
Abstract
The present work is a comparative study that matches between carriers and techniques used to prepare solid mixtures with glimepiride. The study is directed towards elucidation of the most promising carrier capable of highly improving drug dissolution along with the most successful technique used for drug formulation. Mixtures were tested for drug content and dissolution. The most optimum formulae were characterized by DSC, IR and XRPD. Kinetic treatment of dissolution data was performed for physical and co-ground mixtures, solid dispersions and their adsorbates, triple solid dispersions and their adsorbates, microwave generated or treated solid dispersions. Results revealed that enhancing effect mostly reached maximum with ternary solid dispersion adsorbate (TSDads). The latter technique demonstrated a dramatic increase in drug dissolution rate which was reflected in the shortest half-life for most carriers at variable degrees. The highest dissolution rate was attained with pregelatinized starch and decreased to variable degrees with remaining carriers. Differences were ascribed to chemical nature as well as relative water solubility of carriers. The combined effects of incorporating surfactants, polymers and adsorbents to glimepiride contributed together to improve wetting, reduce crystallinity and caused substantial increase in the surface area which made TSDads the most promising technique for enhancing dissolution of glimepiride.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Presenting a successful drug product to the pharmaceutical industry has been a challenging task for scientists due to bioavailability problems encountered with the formulation of poorly soluble drugs (Pouton 2006; Branchu et al. 2007).
Drugs belonging to BCS II undergo dissolution-rate limited gastrointestinal absorption (Shohin et al. 2011; Reddy et al. 2011). Hence, formulation techniques that accelerate drug dissolution can guarantee a parallel improvement in bioavailability (Wei and Löbenber 2006; Kawabata et al. 2011; Kawakami 2012).
Drug dissolution enhancement has been extensively studied over the past decade through different techniques (Ahuja et al. 2007; Blagden et al. 2007; Cilurzo et al. 2008). Solid dispersion was one of the well known methods that disperse drugs at molecular level improving their wettability and dissolution characteristics through amorphisation of the solid matrix (Leuner and Dressman 2000; Vasconcelos et al. 2007; Van den Mooter 2012). Moreover, surfactants were successfully added to the binary drug carrier combination to constitute ternary solid dispersions (Goddeeris et al. 2008; Al-Obaidi et al. 2011; Martins et al. 2012; Li et al. 2012). Many drawbacks could be overcome by the use of the new triple system, beside a great potentiation in drug dissolution. As technology improves day after day, a new approach to prepare solvent free solid dispersion arose. The technique depended on the heat generated from microwaves to prepare solid dispersions. This approach gained much interest due to the avoidance of any risk originating from organic solvents. An improvement in drug dissolution could thus be achieved and the process could be scaled up on industrial scale production (Bergese et al. 2003; Moneghini et al. 2008; Moneghini et al. 2009).
Glimepiride is one of the famous drugs belonging to BCS II, its poor aqueous solubility usually caused poor dissolution and unpredicted bioavailability (Frick et al. 1998). Literature available on the enhancement of dissolution rate of glimepiride was limited to the study of separate techniques. The most famous were formation of inclusion complex with cyclodextrin (Ammar et al. 2006) or preparation of solid dispersions using either water soluble carriers (Boregowda et al. 2011; Rajpurohit et al. 2011), insoluble carriers (Kiran et al. 2009; Reven et al. 2010; Vidyadhara et al. 2011) or preparation of spray congealed microparticles (Ilić et al. 2009). Solid dispersion was compared to micronization techniques, where both methods proved equal ability for dissolution improvement (Ning et al. 2011). Other researchers added the carriers in combination with the drug in the form of liquid solid compacts (Singh et al. 2011). As it was clearly demonstrated, the enhancement in dissolution illustrated by previous researches was mostly studied through the choice and management of single techniques. Hence the obtained results were thought to be successful relative to the plain drug in question. However, no one can confirm the excellence of a certain technique by itself unless it is matched with others so as to give a complete and precise report about relative effectiveness.
Our strategy in the present work, aimed to widen the scope of research through a comparative study that matched the effectiveness of different polymeric carriers with different physicochemical properties and characteristics. After a proper suggestion of the most optimum carrier, the study was then directed to sum up the benefits of different techniques in a new formulation that guaranteed maximum acceleration in glimepiride dissolution rate and extent.
Materials and methods
Materials
Glimepiride (Sedico Pharmaceuticals, Giza, Egypt), Sodium lauryl sulphate (El-Nasr Pharmaceutical Chemical Co., Cairo, Egypt), Sodium carboxymethyl cellulose (Na CMC; Fluka-Biochemica, Switzerland), Pregelatinized starch (PreGelSt; Colorcon Limited, UK), Crosscarmellose sodium (Ac-Di-Sol; E. Merck, Germeny), Crospovidone XL (CP; FMC Corporation, Philadelphia), and Gelucire 44/14 and 50/13 (Gattefosé, France) were used. Glycolys, Starlac (lactose and maize starch) and Pearlitol flash (mannitol and maize starch) were purchased from Roquette (France). Dimethylformamide (DMF) and Dichloromethane (methylene chloride) were obtained from Fine-Chem Limited (Mumbai).
Methods
Physical mixtures (PM), co-ground mixtures (CGM), solid dispersions (SD) and other techniques based on solid dispersion were prepared using glimepiride and the following carriers viz: sodium carboxymethyl cellulose (NA CMC), pregelatinized starch (PreGelSt), Ac-Di-Sol, Glycolys, Crospovidone (CP), Starlac, Pearlitol flash, Gelucire 44/14, and Gelucire 50/13.
Preparation of physical mixtures (PM)
Each carrier was mixed with the drug at drug to carrier ratios 1:1, 1:3 and 1:5, respectively for 5 min in a mortar till homogenous mixture was obtained.
Preparation of co-ground mixtures (CGM)
Previously ground drug and carrier each alone, were further co-ground together at drug to carrier ratios 1:1, 1:3 and 1:5, respectively, for 15 min using a mortar and pestle till homogenous mixture was obtained.
Preparation of solid dispersions (SD)
(a) Modified solvent evaporation method.
All carriers except Gelucire 44/14 and Gelucire 50/13 were tried by this method at drug to carrier ratios 1:1, 1:3, and 1:5, respectively. Drug was dissolved in DMF. Carrier was dissolved or dispersed in distilled water. Drug solution was poured all at once on the carrier dispersion and the whole was evaporated under vacuum at 70 °C in a rotavap (Heidolph, VV 2000, Germany). The formed solid mass was then sieved (20 mesh, ≤850 um).
(b) Melting method.
Gelucire 44/14 and Gelucire 50/13 were prepared by this method at drug to carrier ratios 1:1, 1:3, and 1:5, respectively. Each wax was heated in a water bath (thermostatically controlled shaker: Memmert, WNB22, Germany) to about 5 °C above its melting point. The drug was added to the molten wax with continuous stirring. The mixture was then allowed to cool on an ice bath till complete solidification. The formed solid mass was then sieved (20 mesh, ≤850 um).
Preparation of ternary solid dispersions (TSD)
Glimepiride TSD with the aforementioned carriers was prepared by the melting method using Gelucire 50/13 as surfactant, at drug to carrier to surfactant ratio 1:5:15, respectively. The drug and carrier were added consecutively with continuous stirring in the molten Gelucire till a homogenous dispersion was obtained. The mixture was then allowed to cool on an ice bath till solidification. The formed solid mass was then sieved (20 mesh, ≤850 um).
Preparation of solid dispersion adsorbates (SDads)
Melt adsorption technique described by Dureja and Madan (2007) and Parmar et al. (2011) was used to prepare SDads of glimepiride with Gelucire 44/14 and Gelucire 50/13. Drug was dispersed in the molten wax as previously described. The molten dispersion was added dropwise to lactose powder (preheated to70 °C) with continuous stirring using glass dropper at drug to wax to adsorbent ratio 1:5:15. The respective SDads was then allowed to cool to room temperature, where it remained having the appearance of free flowing powder.
Preparation of ternary solid dispersion adsorbates (TSDads)
Each TSD with the previously mentioned carriers was dropped (while in the molten state) onto lactose powder (preheated to 70 °C) with continuous stirring to obtain the respective TSDads at drug to carrier to surfactant to adsorbent ratio 1:5:15:30. Mixtures were allowed to cool to room temperature where they remained having the appearance of free flowing powder.
Preparation of microwave generated (MwGSD) or microwave treated solid dispersions (MwTSD)
PM of the drug with the prementioned carriers (except for Gelucire 50/13 and Gelucire 44/14) or SD with the same carriers at 1:5 drug to carrier ratio were further treated in the microwave to obtain their respective MwGSD or MwTSD as follows. Fixed amount of each solid mixture was subjected to microwave irradiation for 2 min (longer treatment caused charring of samples) at a power of 850 W in a domestic microwave oven (KM11VL8W-White-Westinghouse®, USA). Samples were placed in small porcelain dishes; one dish was placed at a time in a fixed place inside the oven. Respective MwGSD or MwTSD were obtained and allowed to cool out to room temperature. PM and SD with either Gelucire types left charred residues when placed for the specified time inside the microwave, so they were omitted from this experiment. All solid mixtures prepared with any of the above techniques were stored in tightly closed vials in a desiccator for further evaluation and/or characterisation.
Physicochemical evaluation of the prepared solid mixtures
Determination of drug content
A specified weight from each formula was dispersed in 50 ml dichloromethane by the aid of magnetic stirring for 15 min. Solutions were filtered and the absorbance of glimepiride was measured spectrophotometrically at λ max 228 nm (Spectrophotometer: Shimadzu, UV-2401 PC, Australia) after doing the appropriate dilution. Results were mean of three determinations.
In vitro drug dissolution
The dissolution profile of the drug in prepared solid mixtures with different carriers was determined using USP dissolution tester (Hanson Research, 64-705-045, USA). An accurately weighed amount of each formula equivalent to 3 mg drug was placed in a rotating basket (at 100 rpm) covered by standardized mesh packet. Dissolution was carried out at 37 °C in 900 ml 0.5 % aqueous solution of sodium lauryl sulphate (Shah et al. 1989; de Waard et al. 2008). Two ml samples were withdrawn at different time intervals (20, 40, 60, 80, 100 and 120 min) consecutively and replaced by fresh media. Absorbance of the samples was measured spectrophotometrically at λ max 228 nm. Results were mean of three determinations.
Kinetic analysis of dissolution data
Data obtained from dissolution experiments were treated statistically according to linear regression analysis. Data were fitted to zero order, first order and Higuchi diffusion model.
Physicochemical characterization of drug in optimized solid mixtures
X-ray powder diffraction (XRPD)
The X-ray diffraction pattern for the pure drug, pure excipients and formulations in question were recorded at room temperature using X-Ray diffractometer: Model XGEN-4000, X1-advanced diffraction systems; Scintag Corp., USA at 45 kV and 40 mA current. The scanning rate was 2 °C/min over a diffraction angle (2θ°) range of 5–70 °C.
Differential scanning colorimetry (DSC)
Differential scanning colorimetry (DSC) was performed for the pure drug, pure excipients and the optimized formulae using differential scanning calorimeter: Model DSC-60; Shimadzu, Kyoto, Japan. Samples of 3–4 mg were placed in flat bottomed aluminum pan and heated in an atmosphere of nitrogen at a temperature range of 20–250 °C at a rate of 10 °C/min.
FT-IR spectroscopy
FT-IR spectroscopy was used to investigate the probability of chemical interactions between ingredients of optimized formulae using infrared spectrophotometer: Shimadzu IR-435, Kyoto, Japan. The scanning was performed within a wave number of 4,000–500 cm−1.
Scanning electron microscopy
The surface morphology of glimepiride and optimized formulae based on solid dispersion with the drug were visualized by scanning electron microscopy (SEM, JSM-6390 LV, JEOL, Tokyo, Japan) at a working distance of 20 mm and an accelerated voltage of 5 kV. Samples were gold coated with a sputter coater (Desk V, Denton Vacuum, NJ, USA) before SEM observation under high vacuum of 45 mTorr and high voltage of 30 mV.
Results and discussions
Drug content
Glimepiride assay in all prepared formulae showed 95–100 % content confirming the absence of drug loss during the dispensing procedures.
Comparison between PM, CGM and SD with different carriers at variable drug: carrier ratios with respect to their effect on dissolution half-life:
Effect of the used technique
Many researches showed that dissolution rate enhancement was more pronounced in case of SD than in case of corresponding physical mixtures of insoluble drugs with different carriers (Vippagunta et al. 2002; Zajc et al. 2005; Shah et al. 2009; Biswal et al. 2009; Nakanishi et al. 2011). In our study, this fact did not hold true for all tested carriers as follows
Ac-Di-Sol (cross linked sodium carboxy methyl cellulose) is a water insoluble polymer characterized by its great ability to absorb water from the environment with subsequent expansion and swelling of particles. Therefore; drug embedded in the respective swollen matrix had more chance for being wetted. It seemed that the wicking and swelling action could better operate when Ac-Di-Sol was present in the solid state, in the form of micronized particles with greater surface area available for water penetration (as in case of CGM) than in the molecular dispersion state (in case of SD). Thus it could be reasonable to obtain the least dissolution t 1/2 for the CGM at the three tested ratios (Fig. 1).

Comparison between PM, CGM and SD with water insoluble carriers with respect to their effect on dissolution half-life
The same effect was shown in case of Glycolys (sodium carboxy methyl starch) where carboxymethylation increased the ability of starch to absorb water (Roquette Pharma and Personal-Care 2012). The water insoluble polymer worked more efficiently when found in the micronized state with the drug giving least values for t 1/2 for CGM followed by SD at the three respective drug to carrier ratios (Fig. 1).
As the pretested water insoluble polymers, CGM of the drug with Crospovidone (CP) showed better dissolution enhancement than the respective SD at 1:1 and 1:3 drug to carrier ratios (Fig. 1). However, increasing the ratio of the polymer to fivefold the drug reversed the relation and gave least t 1/2 value for SD.
In contrast to Ac-Di-Sol (cross linked CMC), solid dispersion of the drug with linear water soluble CMC, showed the least dissolution t 1/2 if compared with the corresponding PM and CGM (Fig. 2). This could be a result of preferential enhanced drug wetting in the molecular dispersion state within the water soluble polymer.

Comparison between PM, CGM and SD with water soluble to partial water soluble carriers with respect to their effects on dissolution half-life
The pregelatinization process involved physical modification of the starch resulting in the combined benefits of the soluble and insoluble functionality of starch. The fact that it could be hydrated with cold water to produce viscous slurries resulted in better wetting ability when molecularly dispersed with the drug. Thus SD of the drug with this polymer gave better dissolution enhancement than the corresponding PM and CGM (Fig. 2).
Starlac consists of 85 % α-lactose monohydrate (freely but slowly water soluble portion) and 15 % maize starch (water insoluble but dispersible portion). As the greater percent of the product consisted of a water soluble ingredient; it would be expected to behave as water soluble polymers giving least t 1/2 with prepared SD followed by CGM then finally PM (Fig. 2).
Pearlitol which is spray dried mannitol has good water solubility and hence had the same enhancing strategy as the prementioned water to partial water soluble polymers (Fig. 2).
Regarding Gelucire 50/13 and 44/14, both being water dispersible surfactants, they act as solubilizers and wetting agents. Their surface power improved wettability of the drug when the latter was molecularly dispersed within their matrices. Thus, SD of glimepiride with both grades gave better dissolution t 1/2 than the corresponding PM and CGM (Fig. 3).

Comparison between PM, CGM and SD of two type Gelucires with respect to their effect on dissolution half-life
Effect of the drug: carrier ratio for the three used techniques
Whatever the order of enhancement for the three studied techniques, it was remarkable that for most of the tested carriers, t 1/2 value decreased as the drug: carrier ratio increased suggesting that, dissolution enhancement correlated with the increase in the ratio of polymer in any of the prepared solid mixtures. However, exceptions occurred for some tested polymers. For CP (Fig. 1), t 1/2 in case of CGM reincreased at drug: carrier ratio 1:5. Therefore, it could be suggested that the enhancing in rate of drug dissolution could be partially limited by the swelling capacity of CP. This might attain equilibrium at 1:3 ratio. However, this hypothesis did not match with PM at the same tested ratios. Larger unmicronized CP particles in PM were assumed to possess greater swelling volume (Bühler 2005) and hence could contribute to the higher efficiency in dissolution enhancement at the 1:5 drug to polymer ratio.
The relation was also reversed in case of PM of drug with Na CMC, where increasing the drug: carrier ratio was accompanied by a parallel increase in dissolution t 1/2 (Fig. 2). This case could be reasonable since Na CMC had a great affinity to aqueous medium, thus was preferentially dissolved from PM, creating a viscous sheath of boundary layer. The subsequent passage of solid drug into the bulk of solution was thus hindered. Increasing the proportion of the polymer lengthened the pathway of the drug to reach the bulk due to increase in the thickness of the stagnant layer. However, introducing the polymer in a micronized state with the drug might increase its affinity for invasion of greater volume of dissolution medium to the large surface area solid mixture. This could allow early wetting of the drug with an enhanced release to occur accordingly.
Again, another exception occurred with the lower melting point, highly hydrophilic Gelucire 44/14, which might exhibit instantaneous softening in contact with dissolution medium creating a colloidal dispersion in the stagnant layer around the core of the solid mixture. The increase in the thickness of this layer was parallel to the increase in the ratio of the polymer in its PM with the drug, causing a resistance to the passage of the drug to the bulk of dissolution medium. Hence a rising values of t 1/2 was shown with higher drug to carrier ratios. From Fig. 3, it could be also deduced that the emulsifying properties of Gelucire 44/14 were best manifested at the SD with higher drug: carrier ratio (1:5), where excessive emulsified droplets in the medium made an obstacle to drug diffusion into the bulk. Thus, optimum enhancement of drug dissolution with Gelucire 44/14 SD was exceptionally set at the ratio of 1:3.
Effect of microwave on glimepiride dissolution half-life and flush dissolution in comparison to physical mixtures:
Effect on dissolution half-life
It could be remarkable from Fig. 4 that MwGSD were more effective in enhancing drug dissolution (less values for t 1/2) than the corresponding PM (without pretreatment in microwave) for most of the tested carriers. This confirmed the assumption suggested by previous researchers (Bergese et al. 2003; Moneghini et al. 2008, 2009; Waters et al. 2011) in that: The heat generated by microwave was able to entangle drug inside the matrix of the respective carrier to form a new type of molecular dispersion that accelerated its release. However, CP showed an exception where higher records of t 1/2 in case of MwGSD were seen. This slight retardation in dissolution might be interpreted by a probable spacial redistribution of cross linked polymeric patches by the effect of the generated heat which did not favor drug release.

Comparison between MwGSD and PM with respect to effect on half-life and flush dissolution for different carriers
Effect on flush dissolution
MwGSD of most carriers (except for Pearlitol and Ac-Di-Sol) showed higher burst effect than the corresponding PM (Fig. 4) suggesting higher affinity of drug to dissolution medium. This confirmed the occurrence of a certain type of molecular dispersion by the effect of microwave radiation that assisted wetting of drug particles and accelerated their outward release accordingly.
Comparison between solid dispersion and microwave treated solid dispersion utilizing different carriers
Effect on dissolution half-life
Figure 5 showed lower rate of drug release from MwTSD of PreGelSt, Starlac, Pearlitol and Glycolys (higher t 1/2) than the SD of the same carriers suggesting the unsuitability of further treatment of SD inside the microwave. However, this assumption was not true for Na CMC, Ac-Di-Sol and CP. The technique might be a subject for further detailed physicochemical characterization in the future in order to rationalize for the difference in response for the different tested carriers.

Comparison between MwTSD and SD with respect to effect on half-life and flush dissolution for different carriers
Effect on flush dissolution
The lower rate of drug dissolution from MwTSD was parallel to the lower percent of burst effect for Starlac, Pearlitol and Glycolys. An assumption could be made on the possibility of some physicochemical changes to occur inside the SD matrices of these carriers causing some hindrance to the outer drug release.
Comparison between different techniques based on solid dispersion utilizing different carriers
Effect of Gelucire 50/13 and 44/14 on dissolution half-life
Although both types Gelucire were characterized by a high surface activity along with a relatively high hydrophilicity, their molecular dispersion with the drug were not sufficient to enhance its dissolution to a reasonable extend. However, adsorbing the melted dispersion into a high surface area carrier (SDads) caused a substantial improvement in dissolution (Fig. 6; Table 1) (drop in t 1/2 to ~30 min for both grades). This result confirmed the success of the suggested synergistic combination, providing wetting, solubilization along with micronization of the poorly soluble drug over lactose as adsorbent.

Effect of technique on dissolution half-life for different carriers at drug to carrier ratio 1:5
Effect of water insoluble polymers on glimepiride dissolution half-life
As it was previously illustrated (Fig. 1), Ac-Di-Sol, Glycolys and CP worked better as CGM with the drug. In a trial to increase their efficiency as carriers, solid mixtures based on SD were prepared.
For Ac-Di-Sol, the inclusion of Gelucire in the molecular dispersion seemed to enhance the performance of the polymer giving least t 1/2(~2 min) for TSD (Table 2). Addition of lactose to the TSD causes retardation in t 1/2 to ~57 min. However, TSDads with CP succeeded to make a drop in t 1/2 to ~8 min. It could be thus reasonable to interpret that the efficiency of lactose as adsorbent strongly depended on the type of matrix used and on the composition of the polymer included.
For Glycolys, t 1/2 values for all tested techniques were relatively high to be further considered in drug dissolution enhancement (Table 2).
Effect of water soluble to partial water soluble polymers on glimepiride dissolution half-life
As prementioned, these polymers shared in common the superiority of their SD in decreasing dissolution t 1/2 if compared with their respective PM or CGM (Fig. 2). The probable enhanced wetting of the drug inside these polymer matrices led to the idea of further adding a third agent (Gelucire 50/13) to the SD to make TSD and adsorbing the latter on lactose as a carrier. These techniques were set in comparison with MwGSD and MwTSD. Figure 6 showed the superiority of TSDads of Na CMC and Pearlitol in enhancing drug dissolution in term of least recorded t 1/2 (Table 3).
Starlac showed exceptional results (Table 3) in that: (1) Burst effect for TSD exceeded 50 % and hence t 1/2 could not be calculated. (2) SD gave the lowest value for t 1/2 followed by TSDads. The composition of the polymer (85 % lactose, 15 % maize starch) could clarify the result. Drug could have higher affinity for the lactose (as constituent of the polymer) included in the molecular dispersion than for the externally added lactose (as adsorbent). The latter might act as a barrier between the drug and dissolution medium. This situation was probably responsible for fast desorption of surface lactose into the medium followed by gradual release of the drug from its molecular dispersion giving a lower dissolution rate for TSDads.
PreGelSt was set as the optimum excipient, as t 1/2 reached as minimum as 0.9 min (Table 4; Fig. 6). This might be interpreted by the additive effect of combined factors. First a double effect could operate due to the simultaneous wetting of drug by water soluble polymers along with hydrophilic surfactant. In addition, the surfactant acted also upon the wetted drug by solubilizing and disaggregating its particles. Finally, the strongly wetted and partially solubilized drug was presented to the dissolution medium in a fine micronized state through adsorption technique.
Physicochemical characterization for formulations containing PreGelSt as optimized carrier
The formulation of TSDads with PreGelSt was considered the most promising technique owing to its success to deliver glimepiride at the highest recorded rate. Therefore, further physicochemical characterization was achieved for formulations containing PreGelSt as optimized carrier.
FTIR analysis
In order to test for possible intermolecular interaction between drug and other constituents in the various prepared solid mixtures, each constituent was studied alone for presence of characteristic peaks and then compared to the peaks appearing in the formulation combining all ingredients. Figure 7 showed that:

IR spectra of pure glimepiride (a), PreGelSt (b), Gelucire (c), lactose (d), TSD (e), and TSDads (f)
-
(a)
Pure glimepiride displayed two peaks characteristic of N–H stretching vibration at 3,367 and 3,290 cm−1 and two bands of C=O stretching at 1,708 and 1,674 cm−1. As the proportion of glimepiride in TSD and TSDads decreased, the latter became less expressive in the respective charts.
-
(b)
PreGelSt showed two broad bands of OH stretching at 3,417 and 3,394 cm−1 and a characteristic band of CH aliphatic at 2,924 cm−1.
-
(c)
Gelucire 50/13 showed two weak bands of OH stretching at 3,491 and 3,452 cm−1 along with strong bands stretching of CH aliphatic at 2,916, 2,889, 2,850 cm−1. A characteristic C=O stretching peak was also seen at 1,735 cm−1.
-
(d)
Lactose showed broad bands of OH stretching at 3,379, 3,344, 3,267 cm−1.
-
(e)
TSD exhibited the same NH peak of the drug but was overlapped with the OH signal of the PreGelSt. In addition the same three bands of C=O group characteristics of the drug and Gelucire appeared at the same position reported for the single components.
-
(f)
TSDads exhibited all characteristic bands of drug, PreGelSt, Gelucire in addition to that of lactose at the same position reported for each constituent. This suggested the absence of molecular interaction between the components of either formulations studied.
Further matching study between the IR spectra of the pure drug and various formulations (Figs. 8, 9) revealed no sign of chemical interaction either in the region of stretching vibration or in the fingerprint region. All formulae displayed all characteristic peaks of their respective components at the same position appearing for each constituent when analyzed alone.

Comparison of IR spectra of SD at 1:1, 1:3, and 1:5, respectively (a, b, c), PM (d) and CGM (e) at 1:5 of drug to PreGelSt, respectively

Comparison of IR spectra of pure glimepiride (a, a′), PM and SD (b, b′), MwGSD and MwTSD (c, c′) all at drug to carrier ratio of 1:5
Thermal analysis by DSC
Figure 10 showed thermograms for TSD, TSDads and their component excipients. Glimepiride exhibited a well defined melting peak at 215.3 °C indicating its crystalline nature. PreGelSt did not show any peak which proved its amorphous nature. Gelucire 50/13 showed a characteristic sharp endotherm at 47.02 °C. Lactose had two sharp endothermic peaks at 149.8 and 219.17 °C. Curves corresponding to TSD and TSDads showed the complete disappearance of the characteristic peak of glimepiride which suggested the complete amorphisation of the drug as a result of formation of solid solution within their respective carriers. The same curves also illustrated two small peaks at 38.8 and 44.5 °C which were interpreted by the formation of individual polymorph of Gelucire 50/13 when molecularly dispersed within TSD (Khan and Craig 2003).

DSC thermograms of glimepiride (a), PreGelSt (b), Gelucire 50/13 (c), lactose (d), TSD (e) containing PreGelSt and Gelucire, and TSDads (f) containing PreGelSt, Gelucire and lactose
Figure 11 showed that, preGelSt was able to decrease crystallinity of glimepiride up to an optimum drug to carrier ratio 1:3, where complete amorphisation occurred with disappearance of drug endothermic peak and appearance of a broad peak at (117 °C) instead. The new peak might be the result of an interaction between drug and polymer which was only favored under the influence of the additional applied heat from DSC at this specific ratio. However, upon increasing the ratio to 1:5 a very small peak intensity characteristic of drug reappeared suggesting that: Formation of solid solution in which drug is completely dispersed in the polymeric matrix has to be optimized with respect to amount of polymer contribution in the mixture.

DSC thermograms for glimerpiride (a), PreGelSt (b), SD at 1:1, 1:3, 1:5, respectively (c, d, e), PM (c′), and CGM (d′)
It was also illustrated that: at drug: carrier 1:5, PM, CGM and SD decreased peak intensity of glimepiride to the same extent (Fig. 11). This result suggested equal efficiency of the three techniques in influencing the degree of crystallinity of the drug.
No remarkable change occurred in the thermogram upon treating the PM with microwave irradiation. However, MwTSD showed a slight decrease in peak intensity characteristic of the drug if compared to that before irradiation (Fig. 12). This could be a result of repetitive exposure of the formulation to heat, once during the preparation of SD, twice during treatment with microwave and thrice the additive heat associated with DSC analysis. The combined effects might contribute to slight alteration in the crystal habit of the drug.

DSC thermograms for glimepiride (a), PreGelSt (b), PM (c) and SD (c′), MwGSD (d) and MwTSD (d′)
X-ray powder diffraction
The X-ray diffractogram of glimepiride exhibited sharp and intense peaks at 2θ° equivalent to: 18.10°, 19.12°, 22.00°, 25.21°, 26.32°, besides a series of smaller peaks at 6.35°, 14.62°, 17.02°, 22.82° and 23.64°. The above pattern clearly showed the strong crystal habit of the pure drug. A comparison between XRPD of SD at different drug: PreGelSt ratios showed (Fig. 13) a gradual decrease in peak intensities characteristic of the drug, by increasing the ratio of the polymer, attaining the least values at 1:5 ratio. This result was inconsistent with that of DSC analysis, where endothermic peak relative to drug disappeared completely at the intermediate ratio (1:3) (Fig. 11). This was attributed to the additional heating process associated with DSC which might affect the increased affinity of drug dispersion in PreGelSt to the point of annulating crystallinity at this specific ratio.

X-ray diffraction pattern of glimepiride (a), SD at 1:1, 1:3, 1:5 drug to PreGelSt rations, respectively (b, c, d), PM (b′) and CGM (c′)
A comparison of XRPD of pure drug with that of PM, CGM and SD (Fig. 13) showed a similar reduction in peak intensity in all the tested formulae, but drug still retained some of its crystallinity. Thus, although SD has been considered in the past as a successful tool for suppressing crystallinity of poorly soluble drugs (Leuner and Dressman 2000; Van den Mooter 2012), yet it was obvious that binary systems were not alone sufficient for complete amorphisation of glimepiride and hence were not treated as the optimum technique. XRPD of MwGSD and MwTSD did not exhibit significant change from the respective charts of PM and SD, respectively before microwave treatment (Fig. 14). This result (supported by the dissolution kinetic results and DSC analysis of the respective formulae) excluded the usefulness of microwave treatment as an optimum technique for dissolution enhancement of glimepiride.

X-ray diffraction pattern of glimepiride (a), PM and SD, respectively (b, b′), MwGSD and MwTSD respectively (c, c′) all at drug:PreGelSt of 1:5
Figure 15 demonstrated the XRPD pattern of TSD and TSDads comparatively with their constituent components (glimepiride, PreGelSt, Gelucire and lactose). TSD showed peaks characteristic of Gelucire but with lower intensity, while those related to glimepiride disappeared completely. A parallel phenomenon was observed in case of TSDads where the thermogram showed peaks characteristic of lactose, while those of drug were completely absent. Therefore, suggesting TSDads as the optimum formulation could be ascribed to the following reasons:

X-ray diffraction pattern of glimepiride (a), TSD (b), TSDads (c), Gelucire (d), lactose (e) and PreGelSt (f)
-
(1)
They success to convert the drug which processed a strong crystal habit into a totally amorphous one, homogenously dispersed in the blend. This was confirmed by both DSC analysis and XRPD.
-
(2)
As a result of amorphisation and substantial increase in surface area, drug particles acquired high internal energy necessary for their enhanced wetting and dissolution.
-
(3)
Dissolution kinetics was accelerated to the extent of reaching a t 1/2 of 0.9 min in case of PreGelSt as a carrier (Fig. 16).
Fig. 16 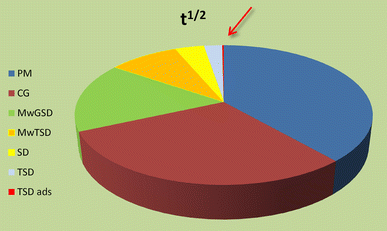
Relation between half-life and technique for solid mixtures containing PreGelSt

Scanning electron microscopy
Surface topography in Fig. 17 clearly showed the difference between the strong crystal habit of glimepiride platelets with distinct sharp edges and the gradual transformation that occurred in different formulae into amorphous structure with smooth to round edges.

SEM micrographs of drug (a), PreGelSt (b), Gelucire 50/13 (c), SD (d), MwGSD (e), MwTSD (f), TSD (g), TSDads (h), and lactose (i)
MwGSD demonstrated the original crystalline structure of the drug physically mixed with preGelSt as this formula originated from a physical mixture of the drug and carrier (without molecular dispersion). SD showed partial smooth structure which was not greatly modified in case of MwTSD. However, the surface of TSD acquired amorphous shape with smooth texture similar to the surface topography of intact Gelucire pellets. This obviously demonstrated the contribution of Gelucire in the final amorphisation of the triple dispersion. TSDads appeared as perfect spherical particles with complete rounded edges coinciding to the surface structure of lactose. Thus, it could be clearly identified that the role of adsorbent did not stop only on disaggregation and micronization of particles but also promoted their spheronization.
Conclusion
Combined effects of wetting, amorphisation, solubilization and micronization could be summed up in one and the same formulation using TSDads with PreGelSt as optimized carrier. The new formulation acquired improved wetting and amorphisation of the drug through preparation of SD in addition to solubilization through incorporation of surfactant in the binary system and micronization through adsorption on high surface area carrier. This provided extra fast dissolution profile and thereby expected enhancement in bioavailability for the poorly soluble drug glimepiride.
Abbreviations
- PM:
-
Physical mixtures
- CGM:
-
Co-ground mixtures
- SD:
-
Solid dispersions
- SDads:
-
Solid dispersion adsorbate
- TSD:
-
Triple solid dispersion
- TSDads:
-
Triple solid dispersion adsorbate
- MwGSD:
-
Microwave generated solid dispersion
- MwTSD:
-
Microwave treated solid dispersion
- PreGelSt:
-
Pregelatinized starch
- CP:
-
Crospovidone
References
Ahuja N, Katare OP, Singh B (2007) Studies on dissolution enhancement and mathematical modeling of drug release of a poorly water-soluble drug using water-soluble carriers. Eur J Pharm Biopharm 65:26–38
Al-Obaidi H, Ke P, Brocchini S, Buckton G (2011) Characterization and stability of ternary solid dispersions with PVP and PHPMA. Int J Pharm 419:20–27
Ammar HO, Salama HA, Ghorab M, Mahmoud AA (2006) Formulation and biological evaluation of glimepiride–cyclodextrin–polymer systems. Int J Pharm 309:129–138
Bergese P, Colombo I, Gervasoni D, Depero LE (2003) Microwave generated nanocomposites for making insoluble drugs soluble. Mater Sci Eng C23:791–795
Biswal S, Sahoo J, Murthy PN (2009) Physicochemical properties of solid dispersions of gliclazide in polyvinylpyrrolidone K90. AAPS PharmSciTech 10(2):329–334
Blagden N, de Matas M, Gavan PT, York P (2007) Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv Drug Deliv Rev 59:617–630
Boregowda SS, Rao BPR, Jayarama RA, Guruchar NLV (2011) Application of water-soluble/dispersible polymeric carriers in drug dissolution modulation. Asian J Pharm Sci 6(1):26–35
Branchu S, Rogueda PG, Plumb AP, Cook WG (2007) A decision-support tool for the formulation of orally active, poorly soluble compounds. Eur J Pharm Sci 32:128–139
Bühler V (2005) Polyvinylpyrrolidone excipients for pharmaceuticals. Povidone, Crospovidone and copovidone. Springer-Verlag, Berlin/Heidelberg
Cilurzo F, Selmin F, Minghetti P, Gennari CGM, Demartin F, Montanari L (2008) Characterization and physical stability of fast-dissolving microparticles containing nifedipine. Eur J Pharm Biopharm 68:579–588
de Waard H, Hinrichs WLJ, Visser MR, Bologna C, Frijlink HW (2008) Unexpected differences in dissolution behavior of tablets prepared from solid dispersions with a surfactant physically mixed or incorporated. Int J Pharm 349:66–73
Dureja DH, Madan AK (2007) Solid dispersion adsorbates for enhancement of dissolution rates of drugs. PDA J Pharm Sci Technol 61(2):97–101
Frick A, Möller H, Wirbitzki E (1998) Biopharmaceutical characterization of oral immediate release drug products. In vitro/in vivo comparison of phenoxymethylpenicillin potassium, glimepiride and levofloxacin. Eur J Pharm Biopharm 46:305–311
Goddeeris C, Willems T, Van den Mooter G (2008) Formulation of fast disintegrating tablets of ternary solid dispersions consisting of TPGS 1000 and HPMC 2910 or PVPVA 64 to improve the dissolution of the anti-HIV drug UC 781. Eur J Pharm Sci 34:293–302
Ilić I, Dreu R, Burjak M, Homar M, Kerč J, Srčič S (2009) Microparticle size control and glimepiride microencapsulation using spray congealing technology. Int J Pharm 381:176–183
Kawabata Y, Wada K, Nakatani M, Yamada S, Onoue S (2011) Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: basic approaches and practical applications. Int J Pharm 420:1–10
Kawakami K (2012) Modification of physicochemical characteristics of active pharmaceutical ingredients and application of supersaturatable dosage forms for improving bioavailability of poorly absorbed drugs. Adv Drug Deliv Rev 64:480–495
Khan N, Craig DQM (2003) The influence of drug incorporation on the structure and release properties of solid dispersions in lipid matrices. J Control Release 93:355–368
Kiran T, Shastri N, Ramakrishna S, Sadanandam M (2009) Surface solid dispersion of glimepiride for enhancement of dissolution rate. Int J PharmTech Res 1(3):822–831
Leuner C, Dressman J (2000) Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm 50:47–60
Li J, Liu P, Liu J, Zhang W, Yang J, Fan Y (2012) Novel Tanshinone II A ternary solid dispersion pellets prepared by a single-step technique: in vitro and in vivo evaluation. Eur J Pharm Biopharm 80:426–432
Martins RM, Siqueira S, Tacon LA, Freitas LAP (2012) Microstructured ternary solid dispersions to improve carbamazepine solubility. Powder Technol 215–216:156–165
Moneghini M, Bellich B, Baxa P, Princivalle F (2008) Microwave generated solid dispersions containing Ibuprofen. Int J Pharm 361:125–130
Moneghini M, Zingone G, De Zordic N (2009) Influence of the microwave technology on the physical–chemical properties of solid dispersion with Nimesulide. Powder Technol 195:259–263
Nakanishi S, Fujii M, Sugamura Y, Suzuki A, Shibata Y, Koizumi N, Watanabe Y (2011) Evaluation of the physicochemical characteristics of Crospovidone that influence solid dispersion preparation. Int J Pharm 413:119–125
Ning X, Sun J, Han X, Wu Y, Yan Z, Han J, He Z (2011) Strategies to improve dissolution and oral absorption of glimepiride tablets: solid dispersion versus micronization techniques. Drug Dev Ind Pharm 37(6):727–736
Parmar KR, Shah SR, Sheth NR (2011) Studies in dissolution enhancement of ezetimibe by solid dispersions in combination with a surface adsorbent. Dissolut Technol 8:55–61
Pouton CW (2006) Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci 29:278–287
Rajpurohit VS, Rakha P, Goyal S, Dureja H, Arorac G, Nagpal M (2011) Formulation and characterization of solid dispersions of glimepiride through factorial design. Iran J Pharm Sci 7(1):7–16
Reddy MS, Fazal ul Haq SM, Apte SS (2011) Solubility enhancement of fenofibrate, a BCS class II drug by self emulsifying drug delivery systems. IRJP 2(11):173–177
Reven S, Grdadolnik J, Kristl J, Zagar E (2010) Hyperbranched poly(esteramides) as solubility enhancers for poorly water-soluble drug glimepiride. Int J Pharm 396:119–126
Roquette Pharma and Personal-Care (2012) Glycolys [on line]. Available at http://www.Roquette-pharma.com/glycolys-superdisintegrant-starch-tablets-capsules-glycolate/. Accessed on 9 Oct 2012
Shah VP, Konecny JJ, Everett RL, McCullough B, Noorizadeh AC, Skelly JP (1989) In vitro dissolution profile of water-insoluble drug dosage forms in the presence of surfactants. Pharm Res 6(7):612–618
Shah J, Vasanti S, Anroop B, Vyas H (2009) Enhancement of dissolution rate of valdecoxib by solid dispersions technique with PVP K 30 & PEG 4000: preparation and in vitro evaluation. J Incl Phenom Macrocycl Chem 63:69–75
Shohin IE, Kulinich JI, Ramenskaya GV, Vasilenko GF (2011) Evaluation of in vitro equivalence for drugs containing BCS class II compound ketoprofen. Dissolut Technol 2:26–29
Singh SK, Prakash D, Srinivasan KK, Gowthamarajan K (2011) Liquisolid compacts of glimepiride: an approach to enhance the dissolution of poorly water soluble drugs. J Pharm Res 4(7):2263–2268
Van den Mooter G (2012) The use of amorphous solid dispersions: a formulation strategy to overcome poor solubility and dissolution rate. Drug Discov Today 9(2):79–85
Vasconcelos T, Sarmento B, Costa P (2007) Solid dispersions as a strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov Today 12(23/24):1068–1075
Vidyadhara S, Babu JR, Sasidhar RLC, Ramu A, Prasad SS, Tejasree M (2011) Formulation and evaluation of glimepiride solid dispersions and their tablet formulations for enhanced bioavailability. Pharmanest 1:15–20
Vippagunta SR, Maul KA, Tallavajhala S, Grant DJW (2002) Solid-state characterization of nifedipine solid dispersions. Int J Pharm 236:111–123
Waters LJ, Bedford S, Parkes GMB (2011) Controlled microwave processing applied to the pharmaceutical formulation of ibuprofen. AAPS PharmSciTech 12(4):1038–1043
Wei H, Löbenber RL (2006) Biorelevant dissolution media as a predictive tool for glyburide a class II drug. Eur J Pharm Sci 29:45–52
Zajc N, Obreza A, Bele M, Srčič S (2005) Physical properties and dissolution behaviour of nifedipine/mannitol solid dispersions prepared by hot melt method. Int J Pharm 291:51–58
Acknowledgments
The authors are grateful for the members of Pharmaceutics Department in National Research Center in Dokki, Cairo, Egypt for their continuous help and support in terminating the experimental work.
Conflict of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Makar, R.R., Latif, R., Hosni, E.A. et al. Optimization for glimepiride dissolution enhancement utilizing different carriers and techniques. Journal of Pharmaceutical Investigation 43, 115–131 (2013). https://doi.org/10.1007/s40005-013-0061-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40005-013-0061-8