to allergens. Allergy 69(7):898–905. https://doi.org/10.1111/all.12414
Yang J, Zhang Z, Yang F, Zhang H, Wu H, Zhu F et al (2021) Computational design and modeling of nanobodies toward SARS-CoV-2 receptor binding domain. Chem Biol Drug Des 98:1–18. https://doi.org/10.1111/cbdd.13847
Acknowledgements
The authors acknowledge the Research and Publication Cell, University of Chittagong, for supporting this research this work under the research project “Natural Therapeutics Against Long Term Effects of Covid-19: An in vitro and Network pharmacological Screening on
Organ-Specific Targeted Pathways.”
Author information
Authors and Affiliations
Contributions
MAR: Research design, planning, supervision, and management. FMS: Data collection, literature review, manuscript preparation. MNU: Network-pharmacological study. SS: Manuscript preparation, literature review. MAH: Molecular docking study, computational analysis. All authors have gone through the manuscript and agreed to submit to In Silico Pharmacology.
Corresponding author
Ethics declarations
Conflict of interest
Authors declare that they don’t have any conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Rahman, M.A., Shorobi, F.M., Uddin, M.N. et al. Quercetin attenuates viral infections by interacting with target proteins and linked genes in chemicobiological models. In Silico Pharmacol. 10, 17 (2022). https://doi.org/10.1007/s40203-022-00132-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40203-022-00132-2