
Abstract
The SARS-CoV-2 deadly virus has infected more than 618 million people and caused nearly 6.5 million deaths worldwide. It contains a single-stranded RNA and polypeptides encoding structural and non-structural proteins; used for recognizing specific receptors followed by their penetration into the human cells. Vaccines and medications to date against the virus show varying levels of efficacy along with severe side effects. So, plant-derived drugs pose favorable alternatives with specific activities and minimum side effects. In this study, the antiviral potential of phytochemicals was investigated with special emphasis on “Flavonoids”'. Based on this, five major flavonoids and four standard synthetic drugs were subjected to molecular docking analysis against the crucial receptor proteins of COVID-19 (3CLpro, Mpro, and ACE2). According to our results, Apigenin (flavone) and Epigallocatechin Gallate (flavanol) show the best docking intensities with our chosen target proteins, proving them to be efficient candidates against the SARS-CoV-2 virus. The docking results were evaluated from their binding free energy values showed comparatively lower binding affinities of the synthetic drugs than the selected phytochemicals. The feasibility, favorable conformation and stability of the receptors were further validated using the Ramachandran plot. The two best flavonoids were then further tested against the paramount Delta and Omicron variants of SARS-CoV-2 virus. The results of the in silico study reflect that Epigallocatechin Gallate and Apigenin had maximum affinity to the target proteins of the Omicron and Delta variants respectively, implying its speculated antiviral potency. This study hence opens up a new avenue of utilizing the compounds mentioned above to disrupt the integrity and virulence of the virus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Coronavirus Disease 19 (COVID-19) global pandemic has infected more than 618 million people and claimed the lives of over 6.5 million people worldwide as reported on September 21, 2022. The scenario in India is alarming itself with more than 44 million cases reported to date and more than 5 lakhs total deaths. This all together led the research enthusiasts to find a potential therapeutic medicine to combat the disease [9]. The Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) has a spherical or pleomorphic shape, encasing a positive-sense single-stranded RNA and four structural proteins, namely Membrane (M) protein, Spike (S) protein, Nucleocapsid (N) protein, and Envelope (E) protein which assemble to form a complete infectious virion. This virus belongs to Beta CoV family and invades mammalian upper to lower respiratory tract and infect the gastrointestinal tract by unknown mechanisms [3, 7, 19]. Patients suffering from respiratory problems along with other comorbidities are more prone to the SARS-CoV-2 virus and thus require rapid medication with potent drugs [24]. The recent mutant variants of the virus (Delta, Omicron) have eventually increased the graphs of daily active cases and mortality rates. Until now, the vaccines and synthetic drugs certified by the World Health Organisation (WHO) against the virus have shown low effectiveness, detrimental side effects, and deaths have been reported even after the completion of vaccination doses.
Plant-derived compounds on the other hand offer higher efficacies, safe and secure, and act as dependable alternatives for treatment with no to minimum side effects [2, 14]. Therefore, our study aims to identify natural antiviral compounds based on molecular docking results to inhibit the high infection rate of COVID-19. Since the traditional drug discovery cycle involves a long time period starting from a search of drugs to its trials, and an even larger sum of money, hence new methodologies are now put to test for faster results. Computational docking is thought to be an effective strategy and a widely used technique in the drug discovery process for understanding the molecular aspects of proteins and protein-ligand interactions. It involves molecular modeling technique that characterizes the role of specific amino acids present in surface contact areas of the SARS-CoV-2 virus and its receptor proteins [24]. Thus, in-silico techniques have been used to find the best flavonoid showing inhibitory effects against viral or host receptor protein, based on their docking scores obtained after evaluation. Different flavonoid classes have been docked to three target receptors and parallelly checked altogether to get antiviral drug formulations as fast as possible [8, 13]. Furthermore, visualization software analyses detailed qualitative assessments of interactions between ligands and receptors by estimating the strength and nature of bonds formed between them.
Target Receptor proteins used in our study
The receptor proteins studied include 3-chymotrypsin-like protease (3CLpro), Main protease (Mpro) of SARS-Cov-2 virus and other accessory proteins like Angiotensin-Converting Enzyme 2 (ACE2) of the host cell. The 3CLpro enzyme is a three-domain cysteine protease with an active site highly conserved in Middle East Respiratory Syndrome (MERS), SARS-CoV, and nowadays SARS-CoV-2. After infection, viral RNA molecules produce polyproteins that undergo cleavage at distinct locations induced by 3CLpro which is essential for viral replication [1, 10, 25, 27]. ACE2 is a dipeptidyl carboxypeptidase and a type 1 transmembrane protein present in the alveolar and bronchial respiratory epithelial cells of the host as a receptor commonly used for SARS-CoV-2 viral entry by binding to the receptor-binding domain (RBD). Amino acids like Leu, Phe, Glu, Ser, Asp, and Tyr present in the RBD domain usually interact with ACE2. Thus, a change in the conformation of the ACE2 catalytic pocket by the bioactive molecule is necessary to disrupt or block the interaction [9, 11, 25, 29]. Mpro, plays an important role in cleaving polyproteins(pp) 1a and 1ab for triggering viral replication and transcription; and is thus used as a potential drug target [7, 14, 16, 24].
The new variants of the virus, Delta (B.1.617.2) and Omicron (B.1.1.529) were first documented in October, 2020, and November, 2021 respectively in multiple countries. The Delta variant had shown a transmissibility rate of 40–60% more than Alpha and twice as the original Wuhan strain of SARS-CoV-2. A Center of Disease Control study showed 9 deaths/1000 cases during the Omicron surge, compared to 13 deaths/1000 cases during the Delta surge. Health experts reported that Omicron causes milder symptoms than previous variants, but it spreads quickly and has infected people very fast.
Flavonoids and its chemistry
Flavonoids are plant phenolic compounds based on a flavan nucleus and have a skeleton consisting of two benzene rings (A and B) linked via a heterocyclic pyrene ring (C ring). They can be divided into various groups, but our study deals with flavones (Apigenin), flavonols (Quercetin, Kaempferol), and flavan-3-ols (Catechin, Epicatechin, Epigallocatechin Gallate) based on their antiviral nature [30]. The number of replacements in hydroxyl groups in their nuclear structure determines the biological potency of flavonoids. It mediates its antioxidant effects by scavenging free radicals and chelating metal ions from hydroxylated phenolic compounds synthesized in plants in response to microbial infection [26]. All flavonoid groups differ in the level of chemical modifications and pattern of substitution of A and B benzene rings. Flavonoids are widely distributed in different plant parts such as nuts, seeds, fruits, and vegetables that people consume frequently [30]. The readily soluble nature of flavonoids plays a major role in therapeutic efficacy. Polyphenols can interrupt the life cycle of viruses and block viral replication, immune-boosting responses, and have anti-inflammatory effects against infections [26]. They have potential biological benefits such as anticancer, antibacterial, antifungal, and antiviral activities [24, 30]. Since flavonoids have extensive health-beneficial properties, they could be of considerable importance for strengthening the host defense mechanism against viral diseases by lowering infection and by downregulation of cytokine production [10, 26]. The antiviral effects of flavonoids were studied in-silico by docking and virtual screening to predict the binding affinity, which may be indicative of the drug’s ability to inhibit the proteolytic activity of the target receptor molecules. Plant-derived drugs are thoroughly evaluated in terms of their potency, efficacy, bioavailability, adverse effects on non-target sites, safety, different stages of preclinical and clinical trials, etc. before being available for public use [12, 28].
Apigenin, a major flavone compound has the ability to sharply decrease interleukin-6 supports the anti-inflammatory nature. It is also reported to have antiviral properties by inhibiting viral coat protein synthesis and disrupting viral RNA association with transcription factors [21, 30]. The hydroxyl group of quercetin recognizes specific amino acid residues on 3CLpro and Mpro of SARS-CoV-2 and theoretically provides 50% inhibitory activity. It is known that Vitamin C and quercetin application together show high anti-inflammatory activities and further inhibit virus entry and block the formation of viral life cycle enzymes [4, 21]. Kaempferol shows inhibitory activity against the 3a ion channel of coronavirus, formed by 3a-coded proteins and it has the potential to inhibit viruses. Docking analysis further demonstrated the potential to inhibit viruses due to the presence of high affinity binding of kaempferol to the ACE2 receptor and regulate the T-cell receptor [21, 26, 30]. Among flavan-3-ols, the antiviral activity of catechin and its derivatives Epicatechin and Epigallocatechin gallate (EGCG) found in tea have been reported [7, 20]. ). EGCG is a well-known antioxidant and shows anti-inflammatory activities. Natural catechins largely appear to have different mechanisms of action such as they may inhibit viral enveloped DNA, (+) or (−) stranded RNA viruses; while EGCG exhibits a high ability to block the early stages of infection by inhibiting reverse transcriptase in vitro and in vivo [13, 15, 16, 31].
Materials and methods
Selection and Preparation of Ligands and Receptors
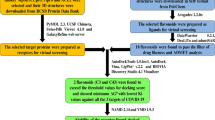
The 3D structures of five potent flavonoids i.e. Epicatechin (PubChem ID:72,276), Epigallocatechin Gallate (PubChem ID:65,064), Quercetin (PubChem ID:5,280,343), Kaempferol (PubChem ID:5,280,863) and Apigenin (PubChem ID:5,280,443) downloaded from PubChem crystal database server (https://pubchem.ncbi.nlm.nih.gov/) in sdf format were docked with important receptor molecules of COVID-19 involved in virus replication and entry. The 3D structures of the original Wuhan strain of SARS-CoV-2 target proteins: 3CLpro (PDB ID: 6M2N); Mpro (PDB ID: 7BRO) and ACE2 complexed with spike glycoprotein (PDB.ID:7DMU) were downloaded from RCSB Protein Data Bank in PDB format (http://www.rcsb.org/) [5, 6]. According to our study, the flavonoids with the best docking scores were further tested for their bonding with the Open state of Delta variant spike protein (PDB ID: 7W92) and Omicron Spike B.1.1.529 (PDB ID: 7QO7) of SARS-CoV-2. The reference articles and physicochemical properties of ligands have been tabulated in supplementary files 1 & 2 respectively. For a better comparative study, Catechin (PubChem ID:9064) as the natural control drug and Chloroquine (PubChem ID:2719) as the synthetic control drug have been used as ligands for docking analysis. Furthermore, three other synthetic drugs, Remdesivir (PubChem ID:121,304,016), Arbidol (PubChem ID:131,411), and Doxycycline (PubChem ID:54,671,203), were put to test with our selected proteins to significantly show the comparison based on the binding energy intensities with natural drugs. The pdb files of ligands were created using the Online SMILES Translator and Structure File Generator (https://cactus.nci.nih.gov/translate/), and later were converted to pdbqt file format using in Auto Dock Tools [7, 11, 13].
Receptor structure validations
PROCHECK and ERRAT web server tools were used to validate the stability, structural quality, and reliability of the protein structures. Good high-resolution structures generally produce quality factors of around 95% or higher. Figures 5a and 6a, and 7a show the Ramachandran plots and Figs. 5b and 6b, and 7b show the Overall Quality Factor of the receptor models. All the structural details supporting the protein models were listed in Supplementary file 3.
Molecular docking and visualisation
Auto Dock Vina software helps us in docking the target protein receptors with the ligands (http://vina.scripps.edu/). The active binding pocket sites and the amino acid residues in it were generated and were run to determine the binding affinities and analyze them by AutoDockVina software [17, 18, 22, 23]. Accordingly, the grid dimensions 40 × 40 × 40Å and center coordinates for docking were set as 12.239, −13.251, and 5.144 for 7BRO; −45.272, −29.842 and 27.319 for 6M2N; 60.86, −56.197, and 45.075 for 7DMU. The lowest Root Mean Square Deviation (RMSD) values were selected to evaluate binding energies [7]. PyMOL and DiscoveryStudioVisualizer visualization software was used to analyze the 3D docked receptor-ligand complexes; and ProteinPlus program to study their interactions.
Results and discussion
The huge number of COVID-19 infections worldwide has compelled us to find a solution to this viral pandemic. This study was designed to determine the binding capacities of several phytochemicals with three crucial receptor proteins. The major plant-derived compounds chosen for our study are Epicatechin, Epigallocatechin gallate, Quercetin, Kaempferol, and Apigenin (Supplementary Fig. 1). The target proteins from the virus are reported to be essential for their growth, infectivity, and survival. The chosen viral targets are 3CLpro, Mpro, and ACE2 (Supplementary Fig. 2). These phytochemicals were put to test based on their medicinal properties, efficacy, etc. and computational docking was carried out to determine their binding to the target proteins. Supplementary Fig. 3 represents the flowchart of the computational docking study. Computational binding deals with the orientation of small flexible ligand to a target receptor protein as well as conformation of ligand-receptor complex when bound to each other. Firstly, the selected protein structures of study are thoroughly analyzed in AutoDockTools and then drug molecules of interest are docked into the binding pockets of the target proteins. The process of evaluation of a particular pose/conformation is done by counting the number of favourable intermolecular hydrogen bond interactions, Vander-Waals interaction and hydrophobic contacts. Secondly, docking study helps to conclude the outcome of a receptor-ligand interaction, which may be either “Tight/High, Moderate/Intermediate or Loose/Low” binding affinity depending on the free binding energies in kcal/mol. The process of classifying which ligands are most likely to interact favourably to a particular receptor is based on the calculation of free binding energy values in kcal/mol from the negative RMSD (Root Mean Square deviation) values done by AutoDockVina software. The binding affinity increases as the free energy value becomes more negative. Negative values indicate favourable formation of ligand-receptor complexes with high affinity. The affinity value with 0 RMSD is considered to be the best value. For the first selected enzyme 3CLpro, Apigenin showed the lowest binding energy value of −8.6 kcal/mol. Then on studying the interaction between them in ProteinsPlus it was seen that efficient hydrogen bonds stabilize the active site pockets (Fig. 1). For ACE2 and Mpro, Epigallocatechin Gallate showed higher interactions, −7.4 kcal/mol, and − 8.8 kcal/mol respectively, as it binds much more tightly to the receptors thus rendering our docking results to be structurally stable and reliable (Figs. 2 and 3). As shown in supplementary Fig. 4, we observe that the binding affinities ranged from (−6.1 to −8.8) kcal/mol, also depicted in Table 1. The lower the binding energy value, the faster the chance of binding of ligands (phytocompounds in this case) to the receptor proteins thus blocking the entry pathway of SARS-CoV-2 or its replication process. In the interaction of flavonoids and the RBD domain of spike protein, bonds interlinking Phe and Leu remained weak, while other residues interact more effectively. Apigenin and Epigallocatechin gallate were observed to be the most effective molecules against all protein targets as they remained in the proximity of the receptor with the least rearrangements.

3D and 2D interactions of flavonoids against 3CLpro receptor (PDB ID: 6M2N) depicting the binding energy values in each case and the best value marked in red

3D and 2D interactions of flavonoids against ACE2 receptor complexes with spike glycoprotein (PDB ID: 7DMU) depicting the binding energy values in each case and the best value marked in red

3D and 2D interactions of flavonoids against the Mpro receptor (PDB ID: 7BRO) depicting the binding energy values in each case and the best value marked in red
Also in our study, two control drugs, Catechin as the natural control drug and Chloroquine as the synthetic control drug, having high effectivity against RNA viruses were utilized to show that our selected phytochemicals are more favorable (Supplementary Fig. 4). Catechin shows binding energy values of (−7.2) kcal/mol, (−6.0) kcal/mol, and (−7.5) kcal/mol and Chloroquine shows (−5.6) kcal/mol, (−5.0) kcal/mol, and (−5.8) kcal/mol against 3CLpro, ACE2, and Mpro respectively. Some other well-known synthetic drugs showing efficient antiviral properties like Remdesivir, Arbidol, Doxycycline, etc have also been tested against the receptor proteins of SARS-CoV-2. These drugs cannot bind efficiently to the active site pockets of the receptors and show an overall average binding energy value of −6.5 kcal/mol. So, these drugs are now removed from the recent treatment protocol guidelines for COVID-19. Multiple measurements to calculate receptor-ligand interaction can also be undertaken using the ‘central tendency’ parameter where average docking score values of each component are considered. Therefore, a comparison of binding energy values of each data set to standard scores of control drugs helps us in ranking them accordingly. The effectivity of the phytochemicals and synthetic drugs against each receptor protein based on the average docking score values in kcal/mol are tabulated in Table 1.
We can observe that Apigenin and catechin derivatives, Epicatechin and Epigallocatechin Gallate can bind much more tightly than flavonols (Quercetin and Kaempferol) to the active site of receptor proteins, thus preventing virus replication and also the entry of the virus into the host. All the drugs are shown to obey “Lipinski’s rule of five”. Also, the drug molecules satisfy the ADMET properties with good metabolic rates and are not reported to show any toxic or carcinogenic effects by the Hazardous Substances Data Bank (HSDB) of the PubChem database. Our selected synthetic drugs do not show efficient docking with our selected receptor proteins as depicted by their binding energy values and also cause environmental hazards and may also induce alterations of metabolic rates in humans and some may serve as irritants.
Some additional supplementary studies have been done to indicate the structural stability and efficacy of the receptor proteins used as targets by the drug molecules. For this, the PROCHECK web server was used to show the different Ramachandran Plot statistics which determine the most favored, allowed, and disallowed regions of the active sites (Supplementary Fig. 5a, 6a, and 7a).On the other hand, the ERRAT web server depicts high-resolution models of all the receptors based on Overall Quality Factor values which have been graphically plotted in supplementary Fig. 5b, 6b, and 7b.
Vander Waals interactions, classical and non-classical hydrogen bonds, and hydrophobic and electrostatic interactions play an important role in receptor-ligand interaction which ultimately helps in the determination of good results by increasing the docking scores. The structural fold of the protein is stabilized by the various inter and intra-hydrogen bonds which play crucial roles to accommodate the ligand at the active site of a protein. So, we next evaluated the structural and thermodynamic stability of the complexes by identifying the pattern of intermolecular interactions. The ProteinsPlus showed efficient hydrogen bond interactions of all the proteins with their selected ligands. Table 2 represents the amino acid compositions and drug score values of the best-docked subpockets of the 3D structure of receptor proteins with the drugs. Also, the hydrogen and hydrophobic bond interaction values are tabulated which helps in stabilizing the active site pockets. The assay and docking results indicate the following – firstly, flavonoids have a wide range of binding affinity to the proteases of SARS-CoV-2 due to the presence of hydrophobic aromatic rings and hydrophilic hydroxyl groups, and secondly, the presence of carbohydrate groups highly influences the binding affinity thus providing best docking scores.
The best two flavonoids i.e., Apigenin and Epigallocatechin gallate, were further tested against the SARS-CoV-2 Delta and Omicron variant Spike proteins, which are regarded to be the most violent and contagious. The average free binding energy values in kcal/mol indicate that not only do Apigenin and Epigallocatechin gallate bind significantly more strongly to those variants than our synthetic drug, Doxycycline, but they also have higher binding affinities to them than our chosen receptors (Table 3). The 3D and 2D interactions of the receptor-ligand complexes were obtained after visualizing in PyMol and DiscoveryStudioVisualizer respectively (Figs. 4 and 5). This provides a new avenue for determining the antiviral potential of natural phytocompounds like flavonoids. These new variants carry some genetic changes that lead to significant community transmission with an increase in the number of daily cases and mortality rates over time. This prominently highlights the disease severity and suggests an emerging risk to global health.

a 3D interactions of Apigenin against the Delta variant (PDB ID: 7W92) visualized in PyMol b 3D interactions of Epigallocatechin Gallate against the Delta variant spike protein (PDB ID: 7W92) visualized in PyMol c 2D diagram depicting the different types of bond interactions between Apigenin and Delta variant spike protein visualized by DiscoveryStudioVisualizer d 2D diagram depicting the different types of bond interactions between Epigallocatechin Gallate and Delta variant spike protein visualized by DiscoveryStudioVisualizer

a 3D interactions of Apigenin against the Omicron spike B.1.1.529 (PDB ID: 7QO7) visualized in PyMol b 3D interactions of Epigallocatechin Gallate against the Omicron spike B.1.1.529 (PDB ID: 7QO7) visualized in PyMol c 2D diagram depicting the different types of bond interactions between Apigenin and Omicron spike B.1.1.529 visualized by DiscoveryStudioVisualizer d 2D diagram depicting the different types of bond interactions between Epigallocatechin Gallate and Omicron spike B.1.1.529 visualized by DiscoveryStudioVisualizer
Conclusive remarks and future outlook
Phytomedicine, the use of medicinal plants for the prevention and treatment of diseases, has emerged worldwide. The potential use of medicinal herb extracts to prevent the spread of the virus by their different mechanisms of action such as inhibiting replication and proliferation of SARS-CoV-2 may offer a reliable solution to this deadly virus without any adverse side effects. It has been reported that natural substrates from Traditional Chinese Medicine (TCM) seemed to have a more positive impact on patients suffering from SARS-CoV, SARS-CoV-2, and influenza viral diseases than conventional therapeutics [6, 25, 30]. A more detailed examination of pharmacological techniques, long-term toxicology, and bioavailability as well as further studies on herbal drug interactions with an emphasis on safety, sustainability, and evidence of efficacy are required in near future for the disclosure of novel antiviral agents. The plant sources chosen in our study produce the different groups of secondary metabolites of flavonoids that have immense medicinal importance and are a major target used to extract and study several unique compounds that have immunomodulatory properties. Also, the receptor proteins selected here are quite common ones which are considered major pillars of COVID-19, involved in envelope formation, virion assembly, and viral pathogenesis, and can be used as a target by many drugs due to their conserved amino acid residues. The molecular docking results of the protein-ligand complexes led to the establishment of structurally compact and strong bonding dynamics. As SARS-CoV, SARS-CoV-2, and MERS-CoV share high similarities due to the presence of several homologous viral proteases, the proposed flavonoids may demonstrate attractive antiviral substrates against the SARS-CoV-2 virus. The combinatory effects of flavonoids, especially when used together with other known standard antiviral drugs may seem to work with better efficiency. A pharmacophore mixture of all the five flavonoids might actively block the majority of virus replication and entry pathways. In vitro, cell-free, and cell-based studies are predominantly compared with studies on animal models to standardize the existing evidence. Furthermore, the appropriate design of clinical trials to demonstrate a prophylactic and therapeutic effect in humans remains challenging. These trials would help to potentially confirm experimental findings into recommendations for use in COVID-19 viral disease. By docking study, we have tried to analyze the potency of the natural drugs to combat receptors for the development of antiviral compounds against coronavirus. As they need several months or years to develop a well-approved therapeutic drug, therefore, for immediate antiviral treatment, flavonoids may serve as favorable agents and help the researchers seeking an effective medication or vaccine production.
Data availability
All the data collected during the study have been provided in the manuscript.
Abbreviations
- ACE2:
-
Angiotensin-converting enzyme 2
- COVID-19:
-
Coronavirus disease 19
- MERS:
-
Middle east respiratory syndrome
- Mpro :
-
Main protease
- RBD:
-
Receptor-binding domain
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- TCM:
-
Traditional Chinese medicine
- 3CLpro :
-
3-Chymotrypsin-like protease
References
Abian O, Ortega-alarcon D, Jimenez-alesanco A, Ceballos-laita L. Structural stability of SARS-CoV-2 3CLpro and identification of quercetin as an inhibitor by experimental screening Olga. Int J Biol Macromol Elsevier. 2020;164:1693–703.
Adhikari B, Marasini BP, Rayamajhee B, Bhattarai BR, Lamichhane G, Khadayat K, Adhikari A, Khanal S, Parajuli N. Potential roles of medicinal plants for the treatment of viral diseases focusing on COVID-19: a review. Phyther Res. 2020;35:1298–312.
Bhowmik D, Nandi R, Jagadeesan R, Kumar N, Prakash A. Identification of potential inhibitors against SARS-CoV-2 by targeting proteins responsible for envelope formation and virion assembly using docking-based virtual screening, and pharmacokinetics approaches. Infect Genet Evol. 2020;84:104451.
Colunga Biancatelli RML, Berrill M, Catravas JD, Marik PE. Quercetin and vitamin C: an experimental, synergistic therapy for the Prevention and Treatment of SARS-CoV-2 related disease (COVID-19). Front Immunol. 2020;11:1–11.
Fu L, Ye F, Feng Y, Yu F, Wang Q, Wu Y, Zhao C, Sun H, Huang B, Niu P, Song H, Shi Y, Li X, Tan W, Qi J, Gao GF. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat Commun. 2020;11:1–8.
Ge X, Liao J. Traditional Chinese medicine network pharmacology study on exploring the mechanism of Xuebijing Injection in the treatment of coronavirus disease 2019. Chin J Nat Med. 2020;18:61–71.
Ghosh R, Chakraborty A, Biswas A, Chowdhuri S. Evaluation of green tea polyphenols as novel coronavirus (SARS CoV-2) main protease (Mpro) inhibitors–an in silico docking and molecular dynamics simulation study. J Biomol Struct Dyn. 2020;39:4362–4374.
Ibrahim MAA, Mohamed EAR, Abdelrahman AHM. Rutin and flavone analogs as prospective SARS-CoV-2 main protease inhibitors: in silico drug discovery study Mahmoud. J Mol Graph Model. 2021;105:107904.
Istifli ES, Netz PA, Sihoglu Tepe A, Husunet MT, Sarikurkcu C, Tepe B. In silico analysis of the interactions of certain flavonoids with the receptor-binding domain of 2019 novel coronavirus and cellular proteases and their pharmacokinetic properties. J Biomol Struct Dyn. 2020;0:1–15.
Jo S, Kim S, Kim DY, Kim MS, Shin DH. Flavonoids with inhibitory activity against SARS-CoV-2 3CLpro. J Enzyme Inhib Med Chem. 2020;35:1539–44.
Joshi T, Joshi T, Sharma P, Mathpal S, Pundir H, Bhatt V, Chandra S. In silico screening of natural compounds against COVID-19 by targeting Mpro and ACE2 using molecular docking. Eur Rev Med Pharmacol Sci. 2020;24:4529–36.
Kapusta K, Kar S, Collins JT, Franklin LM, Kolodziejczyk W, Leszczynski J, Hill GA. Protein reliability analysis and virtual screening of natural inhibitors for SARS-CoV-2 main protease (Mpro) through docking, molecular mechanic & dynamic, and ADMET profiling. J Biomol Struct Dyn. 2020;0:1–18.
Maiti S, Banerjee A. Epigallocatechin gallate and theaflavin gallate interaction in SARS-CoV-2 spike-protein central channel with reference to the hydroxychloroquine interaction: Bioinformatics and molecular docking study. Drug Dev Res. 2021;82:86–96.
Majumder R, Mandal M. Screening of plant-based natural compounds as a potential COVID-19 main protease inhibitor: an in silico docking and molecular dynamics simulation approach. J Biomol Struct Dyn. 2020;0:1–16.
Menegazzi M, Campagnari R, Bertoldi M, Crupi R, Di Paola R, Cuzzocrea S. Protective effect of epigallocatechin-3-gallate (EGCG) in diseases with uncontrolled immune activation: Could such a scenario be helpful to counteract COVID-19? Int J Mol Sci. 2020;21:1–20.
Mouffouk C, Mouffouk S, Mouffouk S, Hambaba L, Haba H. Flavonols as potential antiviral drugs targeting SARS-CoV-2 proteases (3CLpro and PLpro), spike protein, RNA-dependent RNA polymerase (RdRp) and angiotensin-converting enzyme II receptor (ACE2). Eur J Pharmacol. 2021;891:173759.
Mukherjee S, Paul S. In-silico study identifies RO 28-2653 as a novel drug against SARS-CoV2 mutant strains. Int J Comput Biol Drug Des. 2021;14(6):457–80.
Pal A, Pyne N, Paul S. In-silico designing of a multi-epitope vaccine against SARS-CoV-2 and studying the interaction of the vaccine with alpha, beta, delta, and omicron variants of concern. Curr Drug Discov Technol. 2022;20:67–88.
Pan B, Fang S, Zhang J, Pan Y, Liu H, Wang Y, Li M, Liu L. Chinese herbal compounds against SARS-CoV-2: puerarin and quercetin impair the binding of viral S-protein to ACE2 receptor. Comput Struct Biotechnol J. 2020;18:3518–27.
Pandey AK, Verma S. An in-silico evaluation of dietary components for structural inhibition of SARS-Cov-2 main protease. J Biomol Struct Dyn. 2020;0:1–7.
Pastor N, Collado MC, Manzoni P. Phytonutrient and nutraceutical action against COVID-19: current review of characteristics and benefits. Nutrients. 2021;13:1–10.
Paul D, Pyne N, Paul S. Mutation profile of SARS-CoV-2 spike protein and identification of potential multiple epitopes within spike protein for vaccine development against SARS-CoV-2. VirusDis. 2021;32:703–26.
Pyne N, Paul S. Screening of medicinal plants unraveled the leishmanicidal credibility of Garcinia cowa, highlighting norcowanin, a novel anti-leishmanial phytochemical through in-silico study. J Parasit Dis. 2021;46(1):202–14.
Rameshkumar MR, Indu P, Arunagirinathan N, Venkatadri B, El-Serehy HA, Ahmad A. Computational selection of flavonoid compounds as inhibitors against SARS-CoV-2 main protease, RNA-dependent RNA polymerase and spike proteins: a molecular docking study. Saudi J Biol Sci. 2021;28:448–58.
Russo M, Moccia S, Spagnuolo C, Tedesco I, Russo GL. Roles of flavonoids against coronavirus infection. Chem Interact. 2020;328:109211.
Solnier J, Fladerer JP, Flavonoids. A complementary approach to conventional therapy of COVID-19? Phytochem Rev. 2020;20:773.
Tahir ul Qamar M, Alqahtani SM, Alamri MA, Chen LL. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J Pharm Anal. 2020;10:313–9.
Vijayakumar BG, Ramesh D, Joji A, Jayachandra prakasan J, Kannan T. In silico pharmacokinetic and molecular docking studies of natural flavonoids and synthetic indole chalcones against essential proteins of SARS-CoV-2. Eur J Pharmacol. 2020;886:173448.
Williamson G, Kerimi A. Testing of natural products in clinical trials targeting the SARS-CoV-2 (Covid-19) viral spike protein-angiotensin converting enzyme-2 (ACE2) interaction. Biochem Pharmacol. 2020;178:114123.
Zakaryan H, Arabyan E, Oo A, Zandi K. Flavonoids: promising natural compounds against viral infections. Arch Virol. 2017;162:2539–51.
Zhu Y, Xie DY. Docking characterization and in vitro inhibitory activity of Flavan-3-ols and dimeric proanthocyanidins against the main protease activity of SARS-Cov-2. Front Plant Sci. 2020;11:1–14.
Acknowledgements
The authors express their gratitude for the support provided by the UGC-CAS programme, to the Department of Botany, University of Calcutta. The authors also acknowledge the financial.
Funding
The funding was provided by the Council of Scientific and Industrial Research in the form of a CSIR-JRF fellowship to the second author.
Author information
Authors and Affiliations
Contributions
SM performed experimental study, data analysis and manuscript preparation, NP facilitated data interpretation and drafting of the manuscript, SM designed the study, NP amd SM did critical revision and finalized the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no potential conflict of interest in this paper.
Ethical approval
No approval was required for the study.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Corresponding Editor : Manoj Prasad, Reviewers: Parimal Karmakar, Rasmi Abu-Helu, B Datta.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chatterjee, S., Pyne, N. & Paul, S. In silico screening of flavonoids unearthed Apigenin and Epigallocatechin Gallate, possessing antiviral potentiality against Delta and Omicron variants of SARS-CoV-2. Nucleus 67, 359–369 (2024). https://doi.org/10.1007/s13237-023-00431-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13237-023-00431-9