
Abstract
A series of 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]-3-yl)-3-substitued-phenylpropane-1,3-dionederivatives were synthesized using the Gewald synthesis in first step which is followed by Baker−Venkataraman rearrangement to yield title compounds. The FTIR, MS and 1H NMR results of the produced derivatives were validated. The biological potential such as antimicrobial, antioxidant and antimycobacterial activity against a particularly virulent strain of MTB (MTB H37Ra) of the synthesized compounds were examined. Antimicrobial screening outcomes showed that compound S17 turned to be the most effective antibacterial agent against Gram positive bacteria such as Staphylococcus aureus (MIC = 16.87 µM) and Bacillus subtilis (MIC = 9.45 µM) and Gram negative bacteria such as Escherichia coli (MIC = 16.87 µM) and compound S7 against Salmonella typhi (MIC = 9.74 µM) and compound S16 displayed remarkable antifungal activity toward each Candida albicans and Aspergillus niger (MIC = 15.23 µM). The standard drugs, cefadroxil (antibacterial), have MIC value against S. aureus, B. subtilis, E. coli and S. Typhi are 16.40 µM, 32.80 µM, 16.40 µM and 16.40 µM, respectively, and fluconazole (antifungal) has MIC value 20.40 µM against both the C. albicans and A.niger strain. In comparison with ascorbic acid, a standard drug (IC50 44.91 µg/mL), compound S10 demonstrated good antioxidant activity, with an IC50 value of 45.29 µg/mL, according to the results of the antioxidant screening. The results of the in vitro antituberculosis screening showed that compound S23 was found to be effective with an MIC value of 78.125 µg/mL. Molecular docking study of an enzymatic active site of “DprE1-decaprenylphosphoryl-β-D-ribose-2′-epimerase” shows a comparable binding mode to the native ligand with better docking score which contributes in understanding and development of models for ligand–protein interactions. Compound S23 showed better docking score of − 8.516 as compared to the Isoniazid with the docking score of − 6.315 which in future will create the fundamental structural framework for MTB inhibition.
Graphic abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
One of the better strategies for synthesizing new precursor molecules for drug development is to derive known active pharmacophores [1]. Even chemists and biologists find it challenging to discover the lead compounds. To make this easy, molecular docking and web-based software’s were currently being used. Molecules containing thiophene moiety have remarkable biological applications having antimicrobial [2], antitubercular [3], antioxidant [4], anti-inflammatory and analgesic [5], antihypertensive [6] and anticancer activities [7] while they are also used as metal corrosion inhibitors [8, 9] or in the production of light-emitting diodes in materials science [10]. The rationale for the drug design of the synthesized compounds in the manuscript was rooted in the molecular structure of the tetrahydrobenzothiophene derivatives. These compounds were designed with specific pharmacophores, particularly the thiophene moiety, which is known for its diverse biological applications, including antimicrobial, antioxidant activities and antitubercular [11].
Tuberculosis (TB), which is among the most common infectious diseases, continues to be an important global health problem. The widespread rise of Mycobacterium tuberculosis (MTB) strains that are multidrug resistant (MDR) and extensively drug resistant (XDR), including infectious, primary and drug resistance-blocking strains, has made tuberculosis the deadliest illness [12]. Drug-resistant TB, which is notoriously difficult to treat, accounted for about half a million of the 6.4 million new TB cases in 2021 [13]. The number of new chemical entities (17) currently approved for clinical trials alone or in combination with selections from 9 existing anti-TB drugs represents a major improvement on previous years [14, 15]. Although there are first- and second-line medications available to treat the illness, tuberculosis still has a high mortality rate and has grown to be a severe hazard to world health [16]. Due to the emergence of drug testing, significant side effects of existing drugs and drug–drug interactions, there is a need for the development of new antitubercular drugs with low toxicity and effective treatment against MDR and XDR and the underlying pathogens [17]. Based on these facts, efforts to find effective chemotherapy drugs for tuberculosis remain continued.
We are currently able to develop new drugs owing to the recent reports of the antituberculosis activity of numerous novel compounds with good minimum inhibitory concentration (MIC) values [18]. FDA-approved medications such sertaconazole, raloxifene, benocyclidine and zileuton all include the drug-like structure known as the benzo[b]thiophene moiety which have significant pharmacological value [19, 20]. Therefore, while our work on the discovery of antitubercular drugs continues, several 1,3-diketones, flavones and pyrazoles were created from tetrahydrobenzo[b]thiophenecarboxylic acid which showed good inhibitory activity [21,22,23,24].
To reveal possible mechanisms for antitubercular activity of synthesized compounds, protein–ligand binding interaction was visualized at molecular level with the help of molecular docking study [25,26,27]. In silico approaches to molecular docking have proved important to identify the target of different ligands and their thermodynamic intermolecular interactions with target enzymes that control their growth [28, 29]. The selection of DprE1-decaprenylphosphoryl-β-D-ribose-2′-epimerase as the target in the manuscript is justified by its pivotal role in Mycobacterium tuberculosis (MTB) cell wall synthesis. Inhibiting DprE1 disrupts the formation of decaprenylphosphoryl-D-arabinose (DPA), impacting arabinogalactan, a crucial component of the MTB cell wall. This disruption weakens the cell wall, hindering MTB growth and survival. Molecular docking studies enhance the rationale, elucidating ligand–protein interactions crucial for designing effective antitubercular drugs. The selected target’s centrality in MTB viability underscores its potential as a strategic point for drug intervention, justifying its relevance in combating tuberculosis [30]. The active site of DprE1 (decaprenylphosphoryl-β-D-ribose-2′-epimerase) shows similarities to the native ligand in crystal structure by molecular docking study which further helps in understanding the ligand–protein interactions and in the design of basic structure which is required for the inhibition of tubercular bacterium [31].
Result and discussion
Chemistry
Thiophene is a five-membered, sulfur-containing heteroaromatic ring generally used as building block in drugs. It undergoes electrophilic aromatic substitution very readily. Sulfur is the least electron donor as compare to nitrogen and oxygen. Its structural metabolism leads to the formation of reactive metabolites. This compound is widely spread in nature and has diversified application in design of new drug molecule. The Gewald synthesis is a well-established method for the construction of thiophene rings, aromatic heterocyclic compounds featuring a sulfur atom within a five-membered ring. The synthetic route involves the reaction of α,β-unsaturated carbonyl compounds, such as α,β-unsaturated ketones or esters, with elemental sulfur (S8) and α-halo ketones or α-halo esters. The process initiates with the deprotonation of the unsaturated carbonyl compound by a strong base, generating an enolate ion. Subsequent nucleophilic attack by sulfur leads to the formation of a thiolate intermediate. Intramolecular cyclization is facilitated by an α-halo ketone or ester, resulting in the formation of an intermediate that undergoes elimination of a halide ion. The final step yields ethyl-2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylateproduct. Subsequent base hydrolysis, likely employing lithium hydroxide (LiOH), transformed intermediate-1 into benzo[b]thiophene-2-carboxylic acid (Intermediate-2). The pivotal Baker−Venkataraman rearrangement ensued, facilitated by pyridine and substituted acetophenones in POCl3 causing the migration of the aryl group to an adjacent position. This rearrangement mechanism led to the final compounds, 1-(2-amino -2,4,5,6,7,7a-hexahydrobenzo[b] -3-yl)-3-substitued-phenylpropane-1,3-dione derivatives. Thiophene derivatives (S1–S23) were synthesized by following general procedure which were discussed in Synthetic Scheme 1.

Synthesis of 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-phenylpropane-1,3-dione derivatives
Biological evaluation
Antimicrobial activity
According to antimicrobial study findings, compound S17 had the highest antibacterial activity against S. aureus, B. subtilis and E. coli, with MIC values of 16.87, 9.45 and 16.87 µM, respectively. Compounds S7 and S11 had the highest potency against S. typhi, with MIC values of 9.74 and 16.43 µM, respectively. The synthesized compound not only exhibited notable antibacterial activity, but also good antifungal activity, with compound S16 emerging as the most effective antifungal agent against both C. albicans and A. niger (MIC = 15.23 µM), and compound S3 also showing good antifungal activity against A. niger with MIC value 18.85 µM. The results of the overall antibacterial activity (Table 1) showed that compounds S16 and S17 were the most effective antimicrobial agents.
Antioxidant activity
Using the DPPH test at an absorbance of 517, in vitro antioxidant activity of each newly synthesized molecule was examined. The IC50 values of freshly synthesized compounds are shown in Figs. 1, 2 and 3. The entire newly synthesized molecule showed good to moderate antioxidant activity, according to the data. When compared to ascorbic acid (IC50 44.91 µg/mL) as the reference drug, compounds S3 and S10 showed the best antioxidant activity, with IC50 values of 47.70 µg/mL and 45.29 µg/mL, respectively. These substances could serve as a starting point for more research. Table 2 displays the results.

Standard graph of ascorbic acid

Graph of potent antioxidant compounds S3 and S10

IC50 values of S3 and S10 compounds in comparison with ascorbic acid
Antitubercular activity
Avirulent strain of MTB (MTB H37Ra; ATCC 25177) was used to test the derived molecules for in vitro antitubercular potency by assessing growth inhibition using the MABA method. The results of each experiment were run in triplicate, and Table 3 and Fig. 4 show the minimum inhibitory concentrations (MICs) in (µg/mL). A few of the compounds were determined to have good to moderate antitubercular action, according to the outcome. When compared to isoniazid, the standard treatment, compound S23 and compound S22 both showed good antitubercular activity with MIC values of 78.125 µg/mL and 156.25 µg/mL, respectively.

MIC values of compounds compared to standard drug
Molecular docking
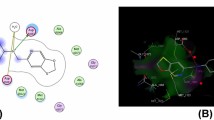
A molecular docking study was carried out in order to identify the best potential binding modes for newly derived molecules with the enzyme (DprE1). It plays a key role in the synthesis of decaprenylphosphoryl-D-arabinose (DPA) which is sole source of D-arabinofuranosyl residues known to be used in the synthesis of arabinogalactan, the fundamental building block of the mycobacterial cell wall core. DprE1 is thus a promising target for antimycobacterial drug design because it is also necessary for cell growth and survival. The Schrodinger suite release 2019–1 49 was used to simulate docking. The most active molecule, S23, had a docking score of − 8.516 while the native ligand had a docking score of − 6.316. All of the compounds’ docking scores are displayed in Table 4. The interacting amino acid residues were identified as Gly55, Gly57, Gly125, Lys418, Tyr 415, Gly117, Gln336 and His132 of DprE1. Isoniazid, compound S13 and compound S23 binding modalities are shown in Fig. 5.

Isoniazid, S23 and S13 binding models with DprE1 target activity
While the NH2 group substituted on tetrahydrobenzothiophene exhibits the interaction with Gln336 and Lys418 and the hydroxyl and amino groups substituted phenyl ring display interaction with the GLY125 and Tyr 415, both oxygen atoms interact with Tyr 415, Gly55, Gly57, Gly125 and His132. Although the binding patterns of all tetrahydrobenzo[b]thiophene-2-carboxylic acid molecules were discovered to be comparable, the molecule S23 stronger binding to the active site of DprE1 is enhanced by the existence of considerably stronger noncovalent interactions.
Molecular dynamic simulation
The complex of compound showed highest docking score (compound S23) with DprE1 undergoes molecular docking simulation study using Desmond MD engine for a period of 100 ns. In order to comprehend the binding mode sustainability as the simulation progressed, an MD simulation was run in order to determine the dynamic nature and alterability of the ligand binding site, the plasticity of the pocket and the contact stability between compound S23 and pocket-specific amino acids. To understand the binding mode sustainability with the progression of the simulation, root mean square fluctuation (RMSF) of compound S23 has been recorded (Fig. 6).We analyzed 1000 frames produced out of 100-ns simulation, and we found contact sustainability with the aforesaid pertinent amino acids, such as Gly55, Arg58, Ser59, Try60, Asn63, Met74, Thr122 and Asn178 (Fig. 7).These contacts were sustained through the mediation of H-bonds, hydrophobic interactions, ionic bonds and water bridges. It was also specifically observed from the simulation study that the protein RMSD value did not cross the 3 Å limit (Fig. 8). This fact signified that the protein structure did not go through a large conformational change. In secondary structure element elucidation, where the reference frame, frame at 50 ns and frame at 100 ns were aligned with each other (Fig. 9). Furthermore, the depiction of Fig. 8 suggested and affirmed the fact that RMSD of compound S23 did not cross the limit of 3 Å and thus suggest the controlled dynamicity of ligand in the orthosteric site of the DprE1. This overall state-of-the-art molecular dynamic simulation confirmed protein–ligand complex stability over the course of the simulation timeline.

Simulation root mean square fluctuation (RMSF) of compound S23

Representation of contact consistency with the key amino acids

Graphical elucidation of the RMSD of the protein and the ligand

Secondary structure element elucidation where alpha sheet is displayed by the deep orange color, and the beta sheet is presented in white
Structure activity relationships (SARs)
From the results of antimicrobial, antioxidant and antitubercular tests of the newly synthesized 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-substituted phenylpropane-1, 3-dione derivatives, it was found that tetrahydrobenzo ring attached at 2 and 3 position of thiophene heterocyclic ring plays a significant role in enhancing all the biological activity. The presence of an electron-withdrawing group (−Cl, NO2 compound S17) in the ortho, meta and para positions of the substituted part increases the antibacterial activity against S. aureus, B. subtilis and E. coli strains, and an electron-releasing group (OH compound S7) in the meta position increases antibacterial activity of the synthesized compound against S. typhi strain. While in case of antifungal activity, the electron-releasing group (OCH3) in the para and the electron-withdrawing group (Cl) in the ortho position (compound S16) of the substituted moiety increases the activity. It was found that electron-releasing group (OCH3, compound S10) in the meta and para position of the substituted part plays a very crucial role in increasing the antioxidant activity. The most potent antimycobacterial activity was found with the compound S23 which has an electron-releasing group in the ortho (OH) and para (CH3) position of the substituted part. Thus, from these results, we can conclude that compounds require different mechanisms to be effective for different targets.
Experimental
General procedure
The investigation employed only laboratory- and analytical-grade materials that were locally sourced. Thin-layer chromatography (TLC) was used to observe the forward reaction stages using commercial silica gel plates (Merck) and silica gel F254 on aluminum sheets. Using the open capillary technique, melting points were examined. Parts per million (5.ppm) downfield tetramethylsilane (internal standard) are used to express the 1H nuclear magnetic resonance spectra, which were obtained using a Bruker Avance 400 NMR spectrometer and the suitable chloroform solvent. Data from the 1H NMR are presented as multiplicity (singlet, doublet, triplet or multiplet) and number of protons. On a Bruker FTIR spectrometer, infrared (IR) spectra were taken.
General procedure for the synthesis of 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-phenylpropane-1,3-dione (S 1–S 23)
Step I: Synthesis of ethyl-2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate: Mixture of ethyl cyanoacetate (5.32 mL, 0.05 mol) and cyclohexanone (5.2 mL, 0.05 mol) was taken in conical flask and allowed to stirred at room temperature followed by the addition of elemental sulfur (1.92 g, 0.06 mol). Then diethylamine (5.26 mL, 0.05 mol) as an amine catalyst was added in it [32]. The reaction mixture was then stirred at 40 °C to 50 °C for 2 h, which undergoes Gewald reaction (the most versatile reaction and involves one-pot cyclocondensation of ketones with activated nitrile derivatives and elemental sulfur to provide 2-aminothiophenes) leading to the formation of intermediate-1(10 mmol).
Step II: Intermediate I was treated with 103 mmol lithium hydroxide in THF (40 ml) for 24 h at 50 °C. The mixture was then cooled to 0 °C and neutralized with acetic acid to pH 7. The precipitate obtained was collected by filtration, washed with water and dried in vacuum. This gives the formation of benzo[b]thiophene-2-carboxylic acid (intermediate 2) [33].
Step III: Synthesis of 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-substitued-phenylpropane-1,3-dione derivatives: “Further, the reaction between intermediate 2 (0.05 mol) and substituted aromatic acetophenone (0.05 mol) in pyridine, KOH and POCl3 provided intermediate esters under refluxed condition. In the next step, these intermediate undergoes Baker−Venkataraman rearrangement (BVR) when it was refluxed for 4 h, affording the formation of 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-substitued-phenylpropane-1,3-dione derivatives. The resulting mixture was cooled, poured onto crushed ice to give a solid precipitate, filtered, washed with 1% potassium bicarbonate and subsequently with water, dried and recrystallized from ethanol.”
Spectral data of newly synthesized thiophene compounds
Compound S1: 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-phenylpropane-1,3-dione, IR, cm−1: 2828 (C–H str.,),1519 (C = C str.,), 1190 (C–N str.,), 3384 (N–H str.,), 1699 (C = O str., carbonyl), 674 (C–S–C str., thiophene ring), 3553 (OH str., aromatic),MS m/z: 302.
Compound S2: 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-(4-chlorophenyl)propane-1,3-dione, IR, cm−1: 2938 (C–H str.,),1521 (C = C str.,), 1257 (C–N str.,), 3306 (N–H str.,), 1679 (C = O str., carbonyl), 685 (C–S–C str., thiophene ring), 779 (C–Cl str., aromatic),MS m/z: 336.
Compound S3: 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-(3-methoxyphenyl)propane-1,3-dione, IR, cm−1: 3040 (C–H str.,),1457 (C = C str.,), 1100 (C–N str.,), 3396 (N–H str.,), 1688 (C = O str., carbonyl), 672 (C–S–C str., thiophene ring), 2995 (OCH3 str., aromatic), 1H NMR (CDCl3, δppm):7.20–7.81 (m, 2H, Ar–H), 6.83 (d, 2H, –NH2), 4.72 (t, 1H, –CH), 3.56 (t, 1H, –CH cyclo), 1.16 (q, 2H, –CH2cyclo), 1.49 (m, 2H, –CH2 cyclo), 2.59 (t, 2H, –CH2cyclo), 3.67 (s, 1H, –CH), 3.68 (s, 3H, –OCH3), MS m/z: 332.
Compound S4: 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-(3-aminophenyl)propane-1,3-dione, IR, cm−1: 3083 (C–H str.,),1531 (C = C str.,), 1259 (C–N str.,), 3511 (N–H str.,), 1685 (C = O str., carbonyl), 675 (C–S–C str., thiophene ring), 3617 (OH str., aromatic),MS m/z: 317.
Compound S5: 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-(p-tolyl)propane-1,3-dione, IR, cm−1: 2800 (C–H str.,),1576 (C = C str.,), 1269 (C–N str.,), 3332 (N–H str.,), 1685 (C = O str., carbonyl), 640 (C–S–C str., thiophene ring), 1354 (CH3 str., aromatic), 1H NMR(CDCl3, δppm):7.19–7.32 (m, 2H, Ar–H), 5.01 (d, 2H, –NH2), 3.68 (t, 1H, –CH), 1.70 (q, 2H, –CH2cyclo), 1.29 (m, 2H, –CH2 cyclo), 1.72 (t, 2H, –CH2cyclo), 3.69 (s, 1H, –CH), 2.66 (s, 3H, –CH3), MS m/z: 316.
Compound S6: 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-(3-nitrophenyl)propane-1,3-dione, IR, cm−1: 2847 (C–H str.,), 1532 (C = C str.,), 1256 (C–N str.,), 3312 (N–H str.,), 1688 (C = O str., carbonyl), 681 (C–S–C str., thiophene ring), 1419 (N–O str., aromatic), 1H NMR (CDCl3, δppm):7.58–7.82 (m, 2H, Ar–H), 5.9 (d, 2H, –NH2), 3.63 (t, 1H, –CH), (t, 1H, –CH cyclo), 1.81 (q, 2H, –CH2cyclo), 1.30 (m, 2H, –CH2 cyclo), 2.57 (t, 2H, –CH2cyclo), 3.64 (s, 1H, –CH), MS m/z: 317.
Compound S7: 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-(2,5-dihydroxyphenyl)propane-1,3-dione, IR, cm−1: 2859 (C–H str.,), 1538 (C = C str.,), 1270 (C–N str.,), 3362 (N–H str.,), 1686 (C = O str., carbonyl), 708 (C–S–C str., thiophene ring), 3600 (OH str., aromatic),MS m/z: 334.
Compound S8: 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-(3-hydroxyphenyl)propane-1,3-dione, IR, cm−1: 2854 (C–H str.,),1593 (C = C str.,), 1285 (C–N str.,), 3337 (N–H str.,), 1680 (C = O str., carbonyl), 688 (C–S–C str., thiophene ring), MS m/z: 318.
Compound S9: 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-(2-hydroxyphenyl)propane-1,3-dione, IR, cm−1: 2949 (C–H str.,),1592 (C = C str.,), 1261 (C–N str.,), 3330 (N–H str.,), 1680 (C = O str., carbonyl), 719 (C–S–C str., thiophene ring), 3658 (OH str., aromatic),MS m/z: 318.
Compound S10: 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-(4-methoxyphenyl)propane-1,3-dione, IR, cm−1: 2956 (C–H str.,),1597 (C = C str.,), 1267 (C–N str.,), 3360 (N–H str.,), 1668 (C = O str., carbonyl), 694 (C–S–C str., thiophene ring), 2810 (O–CH3 str., aromatic), 1H NMR (CDCl3, δppm): 7.25 (m, 2H, Ar–H), 3.74 (d, 2H, –NH2), 3.71 (t, 1H, –CH), 3.58 (t, 1H, –CH cyclo), 1.93 (q, 2H, –CH2cyclo), 1.88 (m, 2H, –CH2 cyclo), 2.54 (t, 2H, –CH2cyclo), 3.73 (s, 1H, –CH), 3.85 (s, 3H, –OCH3), MS m/z: 332.
Compound S11: 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-(4-bromophenyl)propane-1,3-dione, IR, cm−1: 2839 (C–H str.,),1589 (C = C str.,), 1268 (C–N str.,), 3399 (N–H str.,), 1682 (C = O str., carbonyl), 680 (C–S–C str., thiophene ring), 645 (C–Br str., aromatic),MS m/z: 381.
Compound S12: 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-(4-aminophenyl)propane-1,3-dione, IR, cm−1: 2866 (C–H str.,),1521 (C = C str.,), 1265 (C–N str.,), 3497 (N–H str.,), 1648 (C = O str., carbonyl), 664 (C–S–C str., thiophene ring), 1 H NMR(CDCl 3, δppm):7.25 (m, 2H, Ar–H), 4.27 (d, 2H, –NH2), 4.25 (t, 1H, –CH), 4.21 (t, 1H, –CH cyclo), (q, 2H, –CH2cyclo), (m, 2H, –CH2 cyclo), (t, 2H, –CH2cyclo), 4.23 (s, 1H, –CH), 5.91 (t, 2H, –NH2), MS m/z: 317.
Compound S13: 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-(4-nitrophenyl)propane-1,3-dione, IR, cm−1: 2929 (C–H str.,),1587 (C = C str.,), 1262 (C–N str.,), 3396 (N–H str.,), 1687 (C = O str., carbonyl), 696 (C–S–C str., thiophene ring), 1480 (N–O str., aromatic), 1H NMR (CDCl3, δppm):7.25–9.19 (m, 2H, Ar–H), 4.31 (d, 2H, –NH2), 4.28 (t, 1H, –CH), 3.70 (t, 1H, –CH cyclo), 1.95 (q, 2H, –CH2cyclo), 1.32 (m, 2H, –CH2 cyclo), 2.84 (t, 2H, –CH2cyclo), 4.22 (s, 1H, –CH), MS m/z: 347.
Compound S14: 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-(4-hydroxyphenyl)propane-1,3-dione, IR, cm−1: 3075 (C–H str.,),1596 (C = C str.,), 1274 (C–N str.,), 3400 (N–H str.,), 1687 (C = O str., carbonyl), 668 (C–S–C str., thiophene ring), 3604 (OH str., aromatic),MS m/z: 318.
Compound S15: 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-(m-tolyl)propane-1,3-dione, IR, cm−1: 3019 (C–H str.,),1648 (C = C str.,), 1002 (C–N str.,), 3516 (N–H str.,), 1696 (C = O str., carbonyl), 672 (C–S–C str., thiophene ring), 1420 (CH3 str., aromatic), 1H NMR (CDCl3, δppm):7.25 (m, 2H, Ar–H), 5.91 (d, 2H, –NH2), 4.27 (t, 1H, –CH), 4.21 (t, 1H, –CH cyclo), 1.77 (q, 2H, –CH2cyclo), 1.30 (m, 2H, –CH2 cyclo), 2.46 (t, 2H, –CH2cyclo), 4.25 (s, 1H, –CH), 2.68 (s, 3H, –CH3), MS m/z: 316.
Compound S16: 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-(3-bromo-5-methoxyphenyl)propane-1,3-dione, IR, cm−1: 3019 (C–H str.,),1526 (C = C str.,), 1262 (C–N str.,), 3347 (N–H str.,), 1679 (C = O str., carbonyl), 673 (C–S–C str., thiophene ring), 618 (C–Br str., aromatic), 2877 (O–CH3 str., aromatic), MS m/z: 411.
Compound S17: 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-(2,4-dichlorophenyl)propane-1,3-dione, IR, cm−1: 2932 (C–H str.,),1580 (C = C str.,), 1262 (C–N str.,), 3321 (N–H str.,), 1679 (C = O str., carbonyl), 742 (C–S–C str., thiophene ring), 819 (C–Cl str., aromatic), MS m/z: 371.
Compound S18: 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-(2,4-dihydroxyphenyl)propane-1,3-dione, IR, cm−1: 2772 (C–H str.,),1519 (C = C str.,), 1261 (C–N str.,), 3467 (N–H str.,), 1679 (C = O str., carbonyl), 673 (C–S–C str., thiophene ring), 3503 (OH str., aromatic),MS m/z: 334.
Compound S19: 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-(4-fluorophenyl)propane-1,3-dione, IR, cm−1: 2839 (C–H str.,),1591 (C = C str.,), 1275 (C–N str.,), 3345 (N–H str.,), 1684 (C = O str., carbonyl), 685 (C–S–C str., thiophene ring), 1041 (C–F str., aromatic), MS m/z: 320.
Compound S20: 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-(3-chlorophenyl)propane-1,3-dione, IR, cm−1: 2826 (C–H str.,),1541 (C = C str.,), 1261 (C–N str.,), 3398 (N–H str.,), 1678 (C = O str., carbonyl), 752 (C–S–C str., thiophene ring), 677 (C–Cl str., aromatic), 3398 (OH str., aromatic), MS m/z: 336.
Compound S21: 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-(2-chloro-3-methylphenyl)propane-1,3-dione, IR, cm−1: 3008 (C–H str.,),1588 (C = C str.,), 1227 (C–N str.,), 3486 (N–H str.,), 1682 (C = O str., carbonyl), 721 (C–S–C str., thiophene ring), 635 (C–Cl str., aromatic), 1356 (CH3 str., aromatic), 1H NMR (CDCl3, δppm):7.25 (m, 2H, Ar–H), 5.91 (d, 2H, –NH2), 4.27 (t, 1H, –CH), 4.21 (t, 1H, –CH cyclo), 1.78 (q, 2H, –CH2cyclo), 1.30–1.33 (m, 2H, –CH2 cyclo), 2.46 (t, 2H, –CH2cyclo), 4.23 (s, 1H, –CH), 2.48 (s, 3H, CH3), MS m/z: 350.
Compound S22: 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-(3,5-dichloro-2-hydroxyphenyl)propane-1,3-dione, IR, cm−1: 3085 (C–H str.,),1508 (C = C str.,), 1276 (C–N str.,), 3391 (N–H str.,), 1691 (C = O str., carbonyl), 682 (C–S–C str., thiophene ring), 3654 (OH str., aromatic), 788 (C–Cl str., aromatic), 1H NMR (CDCl3, δppm):7.25 (m, 2H, Ar–H), 5.91 (d, 2H, –NH2), 4.25 (t, 1H, –CH), 2.7 (t, 1H, –CH cyclo), 1.76 (q, 2H, –CH2cyclo), 1.57 (m, 2H, –CH2 cyclo), 2.46 (t, 2H, –CH2cyclo), 4.21 (s, 1H, –CH), 4.27 (s, 1H, –OH), MS m/z: 387.
Compound S23: 1-(2-amino-2,4,5,6,7,7a-hexahydrobenzo[b]thiophen-3-yl)-3-(5-chloro-2-hydroxy-4-methylphenyl)propane-1,3-dione, IR, cm−1: 2803 (C–H str.,),1541 (C = C str.,), 1229 (C–N str.,), 3395 (N–H str.,), 1692 (C = O str., carbonyl), 756 (C–S–C str., thiophene ring), 3742 (OH str., aromatic), 1341 (CH3 str., aromatic), 650 (C–Cl str., aromatic), 1H NMR (CDCl3, δppm): 7.25 (m, 2H, Ar–H), 5.70 (d, 2H, –NH2), 4.08 (t, 1H, –CH), 3.59 (t, 1H, –CH cyclo), 1.68 (q, 2H, –CH2cyclo), 1.37 (m, 2H, –CH2 cyclo), 2.57 (t, 2H, –CH2cyclo), 4.07 (s, 1H, –CH), 4.11 (s, 1H, –OH), 2.06 (s, 3H, –CH3), MS m/z: 366.
Screening for antimicrobial activity
MIC (minimal inhibitory concentration) determination
The synthesized compounds were evaluated by microbroth dilution method using 96 flat bottom microtiter plates as described by CLSI guideline, 2020 [34]. Fluconazole (an antifungal) and cefadroxil (an antibacterial) were used as standard drugs. The selected strains for screening were as “Gram positive bacteria: Staphylococcus aureus (MTTC 3160), Bacillus subtilis (MTCC 441), Gram negative bacteria Escherichia coli (MTCC 443), Salmonella typhi (MTCC 3216) and fungal strains Aspergillus niger (MTTC 281) and Candida albicans (MTCC 227)”[35]. The synthesized compounds were diluted first in 10 wells (from 1000 μg/mL to 1.9 μg/mL) and 11th well was kept as positive control and 12th well was left as media control. The overnight grown culture was adjusted to 0.5 McFarland turbidity standards. In each well 10 μL broth was added except 12th well. The presence of turbidity shows the bacterial growth, and the absence of turbidity was interpreted as the MIC. By the addition of p-iodonitrotetrazolium violet dye (INT; 0.2 mg/mL), viability of microorganisms was also confirmed.
Screening for antioxidant activity by DPPH (1,1-diphenyl-2-picrylhydrazyl) method
The newly synthesized substance’s antioxidant activity was assessed spectrophotometrically utilizing the DPPH free radical scavenging technique. When DPPH reacts with hydrogen donors, it is reduced to the equivalent hydrazine and its dark purple color turns to yellow, indicating a considerable reduction in absorption at 517 nm. DPPH is a stable free radical with an absorption maximum at 517 nm. Solution of DPPH (3 g/mL) in methanol was prepared. As a blank control, methanol and DPPH (1:1) solution were utilized. Each synthetic molecule and the standard (ascorbic acid) was diluted in methanol to four different concentrations (25 g/mL, 50 g/mL, 75 g/mL and 100 g/mL), and one milliliter of each concentration was added to one milliliter of the DPPH solution. The mixture was vigorously agitated and left at room temperature in the dark for 30 min, after which the UV absorbance at 517 nm was measured [36]. The percent (%) inhibition of free radical DPPH is calculated as:
ABlank = absorbance of the blank reaction, ASample = absorbance of the test compounds.
The graph plotting percent inhibition and various concentrations of produced compounds served as the basis for calculating the IC50 value.
Screening for antitubercular activity
The antimycobacterium activity of compounds was assessed against M. tuberculosis using the microplate Alamar blue assay. This method uses a thermally stable reagent, has a good correlation with proportionate and is nontoxic. All of the sterile 96-well plates’ outside perimeter wells received 200 µL of sterile deionized water in order to reduce the amount of medium that dries out in the test wells during incubation. 100 µl of the Middlebrook 7H9 broth were added to the 96-well plate, and compounds were serially diluted right there on the plate. The range of final drug testing was 100–0.2 g/ml. The plate was incubated for five days at 37 °C with Parafilm wrapping and sealing it. After that, 25 µL of a freshly prepared 1:1 solution of 10% tween 80 and Alamar blue reagent was added to the plate. While a blue color in the well indicated no bacterial growth, a pink color indicated growth. The MIC is the lowest drug concentration that prevents the color shift from blue to pink [37].
Computational study
The Schrodinger suite release 2019–1 49 was employed to carry out molecular docking of certain legends into the active binding regions of protein on default setting. The right 2D orientation of the chemical structures of the chosen ligands was shown using the ChemOffice program "Chem Draw 16.0," and ChemBio3D was used to lower the energy required for each molecule. The molecules with the least amount of energy were chosen to perform a docking. The three-dimensional structure of the receptor protein was retrieved from https://www.rcsb.org/with a PDB ID of 4FDO (Resolution: 2.40 Å.
R-Value Free: 0.205, R-Value Work: 0.167, R-Value Observed: 0.169) by doing a search on the protein data bank repository. The co-crystallized ligand, along with a few water molecules and cofactors, had to be released in order to effectively prepare the target protein in line with the standard protocol [38]. Target protein preparation was followed by rendering utilizing the glide grid module for grid creation. Grid is a particular location on the receptor protein where the medication will bind. The macromolecule’s target area was placed inside the grid so that it covered the whole structure. It was determined that the glide dock module of the Schrodinger suit provided the docking technique that could be used to dock the ligand and protein most successfully. Each ligand was examined in up to nine different conformations throughout the docking process. Investigating ligand-receptor interactions in both 3D and 2D was done using the Discovery studio visualizer. The target receptor-friendly (lower) free binding energy conformations were those that were selected using this method. The ligands are depicted in various colors, and the residues that interact with H-bonds and one another are indicated as balls and sticks [39].
Docking validation
To confirm the Docking procedure, the co-crystallized ligand (PDB ID: 4FDO) was extracted from its crystal structure and docked again into the active site of enzyme. The computed RMSD value between the co-crystallized ligand and the top-ranked docked conformation was 0.132, as predicted.
Molecular dynamic simulation
Molecular dynamic simulation was performed on Desmond version 2022.4. The complex of compound S23 with DprE1 was taken for the system buildup for MD simulation. System ionization was carried out by use of the default force field, OPLS4. In relation to transmembranes (TMs), the palmitoyl-oleoyl-phosphatidylcholine (POPC) bilayer membrane was positioned by adhering to the OPM information source. The system was neutralized by adding Na + and Cl– ions after it was solvated using the TIP3P water model4 inside the 10 Å orthorhombic box. In particular, the addition of ions was rationally avoided in the 4 Å space surrounding the compound S23. The System Builder program for the Desmond MD engine was used to set up the system. Following its conclusion, a 100-ns MD simulation using the chosen ensemble of NPγT was run in an isothermal-isobaric thermodynamic environment. Throughout the simulation, 1.01325 bar of applied pressure and 300 K of temperature were maintained. Using VMD and the Desmond MD engine’s simulation interaction diagram module, the MD trajectory was further examined after the MD simulation was finished.
Discussion
A series of 1-(2-amino -2,4,5,6,7,7a-hexahydrobenzo[b] -3-yl)-3-substitued-phenylpropane-1,3-dione derivatives were synthesized using the Gewald synthesis in the first step, followed by Baker−Venkataraman rearrangement to yield title compounds. The FTIR, MS and 1H NMR results of the produced derivatives were validated. The biological potential of the synthesized compounds was examined in vitro using various techniques, including the tube dilution method for testing antimicrobial activity, the DPPH method for testing antioxidant activity and the microplate Alamar blue assay (MABA) method for testing antimycobacterial activity against a particularly virulent strain of MTB (MTB H37Ra). Antimicrobial screening outcomes showed that compound S17 turned out to be the most effective antibacterial agent against Staphylococcus aureus (MIC = 16.87 µM), Bacillus subtilis (MIC = 9.45 µM) and Escherichia coli (MIC = 16.87 µM), while compound S7 exhibited outstanding activity against Salmonella typhi (MIC = 9.74 µM), and compound S16 displayed remarkable antifungal activity toward both Candida albicans and Aspergillus niger (MIC = 15.23 µM). Compound S10 demonstrated good antioxidant activity (IC50 = 45.29 µg/mL), outperforming ascorbic acid. From the results of antitubercular activity, compound S23 exhibited promising efficacy with an MIC value of 78.125 µg/mL. Molecular docking studies were conducted to elucidate the binding modes of the synthesized compounds with the enzymatic active site of “DprE1-decaprenylphosphoryl-β-D-ribose-2′-epimerase.” The docking results revealed a comparable binding mode to the native ligand with a favorable docking score, shedding light on the potential of these compounds as inhibitors of DprE1 and contributors to the development of effective antitubercular drugs. The synthesized compounds showed very good binding affinity in comparison with a positive control (Isoniazid). Compound S23 was found to have very good docking score of −8.516 as compared to the Isoniazid with the docking score of − 6.315. So from the study of both docking and as well as in vitro analysis of synthesized compounds it gives the clear idea that compound S23 was found to be very effective as antitubercular drug. This study underscores the significance of understanding ligand–protein interactions at the molecular level for rational drug design against Mycobacterium tuberculosis.
Conclusion
In summary, we can say that new tetrahydrobenzothiophene compounds were created, forming a new class of inhibitors with strong antibacterial, antioxidant and antitubercular characteristics. Compound S23 was discovered to be the most effective against a variety of Gram positive and Gram negative bacterial strains with electron-releasing groups; however, substitution with electron-releasing groups increased the antibacterial activity against S. Typhi strain. Because of presence of methoxy group at the para position and a chloro group at the ortho position, compound S16 has become the most potent antifungal agent. Due to the presence of an electron-releasing group, compound S10 was found to be more powerful in antioxidant test results. Significant antitubercular action was shown by compound S23 against the MTB H37Ra strain. According to the results of the antitubercular screening, the produced compounds with electron-releasing groups (o–OH, p–CH3) on the benzylidene part were found to have substantial action. Molecular docking study reveals that tetrahydrobenzo[b]thiophene-2-carboxylic acid derivatives shown high binding affinity against active site of the DprE1enzymes because of hydrogen bonding with the glycine 55, glycine 57 and glycine 125 residue of receptor protein. The docking score of synthesized compound S23 was more than that of standard drug (Isoniazid). This provides a solid foundation for the development of the lead compounds for this series that will constitute effective antitubercular medicines. Therefore, these thiophene derivatives undoubtedly have a higher chance of being discovered as a lead molecule for the development of new medicinal medicines.
Data availability
All data are provided in the manuscript or cited in the references.
Abbreviations
- MDR:
-
Multidrug resistant
- XDR:
-
Extensively drug resistant
- TB:
-
Tuberculosis
- MTB:
-
Mycobacterium tuberculosis
- µM:
-
Micromolar
- µg:
-
Microgram
- MIC:
-
Minimum inhibitory concentrations
- FTIR:
-
Fourier transform infrared spectroscopy
- MS:
-
Mass spectrometry
- 1H NMR:
-
Proton nuclear magnetic resonance
- DPPH:
-
2,2-Diphenyl-1-picrylhydrazyl
- MABA:
-
Microplate Alamar blue assay
- DprE1:
-
Decaprenylphosphoryl-β-D-ribose-2′-epimerase
- BVR:
-
Baker–Venkataraman rearrangement
- DPA:
-
Decaprenylphosphoryl-D-arabinose
- TLC:
-
Thin-layer chromatography
- UV:
-
Ultraviolet
References
N. Karaman, Y. Sicak, T. Taskin-Tok, M. Ozturk, A. Iyidogan, M. Dikmen, B. Kaymakcioglu, E. Oruc-Emre, J. Eur, Med. Chem. 124, 270 (2016)
R. Shah, P.K. Verma, BMC Chem. 13, 54 (2019)
P.S. Mahajan, M.D. Nikam, L. Nawale, V.M. Khedkar, D. Sarkar, C.H. Gill, ACS Med. Chem. Lett. 8, 751 (2016)
H.M. Metwally, N.A. Khalaf, E. Abdel-Latif, M.A. Ismail, BMC Chem. 17, 6 (2013)
A.D. Pillai, P.D. Rathod, F.P. Xavier, H. Pad, V. Sudarsanam, K.K. Vasu, Bioorg. Med. Chem. 13, 6685 (2005)
R.K. Russell, J.B. Press, R.A. Rampulla, J.J. McNally, R. Falotico, J.A. Keiser, D.A. Bright, A. Tobia, J. Med. Chem. 31, 1786 (1988)
M. Zhao, Y. Cui, L. Zhao, T. Zhu, R.J. Lee, W. Liao, F. Sun, Y. Li, L. Teng, ACS Omega 4, 8874 (2019)
M. Benabdellah, A. Aouniti, A. Dafali, B. Hammouti, M. Benkaddour, A. Yahyi, A. Ettouhami, Appl. Surf. Sci. 252, 8341 (2006)
K.G. Senthil, C. Umarani, A. Ramachandran, J. Indian Chem. Soc. 98, 100079 (2021)
C. Kim, K.S. Choi, J.H. Oh, H.J. Hong, S.H. Han, S.Y. Kim, Sci. Adv. Mater. 7, 2401 (2015)
M. Juhas, V.K. Pallabothula, K. Grabrijan, M. Simovicova, O. Jandourek, K. Konecna, P. Barta, P. Paterova, S. Gobec, I. Sosic, J. Zitko, Bioorg. Chem. 118, 105489 (2022)
B.M. Sahoo, B.K. Banik, A.K. Mahato, C.N. Shanthi, B.C. Mohantad, Green Approaches Med Chem. Sustain. Drug Des. (2020). https://doi.org/10.1016/B978-0-12-817592-7.00024-1
World Health Organization, WHO consolidated guidelines on tuberculosis. module 4: treatment-drug-resistant tuberculosis treatment 2022 Update. (Geneva, Switzerland: World Health Organization, 2022).
B.D. Edwards, S.K. Field, Drugs 82, 1695 (2022). https://doi.org/10.1007/s40265-022-01817-w
G.F. Fernandes, A.M. Thompson, D. Castagnolo, W.A. Denny, J.L. Dos Santos, J. Med. Chem. 65, 7489 (2022). https://doi.org/10.1021/acs.jmedchem.2c00227]
M. Tapera, H. Kekeçmuhammed, K. Sahin, V.S. Krishna, C. Lherbet, H. Homberset, M. Chebaiki, T. Tonjum, L. Mourey, Y. Zorlu, S. Durdagi, E. Saripinar, J. Mol. Struct. 1270, 133899 (2022)
A. Chaudhary, K.K. Jha, S. Kumar, J. Adv. Sci. Res. 3, 03 (2012)
R. Shah, M. Shah, V. Kamboj, P. Verma, J. Heterocycl. Chem. (2022). https://doi.org/10.1002/jhet.4557]
R. Shah, P. Verma, Chem. Cen. J. (2018). https://doi.org/10.1186/s13065-018-0511-5]
V. Anton, H. Artigas, L. Lomba, B. Giner, C. Lafuente, J. Therm. Anal. Calorim. 125, 509 (2016)
N. Kausar, S. Murtaza, M.N. Arshad, R. Munir, R.S. Saleem, H. Rafique, A. Tawab, J. Mol. Struct. 1244, 130983 (2021)
S.R. Alsayed, S. Lun, G. Luna, C.C. Beh, A.D. Payne, N. Foster, W.R. Bishai, H. Gunosewoyo, R. Soc. Chem. 10, 7523 (2020)
S. Srinivasarao, A. Nandikolla, A. Suresh, K.V. Calster, V.L. De, D. Cappoen, B. Ghosh, A. Himanshu, S. Murugesan, S. Chandra, Roy. Soc. Chem. Adv. 10, 12272 (2020)
R. Mishra, P.K. Sharma, J. Int, Eng. Al. Sci. 1, 46 (2015)
S. Ghosh, S.N. Mali, D.N. Bhowmick, A.P. Pratap, J. Indian Chem. Soc. 98, 100088 (2021)
S.N. Mali, A. Pandey, J. Indian Chem. Soc. 298, 100082 (2021)
S.N. Mali, A. Pandey, J. Comput. Biophys. Chem. 21, 857 (2022)
E.A. Abd, F.E. Mohamed, A.I. Alhussein, M.H. Hanaa, A.O. Mohamed, A.G. Hazem, Mini Rev. Med. Chem. 19, 833 (2019)
A. Abbas, A.B. Basharat, K.M. Khan, J. Iqbal, S. Rahman, S. Zaib, S. Perveen, Bioorg. Chem. 82, 163 (2019)
S.N. Mali, A. Pandey, R.R. Bhandare, A.B. Shaik, Sci. Rep. 12, 16368 (2022)
J. Gawad, C. Bonde, Chem. Cent. J. (2018). https://doi.org/10.1186/s13065-018-0515-1]
E.F. Silva-Junior, E.P.S. Silva, P.B. França, J.N. Silva, E.O. Barreto, E.B. Silva, R.S. Ferreira, C.C. Gatto, D.M. Moreira, J.L. Siqueira-Neto, F.B. Mendonça-Júnior, M.C. Lima, J.H. Bortoluzzi, M.T. Scotti, L. Scotti, M.R. Meneghetti, T.M. Aquino, J.X. Araújo-Júnior, Bioorg. Med. Chem. 24, 4228 (2016)
V.M. Dembitsky, H.A. Ali, M. Srebnik, Stud. Inorg. Chem. 22, 367 (2005). https://doi.org/10.1016/S0169-3158(06)80007-1
CLSI (ed.), Clinical and Laboratory Standards Institute., Performance Standards for Antimicrobial Susceptibility Testing, 30th ed. CLSI Suppl. M100. Wayne, PA Clin. Lab. Stand. Inst. (2020)
J.G. Cappuccino, N. Sherman, In microbiology- a laboratory manual, 4th edn. (Addison Wesley Longman, California, 1996)
P.K. Mukherjee, In Quality control and evaluation of herbal drugs: Evaluating natural products and traditional medicine. Kindle Edition P.N.564 (2012)
A. Farzana, R. Chakraborty, A. Thakur, J. Int, Pharm. Chem. 05, 334 (2015)
S. Narramore, C.M. Stevenson, A. Maxwell, D.M. Lawson, C.G. Fishwick, Bioorg. Med. Chem. 27, 3546 (2019)
F. Shaikh, S.L. Shastri, N.S. Naik, R. Kulkarni, J.M. Madar, L.A. Shastri, V. Sunagar, Chem. Select. 4, 105 (2019)
Acknowledgements
Thanks to Head Prof. Harish Dureja, Department of Pharmaceutical Sciences, M.D.U, Rohtak, for providing library and internet facilities, etc.
Funding
None.
Author information
Authors and Affiliations
Contributions
PKV designed and finalized the scheme; RS did research work; and MS and SK did review work and wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The author(s) have no conflicts of interest.
Consent to participate
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shah, R., Verma, P.K., Shah, M. et al. Design, synthesis, biological evaluation and molecular docking studies of thiophene derivatives. J IRAN CHEM SOC 21, 2501–2515 (2024). https://doi.org/10.1007/s13738-024-03088-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-024-03088-6