
Abstract
Rheumatoid arthritis (RA) is a chronic disease, the etiology of which has yet to be clarified, which causes activation of proinflammatory pathways that bring about joint and systemic inflammation. Although peripheral nervous system anomalies are observed widely in RA, very few case reports on changes in the central nervous system (CNS) have been published. In recent years, the pathophysiology of CNS involvement that can occur in RA has attracted a great deal of attention. Emphasis has focused on the possibility that CNS involvement occurs due to blood–brain barrier (BBB) damage associated with chronic inflammation. The present study was performed to investigate the possible effects of BBB dysfunction and tumor necrosis factor (TNF) blocker therapy on BBB function, which may cause CNS damage in patients with RA. 58 RA patients [47 (81.0%) females, 11 (19.0%) males] and 34 healthy controls [24 (70.6%) females, 10 (29.4%) males] were included in the study. All RA patients were on synthetic DMARD therapy at the beginning. Thirty patients continued DMARD therapy, and 28 patients with high disease activity were started on TNF blocker therapy. All demographic characteristics of the patients were recorded. Disease activity was evaluated using the Disease Activity Score 28-joint count C reactive protein. The Mini-Mental State Examination was used to evaluate cognitive function, and the Fazekas scale was used to assess cranial lesions visualized by magnetic resonance imaging (MRI). Patients’ peripheral blood S100β, glial fibrillary acidic protein (GFAP), claudin, interleukin (IL)-17, and IL-1β levels were measured at the beginning of the study and after 6 months. Demographic characteristics (including sex, age, and body mass index) were similar in the RA and control groups. S100β and GFAP levels were significantly higher in the patient group than in the control group. In the group that was started on TNF blocker therapy, S100β and GFAP levels were significantly decreased 6 months after commencement of treatment. No difference was observed between the RA and control groups in terms of hyperintense lesions seen on cranial MRI. The S100β levels increased with lesions in the deep white matter seen on cranial MRI in patients with RA. In conclusion, next to decreasing disease activity and joint erosions by suppressing inflammation, anti-TNF therapy in RA can also suppress potential CNS involvement linked to BBB (blood–brain barrier) dysfunction. Further studies with broader participation and longer patient follow-up are needed to reinforce this hypothesis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Rheumatoid arthritis (RA) is a chronic disease, the etiology of which has yet to be determined, which causes activation of proinflammatory pathways, resulting in joint and systemic inflammation [1].
Although peripheral nervous system anomalies are observed widely in RA, very few case reports regarding changes in the central nervous system (CNS) associated with the disease have been published. Meningitis, encephalitis, focal neurological symptoms, multiple brain infarcts, and white matter anomalies have been reported [2,3,4,5,6].
Brain damage may occur due to disruption of the blood–brain barrier (BBB) associated with chronic inflammation. The permeability of the BBB was shown to be increased in cases of chronic systemic inflammation [7,8,9,10,11,12]. To assess the damage occurring in the BBB, the levels of glial- and neuronal-derived proteins, i.e., S100β, neuron-specific enolase (NSE), and glial fibrillary acidic protein (GFAP), which can be detected in the peripheral blood, have been measured quantitatively [13,14,15]. A few studies have shown that the levels of these molecules are increased in the peripheral blood, synovial fluid, or cerebrospinal fluid in RA.
One of the prominent cytokines in RA’s pathophysiology is the TNF-alpha. TNF-alpha is one of the molecules that lead to BBB destruction causing tight junctions in the BBB to open [16]. New therapies target cytokines associated with the disease such as TNF-alpha, immune cells and B-cells. In RA, biologicals reduce joint inflammation, limit erosive changes, reduce disability and improve the quality of life [17]. It is unclear how RA affected the blood–brain barrier due to chronic inflammation, and if there is an effect, it changes with the drugs used in RA.
Apart from imaging methods and electrophysiological evaluations in traumatic or atraumatic central nervous system diseases, it is also important that a laboratory evaluation parameter also points to possible central nervous system damage. During the last years, the possibility of evaluating brain damage/activity through quantification of glial and neuronal-derived proteins [such as S100B and neuron-specific enolase (NSE)] in peripheral samples has gained appropriate attention in clinical and experimental settings [28,29,30,31,32,33,34,35]. S100B is a calcium-binding protein physiologically produced and released predominantly by astrocytes.
Several studies have documented elevated serum and cerebrospinal fluid levels of S100β, and its overexpression was shown to increase vulnerability to neurodegeneration [30], cerebral hypoxic–ischemic injury [31], traumatic brain injury [35], CNS infection [33, 34], and severe extracerebral infectious diseases [33]. As the possibility of BBB permeability disturbance due to vasculitis associated with RA cannot be excluded, the quantification of brain-derived proteins, such as glial- and neuronal-derived proteins (e.g., S100β and NSE), may provide sensitive and direct biomarkers of brain damage in RA as well as related neurological and neuropsychological outcomes.
One of the most prominent cytokines involved in the pathophysiology of RA is tumor necrosis factor-alpha (TNF-α), which leads to BBB destruction via opening of tight junctions in the BBB [16]. New therapies target cytokines associated with the disease, such as TNF-α, immune cells and B-cells. In RA, several biologicals have been shown to reduce joint inflammation, limit erosive changes, reduce disability, and improve the quality of life [17]. The way in which RA affects the BBB due to chronic inflammation, and whether any such effect is altered by the drugs used in treatment of RA, remain unclear.
The present study was performed to investigate the possible effects of BBB dysfunction and TNF blocker therapy on BBB function, which may cause CNS damage in patients with RA.
Methods
This study was conducted between July 2015 and March 2016 with approval of the local Ethics Committee and suitable with the Declaration of Helsinki. All individuals provided written informed acceptance.
Participants, demographic and clinical characteristics
This study was designed as an observational, prospective, follow-up study.
58 RA patients diagnosed according to the ACR/EULAR 2010 RA classification criteria and 34 control patients were included in the study. The control group composed of patients who were only admitted to the emergency service once for a headache, whose neurological examinations were normal and who had had a cranial MRI scan. Neurological examination of all healthy controls was normal. All 58 RA patients were on synthetic DMARD therapy in the beginning. The disease duration of RA patients ranged from 1 to 20 years. While 30 patients continued DMARD therapy, 28 patients with high disease activity were started on TNF blocker therapy. TNF blockers were randomly selected from the planned outbreak of biological agent therapy. Those included in the control group were matched to the patients by age and gender. All data of the patients were reevaluated 6 months after a change of treatment. The patients’ age, sex, disease onset age, BMI, comorbidities, smoking status and the drugs they used were recorded.
Inclusion criteria: patient group—patients between 18 and 75 years of age with RA diagnosis according to ACR/EULAR 2010 criteria, receiving synthetic DMARD treatment, TNF blocker treatment being naive patient. Control group—being between the ages of 18–75, being completely healthy from the neurological point of view.
Exclusion criteria: being under 18 years of age; having a rheumatological disease other than RA, such as SLE; having DM and malignant HT; having any neurological or psychiatric illness; being unable to cooperate in the evaluation of cognitive functions; having undergone a neurosurgical cranial operation; being pregnant or being in the lactation period; using medication that can lead to neurotoxicity; having a chronic infection; RA patients with a neurologic deficit in their examination.
The disease activity of the RA patients was assessed using DAS28. Disease Activity Score (DAS 28) remission criteria, involving C reactive protein (CRP), swollen and tender joint counts and patient’s global health assessment were used to determine whether the disease in remission. A score of DAS28 between 2.6 and 3.2 indicates low disease activity, 3.2–5.1 moderate and > 5.1 high disease activity [18].
Evaluation of cognitive functions
The cognitive functions of the patients were evaluated at months 0 and 6 with the Mini-Mental State Examination. Mini-mental test; which is used for global mental status; consists of two parts. The first part covers orientation, memory and attention; the maximum score is 21. The second part tests ability to name, follow verbal and written commands, write a sentence spontaneously, the maximum score is nine. The cut-off score for normal cognitive functions was ≥ 27 [19, 20] Those included in the control group were called at the beginning of the study and at the 6th month, invited to the hospital, and the miniment test was evaluated.
Radiological assessment
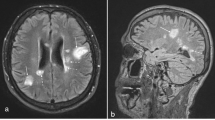
RA patients had contrast-enhanced cranial MRI scans on month 0 (before starting TNF blocker therapy and in the group receiving DMARD therapy). Both groups—the one started on TNF blocker therapy and the one continuing DMARD therapy—had follow-up cranial MRI scans at the end of the 6th month, which were assessed. The patients’ cranial MRI scans were evaluated by an experienced neurologist. The Fazekas scale was used to assess the white matter lesions that are indicators of small vessel disease and neurodegeneration. Based on this scale, the lesions detected in the periventricular white matter and deep white matter in T2-weighted or FLAIR sections were scored between 0 and 3. Fazekas scale on the basis of visual assessment both periventricular (0 = absent, 1 = caps or pencil lining, 2 = smooth halo, 3 = irregular periventricular hyperintensities extending into deep white matter) and subcortical areas (0 = absent, 1 = punctuate foci, 2 = beginning confluence of foci, 3 = large confluent areas). The total Fazekas score was calculated by adding the periventricular and subcortical scores [21, 22]. In the control group, cranial MR was pulled and evaluated using the same method.
Statistical analysis
The data obtained as a result of the study were analyzed with the SPSS (Statistical Program for Social Sciences) for Windows 16.0 statistics software package. In the comparison of group averages, the One Way ANOVA test was used for data with normal distribution and the Wilcoxon Rank test, Mann–Whitney U test and Kruskal–Wallis test were used for data that was not normally distributed. Intergroup comparisons of categorical variables were evaluated with the Chi-square test (where the expected value was found to be under 5 in 2 × 2 tables, Fisher’s exact test was used) and the data were expressed as number and percentage. In the evaluation of interparameter relationships, Spearman’s correlation analysis was used. The results were evaluated within a confidence interval of 95%, and significance was evaluated at a level of 0.05.
Results
58 RA patients [47 (81.0%) females, 11 (19.0%) males] and 34 healthy controls [24 (70.6%) females, 10 (29.4%) males] were included in the study. 30 of the RA patients continued DMARD therapy, and 28 patients were switched to TNF blocker therapy. Demographic characteristics were similar between the two groups, and no statistically significant difference was found between the patient group and the control group in terms of sex, age, and BMI (p > 0.05) (Table 1).
The average age of disease onset in RA patients was 9.28 ± 9.19, their initial average ESR 26.86 ± 10.44 and their average CRP level 13.60 ± 9.77. The initial DAS28-CRP average was 3.23 ± 0.84. RF positivity was determined as 76.2% and anti-CCP positivity as 56.2% in the RA group.
S100 beta, GFAP, Claudin, IL-1 beta and IL-17 mean values in both groups (RA and control group) and the differences between the groups are shown in Table 2.
The scoring of the cranial MRI lesions of both groups according to the Fazekas scale is given in Table 3. A hyperintense lesion was detected in the periventricular white matter of five patients, and the deep white matter of 19 patients out of the 58 patients found in the patient group. A hyperintense lesion was detected in the periventricular white matter of three patients, and the deep white matter of nine patients out of the 34 patients found in the control group. No statistically significant relationship was found between the patient group and the control group in terms of the number of hyperintense lesions observed in the white matter (p > 0.05). While a chronic lacuna was detected in 1 patient in each group, the presence of an acute lacuna was not detected.
The initial and 6th-month levels of S100 beta, GFAP, Claudin, IL-17 and IL-1 beta in the group that continued DMARD therapy and the group that switched to TNF blocker therapy in RA patients are given in Table 4.
The S100 beta, GFAP and Claudin levels of hyperintense lesions in the periventricular white matter and deep white matter of the brains of RA patients according to their Fazekas scores are shown in Table 5. No relationship was determined between the hyperintense lesions observed in the periventricular region in the cranial MRI and each of the three proteins. The S100 beta level was observed to significantly increase as the number of hyperintense lesions seen in the deep white matter increased (p < 0.05).
Discussion
In this study, we found significantly elevated peripheral blood levels of the brain-specific proteins, S100β and GFAP, indicative of BBB dysfunction, in patients with RA relative to controls. In the RA group that was started on TNF blocker therapy, S100β and GFAP levels were significantly decreased 6 months after commencement of therapy compared with levels at the beginning of treatment. Cranial magnetic resonance imaging (MRI) findings did not differ between patients with RA and the control group. In patients with RA, S100β levels increased with the number of lesions in the deep white matter observed on cranial MRI.
With the discovery in studies conducted in recent years, the levels of cytokines such as TNF-alpha, IL-6, IL-1 beta and IL-17 that play a role in systemic inflammation also rise in the central nervous system in some diseases presenting with inflammation. [23, 24]. The permeability of the BBB was shown to be increased in cases of inflammation [7,8,9,10,11]. The peripheral blood levels of some brain-specific proteins (S100β, GFAP, NSE, S100A4) were shown to be high in cases in which the BBB was damaged [13,14,15]. Most of these studies were conducted in experimental animal models; very few studies have been conducted in humans. In one study, the S100β levels measured in the peripheral blood of patients with RA were found to be elevated compared with controls [14]. In this study, we examined the levels of the brain-specific proteins, S100β and GFAP; consistent with the results of the above-mentioned study, we found that levels of these proteins were significantly higher in patients with RA than in controls. We looked at IL-17 and IL-1 beta levels in patients with RA. Unlike Hamed et al., we studied IL-17 and IL-1 beta levels in our study [14]. We found that IL-17 levels were higher in the patient group than in our study. We evaluated S100 beta and GFAP levels 2 times. Another difference is that we also assessed the effect of a given treatment on brain-specific proteins. In our study, we looked at the level of claudin from the tight junction proteins, but we did not find any difference between the two groups.
Another issue that has been discussed is the presence of rheumatoid factor in RA [25]. Case reports on the CNS symptoms of RA have indicated the presence of rheumatoid factor in the cerebrospinal fluid of patients [26, 27]. Recent studies suggested that circulating immune complexes may cause certain neuroinflammatory responses in the brain [28]. In this study, we found no significant difference in the S100β, GFAP, or claudin level between rheumatoid factor and anti-CCP positive and negative patients with RA.
Studies using functional brain imaging indicated that TNF inhibitor therapy can reduce CNS activity associated with pain resulting from inflammation in patients with RA [29].
In one animal study, TNF-α activation was shown to sustain proinflammatory activity in the brain [30,31,32]. Patients with RA undergoing TNF inhibitor therapy (infliximab, etanercept, adalimumab) showed a decreased risk of developing Alzheimer’s disease compared with controls [33]. Exposure to disease-modifying anti-rheumatic drugs (DMARDs) does not affect the risk of Alzheimer’s disease. Detrait et al. [34] reported that the peripheral administration of TNF inhibitors to rats eliminated the amyloid-induced increase and memory loss effects of hippocampal TNF-α. In another study, TNF-α inhibition was shown to have a direct effect on central pain processing before its anti-inflammatory effects were exerted in the periphery [35].
Although all of these studies showed that CNS symptoms in RA are suppressed by inhibition of TNF-α, TNF inhibitor therapy is also known to potentially cause neurological events [35]. The incidence of CNS demyelination after the initiation of anti-TNF-α therapy is unknown. Randomized controlled studies and post-marketing studies have indicated a prevalence ranging between 0.05 and 0.2% for the first three anti-TNF-α agents used clinically (infliximab, etanercept, adalimumab). Bosch et al. [35] investigated 151 demyelinating CNS processes following anti–TNF-α therapy, including optic neuritis and multiple sclerosis (MS) or MS-like diseases. Nozaki et al. [37] reported seven cases, five of which included peripheral neuropathy. Recently, Andreadou et al. [38] reported four additional cases of CNS demyelinating disease in patients receiving anti-TNF therapy.
In this study, we compared the S100β, GFAP, claudin, IL-1β, and IL-17 levels before and after 6 months of TNF inhibitor therapy in patients with RA with those of patients receiving DMARD therapy. Comparison of pre-treatment and post-treatment levels indicated significant decreases in S100β and GFAP levels after 6 months of TNF inhibitor therapy, with no difference in the claudin level and significant increases in the IL-17 and IL-1β levels following TNF inhibitor therapy. In the group that continued to receive DMARD therapy, however, no difference in any of these laboratory parameters between months 0 and 6 was observed. These results suggest that the permeability of the BBB decreases due to the suppression of inflammation with TNF inhibition in RA, and therefore, the levels of brain-specific proteins in the peripheral blood are reduced. In addition, increased IL-17 and IL-1β levels following therapy may be indicators that the inflammation pathway is continuing through other mechanisms due to inhibition of TNF.
MRI provides different benefits using different techniques for the assessment of neurological involvement. Morphological changes in the brain can be assessed in T1-weighted sections, and the microstructural form of the tissue can be assessed in T2-weighted sections. T2-weighted images can be used to identify hyperintense lesions associated with inflammatory and degenerative changes in the white matter, hyperintense lesions that increase with age in normal populations, and hyperintense lesions associated with microvascular disease [39].
Hamed et al. [14] reported hyperintense lesions and small or lacunar ischemic lesions in the deep white matter, periventricular, and gray–white matter junction areas on MR-FLAIR sections in some patients with RA and no neurological deficit. In another quantitative T2-weighted MRI study, no difference was detected between the RA and control groups in terms of white matter lesions [40]. In the present study, hyperintense lesions were detected in the periventricular region in five of 58 patients with RA and in the deep white matter of 19 patients, and a chronic lacunar infarct was detected in one patient. Consistent with other studies, we found no significant difference between the RA and control groups in terms of white matter lesions in the present study. Similar to the study of Hamed et al. [14] we also found that S100β levels increased with the number of hyperintense lesions in the deep white matter observed on cranial MRI in patients with RA.
Hamed et al. [14] found no relationships between cognitive function and S100β levels in patients with RA. We also found no relationship between the MMSE score, which we used to evaluate cognitive function, and the S100β or GFAP level in the present study.
This study has some limitations. It would have been improved by the inclusion of larger numbers of patients in our treatment groups. In addition, we performed cranial MRI assessment in the 6 months after commencement of therapy in patients with RA. We found no difference in hyperintense lesions in the white matter on MRI in the 6 months compared with pre-treatment results. A longer follow-up period may have generated a different result.
Conclusion
In the present study, we showed that subclinical BBB deterioration can occur in patients with RA, that brain-specific protein levels are elevated in the peripheral blood due to BBB dysfunction, and that the levels of these proteins decrease significantly after TNF blocker therapy. Further studies with larger cohorts of patients with RA receiving TNF blocker therapy are required to determine the impact on CNS damage.
References
McInnes IB, Schett G (2011) The pathogenesis of rheumatoid arthritis. N Engl J Med 365:2205–2219
Watson P, Fekete J, Deck J (1977) Central nervous system vasculitis in rheumatoid arthritis. Can J Neurol Sci 4:269–272
Paci R, Giuffrida CM, Marangolo M, Ventura F (1983) Di Paola F (1983) Neuroradiologic picture of cerebral vasculitis in rheumatoid arthritis. Neuroradiology 25:343–345
Ishizuka T, Suzuki K, Hara M, Kitani A, Kawagoe M, Nakamura H, Tsuchiya K (1992) Detection of intracranial lesions of rheumatoid vasculitis with magnetic resonance imaging (MRI). Ryumachi 32:19–26
Chowdhry V, Kumar N, Lachance DH, Salomao DR, Luthra HS (2005) An unusual presentation of rheumatoid meningitis. Neuroimaging 15:286–288
Caballol Pons N, Montalà N, Valverde J, Brell M, Ferrer I, Martínez-Yélamos S (2010) Isolated cerebral vasculitis associated with rheumatoid arthritis. Joint Bone Spine 77:361–363
Banks WA, Robinson SM (2010) Minimal penetration of lipopolysaccharide across the murine blood–brain barrier. Brain Behav Immun 24:102–109
Pan W (2002) Kastin, A.J.TNFa transport across the blood–brain barrier is abolished in receptor knockout mice. Exp Neurol 174:193–200
Nishioku TL, Furusho K, Tomita A, Ohishi H, Dohgu S, Shuto H, Yamauchi A, Kataoka Y (2011) Potential role for S100A4 in the disruption of the blood–brain barrier in collagen-induced arthritic mice, an animal model of rheumatoid arthritis. Neuroscience. 189:286–292
Nishioku TL, Dohgu S, Takata F, Eto T, Ishikawa N, Kodama KB, Nakagawa S, Yamauchi A, Kataoka Y (2009) Detachment of brain pericytes from the basal lamina is involved in disruption of the blood-brain barrier caused by lipopolysaccharide-induced sepsis in mice. Cell Mol Neurobiol 29:309-316
Nishioku T, Yamauchi A, Takata F, Watanabe T, Furusho K, Shuto H, Dohgu S, Kataoka Y (2010) Disruption of the blood–brain barrier in collagen-induced arthritic mice. Neurosci Lett 482:208–211
Ambartsumian N, Klingelhofer J, Grigorian M, Christensen C, Kriajevska M, Tulchinsky E, Georgiev G, Berezin V, Bock E, Rygaard J, Cao R, Cao Y, Lukanidin E (2001) The metastasis associated Mts1(S100A4) protein could act as an angiogenic factor. Oncogene 20:4685–4695
Bao LL, Zhu Y, Elhassan AM, Wu Q, Xiao B, Zhu J, Lindgren JU (2001) Adjuvant induced arthritis: IL-1 beta, IL-6 and TNF-alpha are up-regulated in the spinal cord. Neuroreport 12:3905–3908
Hamed Sherifa A, Selim Zahra I, Elattar Amal M, Elserogy Yasser M, Ahmed Eman A, Mohamed Hanan O (2012) Assessment of biocorrelates for brain involvement in female patients with rheumatoid arthritis. Clin Rheumatol 31:123–132
Goncalves CA, Leite MC, Nardin P (2008) Biological and methodological features of the measurement of S100B, a putative marker of brain injury. Clin Biochem 41:755–763
Germano AF, Tomasello F (2001) Blood–brain barrier permeability changes after subarachnoid haemorrhage: an update. Clinical implications, experimental findings, challenges and future directions. Springer, New York, pp 5–18
Scott DL, Wolfe F, Huizinga TW (2010) Rheumatoid arthritis. Lancet 376:1094–1108
Wells G, Becker JC, Teng J, Dougados M, Schiff M, Smolen J et al (2009) Validation of the 28-joint Disease Activity Score (DAS28) and European League Against Rheumatism response criteria based on C-reactive protein against disease progression in patients with rheumatoid arthritis, and comparison with the DAS28 based on erythrocyte sedimentation rate. Ann Rheum Dis 68:954–960
Al-Rajeh S, Ogunniyi A, Awada A, Daif A, Zaidan R (1999) Preliminary assessment of an Arabic version of the Mini-Mental state examination. Ann Saudi Med 19:150–152
Folstein MF, Folstein SE, McHugh PH (1975) “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12:189–198
Kim GM, Park KY, Avery R et al (2014) Extensive leukoaraiosis is associated with high early risk of recurrence after ischemic stroke. Stroke 45:479–485
Fazekas F, Chawluk J, Alavi A, Hurtig H, Zimmerman R (1987) MR signal abnormalities at 1.5 T in Alzheimer’s dementia and normal aging. Am J Roentgenol 149:351–356
Waisman A, Hauptmann J, Rgen T (2015) The role of IL-17 in CNS disease. Acta Neuropathol 129:625–637
D’Mello C, Le T, Swain MG (2009) Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factor—signaling during peripheral organ inflammation. J Neurosci 29:2089–2102
Mellor R (1959) Cellular origin of rheumatoid factor. J Exp Med 110:875–876
Markenson JA, McDougal JS, Tsairis P, Lockshin MD, Christian CL (1979) Rheumatoid meningitis: a localized immune process. Ann Intern Med 90:786–789
Inan AS, Masatlioglu S, Ozyurek SC, Engin D, Erdem I (2011) Unusual central nervous system involvement of rheumatoid arthritis: success- ful treatment with steroid and azathioprine. Rheumatol Int 31:1383–1385
Teeling JL, Carare RO, Glennie MJ, Perry VH (2012) Intracerebral immune complex formation induces inflammation in the brain that depends on Fc receptor interaction. Acta Neuropathol 2012(124):479–490
Rech J, Hess A, Finzel S, Kreitz S, Sergeeva M, Englbrecht M et al (2013) Association of brain functional magnetic resonance activity with response to tumor necrosis factor inhibition in rheumatoid arthritis. Arthritis Rheum 65:325–333
Skelly DT, Hennessy E, Dansereau MA, Cunningham C (2013) A systematic analysis of the peripheral and CNS effects of systemic LPS, IL. 1beta, TNF-alpha and IL-6 challenges in C57BL/6 mice. PLoS One 2013(8):e69123
Thomson CA, McColl A, Cavanagh J, Graham GJ (2014) Peripheral inflammation is associated with remote global gene expression changes in the brain. J Neuroinflammation 11:73
Chou R, Kane M, Ghimire S, Gautam S (2010) Tumor necrosis factor inhibition reduces the incidence of Alzheimer’s disease in rheumatoid arthritis patients. American College of Rheumatology, Atlanta
Detrait ER, Danis B, Lamberty Y, Foerch P (2014) Peripheral administration of anti-TNF-alpha receptor fusion protein counteracts the amyloid induced elevation of hippocampal TNF-alpha levels and memory deficits in mice. Neurochem Int 72:10–13
Hess A, Axmann R, Rech J, Finzel S, Heindl C, Kreitz S et al (2011) Blockade of TNF-alpha rapidly inhibits pain responses in the central nervous system. Proc Natl Acad Sci USA 108:3731–3736
Kaltsonoudis E, Zikou AK, Voulgari PV, Konitsiotis S, Argyropoulou MI, Drosos AA (2014) Neurological adverse events in patients receiving anti. TNF therapy: a prospective imaging and electrophysiological study. Arthritis Res Ther 16:125
Bosch X, Saiz A, Ramos-Casals M, BIOGEAS Study Group (2011) Monoclonal antibody therapy-associated neurological disorders. Nat Rev Neurol 7:165–172
Nozaki K, Silver RM, Stickler DE, Abou-Fayssal NG, Giglio P, Kamen DL et al (2011) Neurological deficits during treatment with tumor necrosis factor-alpha antagonists. Am J Med Sci 342:352–355
Andreadou E, Kemanetzoglou E, Brokalaki Ch, Evangelopoulos ME, Kilidireas C, Rombos A et al (2013) Demyelinating disease following anti-TNFα treatment: a causal or coincidental association? Report of four cases and review of the literature. Case Rep Neurol Med 2013:671935
King KS, Peshock RM, Rossetti HC, McColl RW, Ayers CR, Hulsey KM et al (2014) Effect of normal aging versus hypertension, abnormal body mass index, and diabetes mellitus on white matter hyperintensity volume. Stroke 45:255–257
Bekkelund SI, Pierre-Jerome C, Husby G, Mellgren SI (1995) Quantitative cerebral MR in rheumatoid arthritis. Am J Neuroradiol 16:767–772
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
About this article

Cite this article
Sağ, S., Sağ, M.S., Tekeoğlu, I. et al. Central nervous system involvement in rheumatoid arthritis: possible role of chronic inflammation and tnf blocker therapy. Acta Neurol Belg 120, 25–31 (2020). https://doi.org/10.1007/s13760-017-0879-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13760-017-0879-3