
Abstract
A new series of naphthyl chalcones (3a–3p) and their pyrazoline derivatives (4a–4h) were synthesized using substituted acetophenones, substituted naphthaldehydes, and hydrazine hydrate as starting materials. All the synthesized compounds were characterized by IR, NMR, and mass spectrometric analysis and screened for antimycobacterial activity against Mycobacterium tuberculosis H37Rv (ATCC 27924) and antibacterial activity against Staphylococcus aureus (MTCC 96), Bacillus subtilis (MTCC 441), Escherichia coli (MTCC 443) and Klebsiella pneumonia (MTCC 109). Compounds 3b and 3p exhibited significant antibacterial activity against all the tested bacterial strains. Amongst the synthesized compounds, compound 4b with 2-hydroxy-5-bromophenyl substitution at 3rd position of pyrazoline showed significant antimycobacterial activity with MIC of 6.25 µM comparable to that of standard isoniazid. The synthesized compounds were further screened for their cytotoxic activity against the MDA-MB-231 and SKOV3 cell lines. The compounds 3a, 3l, 4b, 4c, 4e, and 4h did not exhibit any cytotoxicity, and other compounds exhibited IC50 values higher than 8 and 22 µM against MDA-MB-231 and SKOV3 cell lines, respectively, compared to 1.20 and 1.30 µM shown by standard doxorubicin. To find out the putative binding mode of significantly active and weakly active compounds, a molecular docking study was also performed. In that, the most active compound 4b, displayed a hydrogen bond interaction with docking score of −10.50 kcal/mol and energy of −44.50 weakly active compound 3h did not show any crucial hydrogen bond interaction with the surrounded amino-acid residues and revealed a docking score of −6.74 and docking energy of −42.50.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Tuberculosis (TB) is one of the leading causes of global death, among other infectious diseases. Over the last four decades, there is no much change in the TB treatment regimen. The emergence of drug resistance became thought provoking and demanding the development of regimens (Farah et al. 2015). In recent times, novel drugs like bedaquiline, delamanid, and a few repurposed drugs like clofazimine, linezolid, and carbapenems are being used in TB patients infected with drug-resistant mycobacterium. Effective treatment for TB mostly consists of bactericidal and sterilizing drugs and their combinations for a sufficient duration of action, but in many cases, the current treatment has been failed to show promising antimycobacterial efficacy and permanent cure. However, existing drugs and newly approved leads gradually becoming unsatisfactory due to low efficacy, high toxicity, drug resistance, and prolonged duration of therapy (Sharma et al. 2009).
Chalcones are chemically 1,3-diaryl-2-propen-1-ones, a group of natural or synthetic compounds consisting of open-chain flavonoids, comprises two aromatic rings joined by a three-carbon α, β-unsaturated carbonyl system, precursors for the synthesis of various flavonoids and isoflavonoids (Lin et al. 2002). Chalcone has become a vital lead molecule for its valuable pharmacological activities, including antioxidant (Ahmad et al. 2013), antibacterial (Kumar et al. 2014), antitrypanosomal (Patole et al. 2006), antileishmanial (Lopes et al. 2015), anticancer (Casey et al. 2019), antidiabetic (Zhou et al. 2010), and anti-inflammatory (Kumar et al. 2016) activities. Licochalcone, a naturally occurring flavonoid with chalcone moiety, exhibited potent antimycobacterial activity against several strains of mycobacterium and proved that halogenated chalcones as potent antitubercular agents (Lin et al. 2002). Sivakumar et al. (2007) developed a series of chalcone derivatives and drawn their structure–activity relationship with respect to antitubercular activity. In recent findings, chalcone derivatives showed promising antimycobacterial activity by selectively inhibiting various targets like Dihydrofolate reductase (Hans et al. 2010), Tyrosine phosphatase A (Chiaradia et al. 2012), Enoyl-Acp reductase (He et al. 2006), etc. Gomes et al. reported a furanyl containing chalcone ((2E)-3-(5-nitro-furan-2-yl)-1-[4-(piperidin-1-yl)-phenyl]prop-2-en-1-one) as a promising antitubercular agent with MIC of 0.07 and 0.03 µM against Rifampicin (rmp resistant) and isoniazid (inh resistant) strains (Gomes et al. 2017). Fluorine substituted chalcone, (E)-3-(4-fluorophenyl)-1-(2,4,6-trimethoxyphenyl)prop-2-ene-1-one was reported as a potent antitubercular agent by Burmaoglu et al. (2017). A series of chalcones were synthesized by the reaction of 2,5-dimethylthiophene with various heterocyclic aldehydes and evaluated for antibacterial activity (Khan and Asiri 2017). Among the series, pyrazole containing chalcone showed better antibacterial activity (MIC of 16 µg/ml) against S. typhimurium and Escherichia coli than the reference chloramphenicol.
Naphthalenes have been recognized as a new range of potent antimicrobials effective against an extensive range of human pathogens. They occupy a central place among medicinally essential compounds due to their diverse and exciting antibiotic properties with minimum toxicity as well as antimycobacterial properties (Rokade et al. 2010). Interestingly, a set of naphthyl chalcone-based hybrid molecules were developed and screened by Chiaradia et al. (2012) for their antitubercular activity. A well-organized structure–activity relationship was derived for inhibition of Mycobacterium tyrosine phosphatase A.
In drug discovery and research, heterocyclic compounds occupy a central role, and nitrogen-containing moieties demonstrated a diverse range of pharmacological activities. Pyrazolines, nitrogen-containing heterocyclic compounds have been investigated extensively for the generation of novel antimicrobial agents. Various research findings revealed that pyrazoline derivatives has been proved as an attractive scaffolds in developing promising leads with potent biological activities like antidepressant (Marrapu et al. 2011), anticonvulsant (Zhou et al. 2010), antibacterial (Lopes et al. 2015), antifungal (Figueroa-valverde et al. 2017), antiamoebic (Nayyar et al. 2006), anti-HIV (Sivakumar et al. 2007), and anticancer (Sharma et al. 2009). Ahmed and Husain (2016) synthesized a new set of pyrazoline derivatives and found significant antimycobacterial and antifungal activity.
At present, the scenario of research and focus of Health organizations and funding agencies is toward the finding of novel molecules for safe and effective TB treatment with a shorter duration, and also to encourage researchers toward discovery and development of new antitubercular drugs (Castaño et al. 2019). Because of the above, a new series of naphthyl chalcones and the corresponding pyrazoline derivatives were synthesized by standard protocols, characterized and screened for antibacterial and antitubercular activity. Designing of the target compound is shown in Fig. 1.

Designing approach of the target compounds
Experimental
Materials and methods
All the chemicals and solvents were procured from Sigma-Aldrich Pvt. Ltd. The reactions were monitored with the help of thin-layer chromatography using pre-coated aluminum sheets, and column chromatography was performed on silica gel 60–100, 250–400 mesh (Merk) for purification of the compounds. The solvent system of ethyl acetate: hexane and chloroform: methanol of different ratios was used. The spot were visualized under a UV lamp. Melting points of the synthesized compounds were determined using MEL TEMP electro thermal apparatus. IR spectra were acquired on the Perkin Elmer FT-IR spectrometer. Both 1H NMR and 13C NMR spectra of the synthesized compounds were performed with Bruker Avance-II 400 NMR Spectrometer operating at 400 and 100 MHz, respectively, at Andhra University, Vishakhapatnam; the chemical shifts were reported in ppm units with respect to TMS as the internal standard. Mass spectra of the synthesized compounds were recorded with 6545 QTOF MS/MS at IICT, Hyderabad. Schrodinger software was used for molecular docking studies at BITS Pilani, Pilani Campus.
Procedure for synthesis of naphthyl chalcones (3a–3p)
Chalcones were prepared by base-catalyzed condensation of a mixture of the substituted acetophenones and substituted naphthaldehydes in ethanol. To the ethanolic solution of substituted acetophenone (2) (0.1 mol), an equimolar quantity of substituted naphthaldehyde (1) (0.1 mmol) was added. To the reaction mixture, aqueous potassium hydroxide (5 ml, 60%) was added gradually drop wise with constant stirring, and the reaction was continued at room temperature under stirring for 14–16 h. The progress of the reaction was supervised using TLC. The volume of KOH varied according to the reaction (Lopes et al. 2015). To the reaction mixture, distilled water and 10% hydrochloric acid were added for precipitation of the compounds (3a–p). Resulting solids were filtered and recrystallized from ethanol.
Procedure for synthesis of pyrazolines (4a–4h)
To the solution of appropriate chalcone (3a–3p, 0.01 mol) in ethanol (10 ml), hydrazine hydrate (0.02 mol) was added. To the reaction mixture, the catalytic amount of glacial acetic acid was added and refluxed for 8–10 h. The progress of the reaction was monitored using TLC (Ahmad and Husain 2016). After the end of the reaction, the crude mixture was poured into ice-cold water, and separated precipitate was filtered off. Obtained solid compounds (4a–4h) were purified by column chromatography/recrystallization.
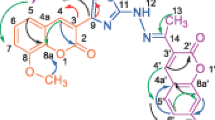
Plausible mechanism involved in the reactions is shown in Figs. 2 and 3.

Plausible mechanism for the napthylchalcones (3a–p)

Plausible mechanism for the napthylpyrazoline (4a–h)
Characterization of synthesized compounds is given below:
1-(5-bromo-2-hydroxyphenyl)-3-(naphthalen-1-yl)prop-2-en-1-one (3a)
Compound 3a obtained as orange solid (yield 70%), mp 58–60 °C. 1H NMR (CDCl3, 400 MHz, δ, TMS = 0): 12.81 (s, 1H, –OH), 8.86–8.82 (d, 1H, H-α, J = 14.8 Hz), 8.30–8.28 (d, 1H, Ar–H, J = 8.4 Hz), 8.08–8.07 (d, 1H, Ar–H, J = 2.4 Hz), 7.95–7.93 (d, 1H, Ar–H, J = 8.0 Hz), 7.70–7.56 (m, 5H, Ar–H & H-β), 7.00–6.98 (d, 1H, J = 8.8 Hz). 13C NMR (CDCl3, 100 MHz, δ, TMS = 0): 192.8, 162.8, 138.6, 135.8, 134.9, 133.6, 133.5, 132.6, 128.8, 128.3, 126.9, 126.5, 126.0, 124.3, 122.9, 122.6, 121.3, 119.7, 116.5. Anal. Calcd. MS: 353.211; found m/z: 353.000 (M+), 355.000 (M+ + 2). IR (KBr, cm−1): 3468.29 (–OH), 2956.23 (C–H, aliphatic), 1656.27 (C=O).
1-(2, 4-dichlorophenyl)-3-(naphthalen-1-yl)prop-2-en-1-one (3b)
Compound 3b obtained as cream solid (yield 68%), mp 62–64 °C. 1H NMR (CDCl3, 400 MHz, δ, TMS = 0): 8.42–8.38 (d, 1H, H-α, J = 15.6 Hz), 8.14–8.12 (d, 1H, Ar–H, J = 8.4 Hz), 7.97–7.95 (d, 1H, Ar–H, J = 8 Hz), 7.92–7.87 (m, 2H, Ar–H), 7.63–7.58 (m, 5H, Ar–H & H-β), 7.43–740 (dd, 1H-Ar, J = 2.0 Hz), 7.285–7.28 (d, 1H, Ar–H, J = 1.6 Hz), 7.24 (s, 1H, Ar–H). 13C NMR (CDCl3, 100 MHz, δ, TMS = 0): 189.6, 146.5, 135.9, 135.1, 133.6, 133.2, 132.7, 132.0, 131.8, 129.5, 128.8, 128.3, 127.5, 126.9, 126.3, 126.1, 124.0, 122.3, 121.5. Anal. Calcd. MS: 327.029; found m/z: 327.000 (M+), 329.000 (M+ + 2). IR (KBr, cm−1): 3056.95 (C–H, aromatic), 2986.36 (C–H, aliphatic), 1648.32 (C=O).
1-(2-hydroxyphenyl)-3-(naphthalen-1-yl) prop-2-en-1-one (3c)
Compound 3c obtained as light yellow solid (yield 63), mp 56–58 °C. 1H NMR (CDCl3, 400 MHz, δ, TMS = 0): 12.89 (s, 1H, –OH), 8.83–8.79 (d, 1H, H-α, J = 15.2 Hz), 8.32–8.29 (d, 1H, Ar–H J = 8.4 Hz), 8.04–7.92 (m, 1H, Ar–H), 7.80–7.76 (d, 1H, H-β, J = 15.2 Hz), 7.66–7.49 (m, 4H, Ar–H), 7.09–7.07 (d, 1H, Ar–H, J = 8.4 Hz), 7.00–6.97 (t, 1H, Ar–H, J = 7.6 Hz). 13C NMR (CDCl3, 100 MHz, δ, TMS = 0): 191.8, 162.9, 135.9, 135.8, 133.8, 133.6, 132.0, 129.8 128.8, 128.3, 126.9, 126.3, 126.0, 124.0, 122.9, 121.8, 121.5, 121.2, 118.0. Anal. Calcd. MS: 274.02; found m/z: 275 (M+ + 1). IR (KBr, cm−1): 3442.2 (OH), 3032 (C–H, aromatic), 1634.47 (C=O).
1-(4-methoxyphenyl)-3-(naphthalen-1-yl) prop-2-en-1-one (3d)
Compound 3d obtained as yellow solid (yield 73%), mp 52–54 °C. 1H NMR (CDCl3, 400 MHz, δ, TMS = 0): 8.65–8.61 (d, 1H, H-α, J = 13.6 Hz), 8.60–8.57 (d, 1H, Ar–H, J = 9.6 Hz), 8.53–8.46 (m, 2H, Ar–H), 8.08–8.05 (d, 1H, H-β, J = 13.2 Hz), 7.98–7.97 (t, 2H, Ar–H, J = 6.8 Hz), 7.65–7.63 (m, 4H, Ar–H), 3.83 (s, 3H, –OCH3). 13C NMR (CDCl3, 100 MHz, δ, TMS = 0): 189.5, 136.9, 135.8, 133.6, 133.6, 132.2, 132.0, 132.0, 128.9, 128.8, 128.3, 126.9, 126.3, 126.0, 124.6, 122.9, 122.3. Anal. Calcd. MS: 288.12; found m/z: 289.000 (M+ + 1). IR (KBr, cm−1): 3020.28 (C–H, aromatic), 1649.28 (C=O).
1-(5-bromo-2-hydroxyphenyl)-3-(2-methoxynaphthalen-1-yl)prop-2-en-1-one (3e)
Compound 3e obtained as yellowish orange solid (yield 74%), mp 60–62 °C. 1H NMR (CDCl3, 400 MHz, δ, TMS = 0): 12.97 (s, 1H, –OH), 10.92 (s, 1H, Ar–H), 9.31–9.29 (d, 1H, Ar–H, J = 7.2 Hz), 8.68–8.65 (d, 1H, H-α, J = 14.8 Hz), 8.28–7.28 (m, 6H, Ar–H), 8.07–8.03 (d, 1H, H-β, J = 14.0 Hz), 6.98–6.97 (d, 1H, Ar–H, J = 7.6 Hz). 4.08 (s, 1H, –OCH3). 13C NMR (CDCl3, 100 MHz, δ, TMS = 0): 192.5, 162.4, 151.2, 145.6, 138.6, 134.5, 130.6, 129.8, 129.7, 128.5, 126.7, 124.2, 122.8, 121.3, 119.7, 119.2, 116.2, 114.8, 56.4. Anal. Calcd. MS: 383.02; found m/z: 383 (M+), 385 (M+ + 2). IR (KBr, cm−1): 3456.28 (OH), 2978.23 (C–H, aliphatic), 1649.58 (C=O).
1-(2-hydroxyphenyl)-3-(2-methoxynaphthalen-1-yl)prop-2-en-1-one (3f)
Compound 3f obtained as white solid (yield 75%), mp 62–64 °C. 1H NMR (CDCl3, 400 MHz, δ, TMS = 0): 10.92 (s, 1H, –OH), 8.67–8.64 (d, 1H, H-α, J = 15.6 Hz), 8.31–8.29 (d, 1H, Ar–H, J = 8.8 Hz), 8.11–8.07 (m, 6H, Ar–H & H-β), 7.93–7.32 (m, 6H, Ar–H), 7.26–7.08 (d, 1H, Ar–H, J = 8.4 Hz), 6.98–6.95 (t, 1H, Ar–H, J = 7.6 Hz), 4.11 (s, 3H, –OCH3). 13C NMR (CDCl3, 100 MHz, δ, TMS = 0): 192.9, 163.2, 150.0, 145.2, 139.6, 138.4, 130.0, 129.8, 129.2, 128.6, 126.7, 124.8, 123.4, 122.7, 121.6, 121.4, 119.8, 116.2, 114.8, 56.3. Anal. Calcd. MS: 304.339; found m/z: 305.000 (M+ + 1). IR (KBr, cm−1): 3428.96 (OH), 3002.32 (C–H, aromatic), 2856.45 (C–H, aliphatic), 1652.39 (C=O).
1-(4-bromophenyl)-3-(2-methoxynaphthalen-1-yl)prop-2-en-1-one (3g)
Compound 3g obtained as white solid (yield 75%), mp 52–54 °C. 1H NMR (CDCl3, 400 MHz, δ, TMS = 0): 10.95 (s, 1H, –OH), 8.65–8.62 (d, 1H, H-α, J = 15.6 Hz), 8.18–8.09 (d, 1H, Ar–H, J = 8.8 Hz), 7.90–7.72 (m, 6H, Ar–H), 7.37–7.22 (m, 4H, Ar–H & H-β), 7.16–7.05(d, 1H, Ar–H, J = 8.4 Hz), 6.98–6.95 (t, 1H, Ar–H, J = 7.6 Hz), 3.96 (s, 3H, –OCH3). 13C NMR (CDCl3, 100 MHz, δ, IMS = 0): 192.6, 163.4, 150.5, 145.2, 139.7, 138.5, 130.4, 129.7, 129.1, 128.6, 126.7, 124.6, 123.8, 122.6, 121.4, 119.35, 116.2, 114.5, 56.3. Anal. Calcd. MS: 367.24; found m/z: 367.000 (M+), 369.00 (M+ + 2). IR (KBr, cm−1): 3028.92 (C–H, aromatic), 1649.63 (C=O).
3-(2-methoxynaphthalen-1-yl)-1-(4-methoxyphenyl)prop-2-en-1-one (3h)
Compound 3h obtained as cream solid (yield 65%), mp 56–58 °C. 1H NMR (CDCl3, 400 MHz, δ, TMS = 0): 8.51–8.47 (d, 1H, H-α, J = 16.0 Hz), 8.30–8.28 (d, 1H, Ar–H J = 8.4 Hz), 8.11–8.09 (d, 2H, Ar–H, J = 8.8 Hz), 7.92–7.88 (m, 2H, Ar–H & H-β), 7.84–7.82 (d, 1H, Ar–H, J = 8.0 Hz), 7.56–754 (t, 1H, Ar–H, J = 8.0 Hz), 7.44–7.40 (t, 1H, Ar–H, J = 7.6 Hz), 7.36–7.34 (d, 1H, Ar–H, J = 9.2 Hz), 7.03–7.01 (d, 2H, Ar–H, J = 8.8 Hz), 4.07 (s, 3H, –OCH3), 3.92 (s, 3H,–OCH3). 13C NMR (CDCl3, 100 MHz, δ, TMS = 0): 190.8, 163.5, 158.5, 143.3, 132.9, 131.8, 131.8, 129.5, 127.9, 127.3, 126.9, 123.8, 122.9, 122.8, 122.7, 118.8, 110.3, 55.9, 55.8. Anal. Calcd. MS: 318.37; found m/z: 319.000 (M+ + 1). IR (KBr, cm−1): 3067.45 (C–H, aromatic), 2964.52 (C–H, aliphatic) 1652.23 (–C=O).
1-(5-bromo-2-hydroxyphenyl)-3-(4-methoxynaphthalen-1-yl) prop-2-en-1-one (3i)
Compound 3i obtained as orange solid (yield 74%), mp 62–64 °C. 1H NMR (CDCl3, 400 MHz, δ, TMS = 0): 12.97 (s, 1H, O–OH), 10.23 (s, 1H, Ar–H), 9.34–9.31 (d, 1H, Ar–H, J = 8.8 Hz), 8.83–8.79 (d, 1H, H-α, J = 15.2 Hz), 8.32–8.31 (m, 1H), 8.29–8.27 (d, 1H, Ar–H, J = 8.4 Hz), 8.07 (s, 1H, Ar–H), 8.02–8.00 (d, 1H-Ar, J = 8.0 Hz), 7.96–7.94 (d, 1H, Ar–H, J = 8.0 Hz), 7.74–7.56 (m, 3H, Ar–H & H-β), 4.13 (s, 3H, –OCH3). 13C NMR (CDCl3, 100 MHz, δ, TMS = 0): 192.8, 162.8, 153.2, 138.8, 135.8, 134.9, 131.2, 126.5, 125.5, 125.3, 124.5, 124.0, 123.3, 123.1, 121.0, 119.7, 116.2, 103.0, 56.2. Anal. Calcd. MS: 383.24; found m/z: 383 (M+), 385 (M+ + 2). IR (KBr, cm−1): 3442.42 (OH), 3067.45 (C–H, aromatic), 2956.54 (C–H, aliphatic), 1646.16 (C=O).
1-(4-bromophenyl)-3-(4-methoxynaphthalen-1-yl) prop-2-en-1-one (3j)
Compound 3j obtained as light cream solid (yield 75%), mp 62–64 °C. 1H NMR (CDCl3, 400 MHz, δ, TMS = 0): 8.68–8.64 (d, 1H, H-α, J = 15.2 Hz), 8.36–8.31 (m, 1H, Ar–H), 8.26–8.24 (d, 1H, Ar–H, J = 8.4 Hz), 8.08–8.06 (d, 1H, Ar–H, J = 8.8 Hz), 7.96–7.92 (t, 2H, Ar–H, J = 8.0 Hz), 7.81–7.79 (d, 1H, Ar–H, J = 8.4 Hz), 7.69–7.62 (m, 3H, Ar–H & H-β), 3.98 (s, 3H, –OCH3). 13C NMR (CDCl3, 100 MHz, δ, TMS = 0): 189.7, 153.2, 136.9, 135.8, 132.2, 131.8, 130.2, 128.0, 126.5, 125.4, 125.2, 124.0, 123.6, 123.6, 121.0, 116.8, 103.9, 56.3.Anal. Calcd. MS: 367; found m/z: 367.000 (M+), 369.000 (M+ + 2). IR (KBr, cm−1): 3002.86 (C–H, aromatic), 2989.56 (C–H, aliphatic), 1652.23 (C=O).
3-(4-methoxynaphthalen-1-yl)-1-(4-methoxyphenyl) prop-2-en-1-one (3k)
Compound 3k obtained as light yellow solid (yield 78%), mp 62–64 °C. 1H NMR (CDCl3, 400 MHz, δ, TMS = 0): 8.64–8.62 (d, 2H, Ar–H, J = 5.6 Hz), 8.61–8.58 (d, 2H, Ar–H, J = 9.2 Hz), 8.32–8.28 (d, 1H, H-α, J = 15.2 Hz), 7.98–7.95 (d, 1H, H-β, J = 14.8 Hz), 7.56–7.53 (d, 4H, Ar–H, J = 10.4 Hz), 7.280–7.256 (d, 1H, Ar–H, J = 9.6 Hz), 7.23–7.21 (d, 1H, Ar–H, J = 8.8 Hz), 3.83 (s, 3H, –OCH3), 3.78 (s, 3H, –OCH3). 13C NMR (CDCl3, 100 MHz, δ, TMS = 0): 189.5, 166.6, 153.2, 135.8, 130.0, 130.2, 126.6, 125.3, 125.3, 125.1, 124.2, 123.6, 121.5, 121.0, 116.5, 114.8, 103.2, 56.2, 55.6. Anal. Calcd. MS: 318.37; found m/z: 319.000 (M+ + 1). IR (KBr, cm−1): 3002.56 (C–H, aromatic), 2906.56 (C–H, aliphatic), 1643.89 (C=O).
1-(2-hydroxyphenyl)-3-(4-methoxy naphthalene-1-yl) prop-2-en-1-one (3l)
Compound 3l obtained as yellow solid (yield 74%), mp 60–62 °C. 1H NMR (CDCl3, 400 MHz, δ, TMS = 0): 11.02 (s, 1H, –OH), 8.67–8.64 (d, 1H, H-α, J = 15.6 Hz), 8.31–8.29 (d, 1H, Ar–H, J = 8.8 Hz), 8.11–8.07 (m, 6H, Ar–H & H-β), 7.93–7.32 (m, 6H, Ar–H), 7.26–7.08 (d, 1H, Ar–H, J = 8.4 Hz), 6.98–6.95 (t, 1H, Ar–H, J = 7.6 Hz), 3.82 (s, 3H, –OCH3). 13C NMR (CDCl3, 100 MHz, δ, TMS = 0): 192.6, 163.5, 153.2, 135.5, 132.9, 131.8, 129.8, 126.9, 125.7, 125.6, 125.1, 124.5, 123.2, 121.8, 121.6, 121.1, 118.3, 116.5, 103.8, 56.2. Anal. Calcd. MS: 304.339; found m/z: 305.000 (M+ + 1). IR (KBr, cm−1): 3020.56 (C–H, aromatic), 1652.56 (C=O).
1-(5-bromo-2-hydroxyphenyl)-3-(6-methoxynaphthalen-2-yl)prop-2-en-1-one (3m)
Compound 3m obtained as orange solid (yield 75%), mp 64–66 °C. 1H NMR (CDCl3, 400 MHz, δ, TMS = 0): 11.02 (s, 1H, –OH), 8.65–8.32 (d, 1H, H-α, J = 14.6 Hz), 8.21–8.09 (d, 1H, Ar–H, J = 8.6 Hz), 8.01–7.78 (m, 6H, Ar–H), 7.65–7.32 (m, 6H, Ar–H & H-β), 7.26–7.08 (d, 1H, Ar–H, J = 8.4 Hz), 6.88–6.75 (t, 1H, Ar–H, J = 7.6 Hz), 3.78 (s, 3H, –OCH3). 13C NMR (CDCl3, 100 MHz, δ, TMS = 0): 189.7, 166.5, 157.3, 145.2, 135.9, 131.2, 130.9, 130.2, 129.6, 129.3, 127.5, 127.2, 126.6, 121.5, 119.5, 114.8, 114.2, 105.7, 55.8. Anal. Calcd. MS: 383.24; found m/z: 383 (M+), 385 (M+ + 2). IR (KBr, cm−1): 3452.88 (OH), 3089.28 (C–H, aromatic), 2923.63 (C–H, aliphatic), 1662.5 (C=O).
3-(6-methoxynaphthalen-2-yl)-1-(4-methoxyphenyl)prop-2-en-1-one (3n)
Compound 3n obtained as pale yellow solid (yield 78%), mp 66–68 °C. 1H NMR (CDCl3, 400 MHz, δ, TMS = 0): 7.90 (s, 1H, Ar–H), 7.79–7.73 (m, 4H, Ar–H & H-α), 7.69–7.65 (m, 3H, Ar–H & H-β), 7.214–7.16 (m, 3H, Ar–H), 6.83–6.39 (d, 1H, Ar–H, J = 6.4 Hz), 3.96 (s, 3H, –OCH3), 3.43 (s, 3H, –OCH3). 13C NMR (CDCl3, 100 MHz, δ, TMS = 0): 189.9, 166.8, 157.2, 145.2, 138.2, 131.2, 130.9, 130.2, 129.5, 127.6, 126.3, 121.5, 119.5, 114.2, 105.6, 55.8, 55.2. Anal. Calcd. MS: 318.37; found m/z: 319.000 (M+ + 1). IR (KBr, cm−1): 3025.32 (C–H, aromatic), 2856.95 (C–H, aliphatic), 1648.32 (C=O).
1-(4-bromophenyl)-3-(6-methoxynaphthalen-2-yl)prop-2-en-1-one (3o)
Compound 3o obtained as yellowish orange solid (yield 75%), mp 54–56 °C. 1H NMR (CDCl3, 400 MHz, δ, TMS = 0): 8.00–7.93 (m, 4H, Ar–H), 7.81–7.78 (m, 3H, Ar–H), 7.69–7.67 (d, 2H, Ar–H, J = 8.4 Hz), 7.58–7.54 (d, 1H, H-α, J = 15.6 Hz), 7.22–7.17 (m, 2H, Ar–H & H-β), 3.97 (s, 3H, –OCH3). 13C NMR (CDCl3, 100 MHz, δ, TMS = 0): 189.65, 158.20, 145.65, 138.28, 136.92, 132.25, 131.02, 130.06, 129.65, 128.52, 127.50, 127.05, 126.52, 121.30, 119.56, 114.85, 55.8. Anal. Calcd. MS: 367.24; found m/z: 367.00 (M+), 369.00(M+ + 2). IR (KBr, cm−1): 3023.56 (C–H, aromatic), 1646.58 (C=O).
3-(6-methoxynaphthalen-2-yl)-1-(4-nitrophenyl)prop-2-en-1-one (3p)
Compound 3p obtained as light brown solid (yield 74%), mp 60–62 °C. 1H NMR (CDCl3, 400 MHz, δ, TMS = 0): 8.89 (s, 1H, Ar–H), 8.48–8.46 (d, 1H, Ar–H, J = 8.4 Hz), 8.41–8.39 (d, 1H, Ar–H, J = 7.6 Hz), 8.08 (s, 1H, Ar–H), 8.05–8.03 (d, 1H, Ar–H, J = 5.2 Hz), 7.84–7.33 (m, 4H, Ar–H), 7.64–7.60 (d, 1H, H-α, J = 15.6 Hz), 7.24–7.19 (m, 2H, Ar–H & H-β), 3.98 (s, 3H, –OCH3). 13C NMR (CDCl3, 100 MHz, δ, TMS = 0): 189.79, 157.65, 153.28, 145.25, 144.32, 138.90, 131.23, 130.95, 129.83, 127.56, 127.08, 126.56, 124.85, 124.42, 121.35, 119.65, 105.85, 55.68. Anal. Calcd. MS: 333.34; found m/z: 334.23 (M+ + 1). IR (KBr, cm−1): 3016.62 (C–H, aromatic), 2978.32 (C–H, aliphatic), 1642.38 (C=O).
3-(4-methoxyphenyl)-5-(naphthalen-1-yl)-4,5-dihydro-1H-pyrazole (4a)
Compound 4a obtained as light brown solid (yield 65%), mp 114–116 °C. 1H NMR (CDCl3, 400 MHz, δ, TMS = 0): 8.36–8.34 (d, 2H, Ar–H, J = 8.8 Hz), 8.02–7.99 (d, 2H, Ar–H, J = 11.6 Hz), 7.85–7.11 (t, 1H, Ar–H, J = 8.8 Hz), 7.44–7.41 (m, 3H, Ar–H), 7.26–7.24 (d, 2H, Ar–H, J = 8.4 Hz), 6.95 (1H, –NH-pyrazoline), 4.26–4.21 [1H, H-x (pyrazoline), J = 8.8 Hz], 3.92 (s, 3H, –OCH3), 3.52–3.47 (dd, 1H, Hb, J = 8.8, 15.2 Hz), 3.215–3.168 (dd, 1H, Ha, J = 9.6, 15.2 Hz). 13C NMR (CDCl3, 100 MHz, δ, TMS = 0): 163.2, 151.2, 137.8, 130.9, 130.5, 128.9, 128.7, 128.5, 126.4, 125.8, 125.6, 122.9, 122.6, 114.6, 55.6, 48.9, 43.2. Anal. Calcd. MS: 302.37; found m/z: 303 (M+ + 1). IR (KBr, cm−1): 3258.56 (NH), 2924.54 (C–H, aliphatic), 1589.34 (C=N), 1398.56 (C–N).
4-bromo-2-[5-(naphthalen-1-yl)-4,5-dihydro-1H-pyrazol-3-yl]phenol (4b)
Compound 4b obtained as light cream solid (yield 68%), mp 124–126 °C. 1H NMR (CDCl3, 400 MHz, δ, TMS = 0):12.81 (s, 1H, –OH), 10.48 (s, 1H, Ar–H), 8.11–7.75 (m, 5H, Ar–H), 7.46–7.39 (m, 4H, Ar–H & pyrazoline-NH), 3.81–3.74 [dd, 1H, H-x (pyrazoline), J = 8.4 Hz], 3.44–3.41 (dd, 1H, Hb, J = 8.4, 14.2 Hz), 3.23–3.17 (dd, 1H, Ha, J = 10.4, 14.2 Hz). 13C NMR (CDCl3, 100 MHz, δ, TMS = 0): 161.0, 152.7, 137.8, 135.6, 134.2, 130.9, 130.6, 128.8, 128.6, 126.7, 125.6, 125.4, 122.9, 122.6, 121.4, 119.3, 49.6, 43.3. Anal. Calcd. MS: 367.24; found m/z: 367.00 (M+), 369.00(M+ + 2). IR (KBr, cm−1): 3736.29 (OH), 3398.24 (NH), 3042.54 (C–H, aromatic), 1585.58 (C=N), 1392.45 (C–N).
3-(4-bromophenyl)-5-(2-methoxynaphthalen-1-yl)-4,5-dihydro-1H-pyrazole (4c)
Compound 4c obtained as white solid (yield 70%), mp 118–120 °C. 1H NMR (CDCl3, 400 MHz, δ, TMS = 0): 8.02–8.01 (d, 1H, Ar–H, J = 7.2 Hz), 7.99–7.97 (d, 1H, Ar–H, J = 8.0 Hz), 7.72–7.70 (d, 2H, Ar–H, J = 7.2 Hz), 7.58–7.56 (d, 2H, Ar–H, J = 7.2 Hz), 7.54–7.52 (d, 1H, Ar–H, J = 8.0 Hz), 7.46–7.44 (d, 1H, Ar–H, J = 8.8 Hz), 7.04–7.01 (m, 2H, Ar–H), 6.92 (s, 1H, –NH-pyrazoline), 4.25–4.19 [dd, 1H, H-x (pyrazoline), J = 7.8 Hz], 3.96–3.90 (dd, 1H, Hb, J = 7.8, 11.2 Hz), 3.83 (s, 3H, –OCH3), 3.21–3.16 (dd, 1H, Ha, J = 8.4, 11.2 Hz). 13C NMR (CDCl3, 100 MHz, δ, TMS = 0): 153.2, 151.7, 135.6, 132.6, 131.4, 129.2, 128.6, 128.0, 126.3, 125.4, 124.6, 123.5, 118.4, 111.6, 56.2, 43.5, 43.3. Anal. Calcd. MS: 381.27; found m/z: 381 (M+), 383 (M+ + 2). IR (KBr, cm−1): 3442.96 (NH), 3070.63 (C–H, aromatic), 1566.44 (C=N), 1376.98 (C–N).
4-bromo-2-(5-(2-methoxynaphthalen-1-yl)-4,5-dihydro-1H-pyrazol-3-yl)phenol (4d)
Compound 4d obtained as pale yellow solid (yield 75%), mp 132–134 °C. 1H NMR (CDCl3, 400 MHz, δ, TMS = 0): 11.28 (s, 1H, –OH), 8.48 (s, 1H, Ar–H), 8.39–8.37 (d, 1H, Ar–H, J = 8.4 Hz), 8.00–7.98 (d, 1H, Ar–H, J = 8.0 Hz), 7.83–7.78 (t, 1H-Ar, J = 10.2 Hz), 7.44–7.416 (m, 3H, Ar–H & –NH-pyrazoline), 6.993–6.973 (d, 1H-Ar, J = 8.0 Hz), 6.93–6.89 (t, 1H-Ar, J = 9.2 Hz), 6.80 (s, 1H, –NH), 4.11–4.07 [t, 1H, H-x (pyrazoline), J = 8.4 Hz], 3.95 (s, 3H, –OCH3), 3.75–3.68 (dd, 1H, Hb, J = 11.2, 8.4 Hz), 3.22–3.16 (dd, 1H, Ha, J = 10.8, 11.2 Hz). 13C NMR (CDCl3, 100 MHz, δ, TMS = 0): 162.5, 153.2, 151.6, 135.3, 134.5, 133.6, 129.5, 128.3, 128.0, 126.3, 124.3, 123.5, 121.0, 119.3, 118.5, 115.8, 111.6, 55.8, 44.2, 43.8. Anal. Calcd. MS: 397.27; found m/z: 397 (M+), 399 (M+ + 2). IR (KBr, cm−1): 3286.98 (NH), 2989.56 (C–H, aliphatic), 1554.56 (C=N).
3-(4-bromophenyl)-5-(4-methoxynaphthalen-1-yl)-4,5-dihydro-1H-pyrazole (4e)
Compound 4e obtained as white solid (yield 68%), mp 128–130 °C. 1H NMR (CDCl3, 400 MHz, δ, TMS = 0): 8.11–8.39(m, 10H, Ar–H), 6.19 (s, 1H, –NH-pyrazoline), 5.74–5.69 [t, 1H, H-x (pyrazoline), J = 10.4 Hz], 4.14 (s, 3H, –OCH3), 3.81–3.74 (dd, 1H, Ha, J = 10.4, 16 Hz), 3.23–3.17 (dd, 1H, Hb, J = 8.4, 16 Hz). 13C NMR (CDCl3, 100 MHz, δ, TMS = 0): 156.2, 152.1, 135.6, 133.8, 132.3, 131.7, 129.6, 128.5, 126.2, 125.4, 124.8, 124.0, 123.8, 104.9, 56.3, 49.4, 43.2. Anal. Calcd. MS: 381.27; found m/z: 381 (M+), 383 (M+ + 2). IR (KBr, cm−1): 3328.29 (NH), 2956.65 (C–H, aliphatic), 1562.68 (C=N).
5-(4-methoxynaphthalen-1-yl)-3-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazole (4f)
Compound 4f obtained as cream solid (yield 65%), mp 112–114 °C. 1H NMR (CDCl3, 400 MHz, δ, TMS = 0): 8.47–8.00 (m, 3H, Ar–H), 7.87–7.84 (d, 1H, Ar–H, J = 9.6 Hz), 7.98–7.89 (m, 4H, Ar–H), 7.69–7.67 (d, 1H, Ar–H, J = 7.6 Hz), 7.19–7.17 (d, 1H, Ar–H, J = 8.0 Hz), 6.91 (s, 1H, –NH-pyrazoline), 4.56–4.14 [t, 1H, H-x, pyrazoline, J = 8.4 Hz] 3.96 (s, 3H, –OCH3), 3.93 (s, 3H, –OCH3), 3.49–3.42 (dd, 1H, Hb, J = 8.4, 12.8 Hz), 3.16–3.08 (dd, 1H, Ha, J = 10.4, 12.8 Hz). 13C NMR (CDCl3, 100 MHz, δ, TMS = 0): 162.9, 155.2, 151.7, 133.5, 132.3, 129.6, 128.8, 128.7, 126.1, 124.8, 124.7, 124.0, 123.8, 114.5, 104.9, 56.2, 55.8, 49.4, 43.2. Anal. Calcd. MS: 333.40; found m/z: 334.00 (M+ + 1). IR (KBr, cm−1): 3249.65 (NH), 2987.29 (C–H, aliphatic), 1569.56 (C=N).
3-(4-bromophenyl)-5-(6-methoxynaphthalen-2-yl)-4,5-dihydro-1H-pyrazole (4g)
Compound 4g obtained as pale yellow solid (yield 74%), mp 142–144 °C. 1H NMR (CDCl3, 400 MHz, δ, TMS = 0): 7.76–7.71 (t, 4H, Ar–H, J = 9.2, 9.6 Hz), 7.58–7.40 (m, 5H, Ar–H), 7.18–7.15 (m, Ar–H, –NH-pyrazoline), 5.12–5.07 (q, 1H, H-x J = 10.8, 8.8 Hz), 3.97 (s, 1H, –OCH3), 3.55–3.49 (dd, 1H, Hb, J = 10.8, 16 Hz), 3.15–3.09 (dd, 1H, Ha, J = 8.8, 16 Hz). 13C NMR (CDCl3, 100 MHz, δ, TMS = 0): 157.7, 151.4, 134.1, 132.0, 131.9, 129.7, 129.3, 128.8, 128.7, 126.0, 124.7, 122.8, 119.1, 105.7, 55.3, 52.3, 41.1. Anal. Calcd. MS: 381.24; found m/z: 381 (M+), 383 (M+ + 2). IR (KBr, cm−1): 3442.96 (NH), 3070.63 (C–H, aromatic), 2924.13 (C–H, aliphatic), 1612 (C=N).
4-bromo-2-(5-(6-methoxynaphthalen-2-yl)-4,5-dihydro-1H-pyrazol-3-yl)phenol (II-4h)
Compound 4h obtained as light brown solid (yield 74%), mp 152–154 °C. 1H NMR (CDCl3, 400 MHz, δ, TMS = 0): 11.28 (s, 1H, –OH), 7.76–7.71 (m, 2H, Ar–H), 7.58–7.47 (m, 4H, Ar–H), 7.19–7.14 (m, 3H, Ar–H), 6.86 (s, 1H, –NH-pyrazoline), 5.12–5.07 [t, 1H, H-x (pyrazoline), J = 8.4 Hz], 3.98 (s, 1H, –OCH3), 3.55–3.49 (dd, 1H, Ha, J = 8.4, 12 Hz), 3.15–3.09 (dd, 1H, Hb, J = 9.6, 12 Hz). 13C NMR (CDCl3, 100 MHz, δ, TMS = 0): 161.56, 156.28, 151.75, 135.62, 134.28, 132.80, 132.68, 129.56, 129.35, 128.56, 126.65, 124.20, 121.05, 119.56, 115.85, 105.25, 55.65, 51.28, 42.95. Anal. Calcd. MS: 397.27; found m/z: 397 (M+), 399 (M+ + 2). IR (KBr, cm−1): 3269.56 (NH), 2896.24 (C–H, aromatic), 1572.64 (C=N) (Table 1).
In vitro antibacterial activity
The MIC for antibacterial activity of synthesized compounds was determined by the serial dilution technique using Mueller–Hinton nutrient broth, and ciprofloxacin was used as standard. The stock solution of test compounds (4 mg/ml) was prepared in dimethylformamide (DMF) and sterilized by membrane filtration method using 0.22-µm pore size polycarbonate sterile membrane (Nuclepore) filters. Various concentrations of test compounds were prepared in the range of 1–1000 μg/ml by serial dilution. The total four bacterial strains used for screening antibacterial activity were Staphylococcus aureus (MTCC 96), Bacillus subtilis (MTCC 441), E. coli (MTCC 443), Klebsiella pneumonia (MTCC 109). A standard protocol was followed to evaluate antibacterial activity (Malothu et al. 2015). Test compounds at various concentrations were added to the sterile culture medium in a sterilized borosilicate test tube and bacterial strain inoculated at 106 CFU/ml concentration. The tubes were incubated at 37 °C for 24 h and then examined for the presence or absence of visible growth of the test organisms. The MIC values were determined by considering the lowest concentration of the test compound, where the tubes remained clear, indicating that the bacterial growth was completely inhibited at this concentration. Simultaneously, controls were maintained by employing DMF to observe the solvent effects. SAR is presented in “Results and discussion.”
Antimycobacterial screening assay
Antimycobacterial activity of the synthesized compounds was performed with Mycobacterium tuberculosis strain (American Type Culture Collection (ATCC) 27924) using growth inhibition assay by turbidimetry (Rachakonda et al. 2013). Isolated single colonies of M. tuberculosis from 7H10 agar plate were grown overnight in Middlebrook 7H9 medium (0.47% Middlebrook 7H9 broth base, 10% ADS, 0.2% glycerol, and 0.1% Tween-80) to mid-exponential phase at 37 °C. Subsequently, 5 ml of Middlebrook 7H9 broth were inoculated with the overnight grown culture and allowed to grow at 37 °C to early log phase (OD600 ͌ 03). For the antimycobacterial assay, 98 µl of 1:1000-folds dilution of secondary culture was dispensed into a 96-well microtiter plate. To each well, 2 µl of the test compound in DMSO was added to attain a final concentration in the range of 1–100 µM and allowed to grow at 37 °C for 7 days. 240 µl of sterile water was added to each well of the peripheral rows of 96-well plates to minimize media evaporation during incubation. Bacterial growth was assessed after 7 days of incubation by measuring turbidity at 600 nm OD600 values using TECAN Infinite 200 PROTM (Tecan Instruments, Switzerland). Positive controls were included in every assay plate using stock solutions of INH (10 mg/ml, HiMedia) and rmp (10 mg/ml, HiMedia) to achieve the final concentration of 0.5, 1, 2, 4, 8, and 16 mg/ml for INH and 0.25, 0.5, 1, 2, 4, and 8 mg/ml for rmp. Additional controls DMSO (solvent without compound) and medium without inoculums were included in all the assay plates avoiding intra assay variability. The results were analyzed as the percentage of growth inhibition. All experiments were carried out in triplicates, and results were reported as minimum inhibitory concentration (MIC) in µM.
In vitro cytotoxic studies
Cell lines used in this study were purchased from the ATCC. The synthesized compounds were evaluated for their in vitro antiproliferative activity in two different human cancer cell lines by Resazurin assay (Rachakonda et al. 2013). MDA-MB-231 (breast cancer cell lines) and SKOV3 (ovarian cancer cell lines) were grown in Dulbecco’s modified Eagle medium (containing 10% FBS in a humidified atmosphere of 5% CO2 at 37 °C) containing nonessential amino acids. The cell lines were grown in their respective media and seeded into 96-well microtiter plates in 100 µl aliquots at plating densities depending on the doubling time of individual cell lines. The microtiter plates were incubated at 37 °C, 5% CO2, 95% air, and 100% relative humidity for 24 h before the addition of test compounds and standard. Four concentrations of test compounds (0.1, 1, 10, and 100 µM) were evaluated as per the NCI cell line screening protocol, and each was done in triplicate well. The assay was terminated by the addition of 50 µl of Resazurin solution and incubated for 60 min at 37 °C. The Fluorescence Intensity was read on a multimode plate reader (TECAN Infinite 200 PRO™) at a wavelength of 560 nm excitation/590 nm emission. The potency of compounds is expressed in IC50 and presented in Table 4.
In silico prediction of physicochemical and ADMET parameters
Physicochemical parameters of the designed compounds were in silico predicted using the Qik-prop module of Schrödinger. Various parameters predicted were; molecular weight (M.Wt.), total solvent-accessible volume, number of hydrogen bond donor (HBD), number of hydrogen bond acceptor (HBA), Vander wall polar surface area of nitrogen and oxygen atoms, octanol/water partition coefficient (log P), aqueous solubility (Log S), predicted apparent Caco-2 cell permeability in nm/s (PCaco), apparent Madin Darby Canine Kidney permeability and percentage of human oral absorption. The detailed predicted values were presented in Table 5.
Molecular docking studies
Molecular docking studies were carried out using Schrödinger software (Version 2019-1, Schrodinger) installed on Intel Xenon W 3565 processor and Ubuntu enterprise version 14.04 as an operating system. Targeted ligands were drawn using ChemDraw 18.0. The results of the docking study were analyzed with the help of XP Visualiser (Version 2019-1, Schrödinger).
Ligand preparation
The ligands used as input for docking study were sketched using ChemDraw software and cleaned up the structures for bond alignment. Then the ligands were incorporated into the workstation, and the energy was minimized using OPLS3e force field in Ligprep (Version 2019-1, Schrödinger). This minimization helps to assign bond orders, the addition of hydrogens to the ligands, and conversion of 2D to 3D structure for further docking studies. The generated output file (best conformations of the ligands) was further used for docking studies.
Protein preparation
Protein was prepared using the protein preparation wizard (Version 2019-1, Schrödinger). Hydrogen atom was added to the proteins, and charges were assigned. Generated Het states using Epik at pH 7.0 ± 2.0. Pre-processed the protein and refined, modified the protein by analyzing the workspace; water molecules and other heteroatoms were examined, nonsignificant atoms were excluded from the crystal structure of the protein. Finally, the protein was minimized by using the OPLS3e force field. A grid was created by considering the co-crystallized ligand present in the active site of the selected target of enoyl-acyl carrier protein reductase (ENR) from M. tuberculosis (PDB-4TZK).
Receptor grid generation
A receptor grid was generated around the protein by picking the inhibitory ligand (X-ray pose of the ligand in the protein). The centroid of the ligand was selected to create a grid box around it, and the Vander Waals radius of receptor atoms was scaled to 1.00 Å with a partial atomic charge of 0.25.
Docking studies
Docking studies of the significantly active and weakly active molecules were performed by using the Glide module in Schrödinger. All docking calculations were performed using Extra Precision (XP) mode. A scaling factor of 0.8 and a partial atomic charge of less than 0.15 was applied to the atoms of the protein. Glide docking score was used to determine the best-docked confirmation from the output. The interactions of these docked conformations were investigated further using XP visualizer.
Molecular docking of the titled compounds
The ENR from M. tuberculosis is one of the critical enzymes involved in the mycobacterial fatty acid elongation cycle and has been validated as an effective antimycobacterial target. The crystal structure of ENR from M. tuberculosis (strain ATCC 25618/H37Rv) (PDB-4TZK) (https://www.rcsb.org/structure/4TZK) was retrieved from Protein data bank with a resolution of 1.62 Å. The selected target contains a single-A chain with 269 sequence length residues with one co-crystal ligand (1-cyclohexyl-n-(3,5)-5-oxopyrrolidine-3-carboxamide) and one co-factor NAD. A molecular docking study was performed to understand the putative binding pattern of ligand at the active site of the target protein. The binding interactions of the significantly active and weakly active compounds with the target protein were analyzed. Before the analysis, the protocol was validated by checking the RMSD between docked pose and the X-ray co-crystallized pose.
Validation of the docking protocol
The precision of the docking study was determined by finding the lowest energy conformation of the co-crystallized ligand and resembles an experimental binding mode as determined by X-ray crystallography. The docking procedure was authenticated by removing the co-crystallized ligand from the binding site of the protein and re-docking the same co-crystallized ligand in the active binding site. The hydrogen-bonding interaction and RMSD between the anticipated confirmation and the observed X-ray crystallographic conformation were used for validation. The RMSD for the targeted protein was found to be 0.5 Å (Fig. 4) and indicated that the docking protocol could be reliable for further study to carry out the docking of titled compounds.

Overlay view of re-docked pose of co-crystal ligand (pink) with its X-ray pose (white) inside the enoyl-acyl carrier protein reductase (4TZK) (RMSD-0.5 Å)
Results and discussion
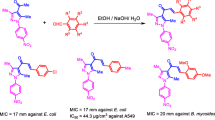
Target compounds (3a–3p and 4a–4h) were synthesized, as mentioned in Scheme 1.

Synthesis of naphthyl chalcones and pyrazolines (3a–p & 4a–h; Nature of substituents, R, R1 used is given in Table 1)
Chalcones were prepared by base-catalyzed condensation of a mixture of the substituted acetophenones and substituted aldehydes (i.e., substituted naphthaldehydes) in alcohol. To a solution of substituted acetophenone (2) (0.1 mol) in ethanol (10 ml), substituted naphthaldehyde (1) (0.1 mol) was added. To this, aqueous solution potassium hydroxide (60%, 5 ml) was added gradually with constant stirring and continued stirring at room temperature for 16–24 h. The separated solid was filtered and washed with ice-cold water (2 × 50 ml) until the swashing was neutral to litmus. Crude chalcones (3a–3p) were recrystallized from ethanol and dried at room temperature. To the solution of appropriate chalcone (3), hydrazine hydrate (0.02 mol) in ethanol was added and refluxed for 8–10 h. The progress of the reaction was monitored using TLC. After completion of the reaction, the mixture was poured into ice-cold water, and the crude product was separated and filtered. The crude products were purified by column chromatography/recrystallization to get the corresponding pyrazolines in pure form (4a–4h).
The structures of the synthesized compounds were confirmed by spectral analysis like FT-IR, 1H NMR, 13C NMR, ESI-MASS. IR spectra of chalcones (3a–3p) showed characteristic absorption bands between 1642–1662 cm−1 due to carbonyl (C=O) stretching. Absorption bands between 2856–2989 cm−1 indicate the presence of aliphatic C–H bonds. Aromatic C–H stretching was observed in the range of 3002–3089 cm−1. One of the olefinic protons of the chalcone appeared as a doublet in the range δ 8.07–8.86 ppm with a J value of 13–18 Hz, and another olefinic proton appeared as a doublet or multiplet in the range of δ 7.21–8.03 ppm. The coupling constant (J) of 12–18 Hz indicates the trans (E) configuration of the chalcone double bond. All the aromatic protons appeared in the range of δ 6.8–10.2 ppm. A singlet observed for one proton in the range of δ 3.72–3.96 ppm indicates the presence of the –OCH3 group in the compounds 3e–3p.Incompound 3a, the –OH proton appeared as a broad singlet at δ 12.88 ppm. 13C NMR spectra showed a signal due to carbonyl (C=O) carbon in the range between δ 189–192 ppm. The ESI-Mass (m/z) spectra of chalcones having halogens showed [M]+ and [M + 2]+ isotopic peaks. The chalcones with bromine (3a, 3e, 3g, 3i, 3j, 3m, and 3o) exhibited [M]+ and [M + 2]+ peaks in 1:1 ratio.
The IR spectra of corresponding pyrazolines (4a–4h) have shown absorption bands at 3286–3420 cm−1 due to the presence of –NH (pyrazoline). Absorption bands between 2924–2989 cm−1 indicate the presence of aliphatic C–H groups, aromatic C–H stretching was observed at 3042–3070 cm−1. Absorption bands observed between 1540–1589 cm−1 were due to –C=N stretching. 1H NMR spectra of pyrazolines showed doublet of doublet in the range of δ 4.12–5.24 ppm assignable to one of the pyrazoline ring proton (Hx). Two doublets of doublets in the range of δ 3.32–3.85 & 3.09–3.43 ppm indicate the presence of two nonequivalent protons of the pyrazoline ring (Hb and Ha). A singlet for one proton in the range of δ 6.8–7.1 ppm was due to the presence of pyrazoline –NH. All other aromatic protons appeared in the range of δ 7.1–10.2 ppm. 13C NMR spectra have shown signals in the range of δ 40–163 ppm. Pyrazoline carbons observed between δ 41–56 ppm. In compounds 4c–4h, signals were observed between δ 54–56 ppm due to the presence of the –OCH3 group. All other carbons of the aromatic ring were observed in the region between δ 102–163 ppm. The mass ESI-MS (m/z) spectra of compounds 4b, 4c, 4d, 4e, 4g have shown isotopic peaks at [M]+ & [M + 2]+ in the ratio of 1:1 due to the presence of bromine. Compound 4a, 4f and 4h have shown molecular ion peaks at [M + 1]+.
In vitro antibacterial activity
All the synthesized compounds were primarily screened for the antibacterial activity for two Gram +ve strains and two Gram −ve strains to ascertain the antibacterial susceptibility of the synthesized molecules. The results (Table 2) indicated that all compounds showed low antimicrobial activity against both the tested strains with MIC values in the range of 14–248 µg/ml when compared to the standard drug ciprofloxacin. Compound 3d & 4e exhibited significant activity against Gram +ve strains and Gram −ve organisms with MIC of 24, 15, and 16, 14 µg/ml, respectively. However, all the compounds showed less potency when compared to the standard, ciprofloxacin. Among all the synthesized chalcones and their pyrazoline derivatives, compound 3d with the 4-methoxy group on phenyl ring exhibited the highest potency with MIC of 24 and 15 µg/ml against S. aureus and B. subtilis. However, with the introduction of an additional –OCH3 groups at 2′ and 4′ positions on naphthalene ring as in compounds 3h and 3l resulted in a decrease in potency with MIC of 71, 74, and 155, 139 µg/ml against the two same organisms. However, the potency of compound 3d showed MIC of 74, 66 µg/ml against Gram −ve bacteria E. coli, and K. pneumoniae, but the additional methoxy group at 2nd position of naphthalene ring (3h) resulted in increase in potency by nearly two times exhibiting MIC of 32 and 36 µg/ml against the two Gram −ve organisms. Compound 3j, with the 4-methoxy group on naphthalene ring, 2-hydroxy substituent on phenyl ring (R = 4-OMe, R′ = 2-OH) showed potency equal to that of 3h (4-OMe and 2′-OMe) against Gram −ve organisms (E. coli and K. pneumoniae) with MIC of 32 and 35 µg/ml. But additional 5-bromo substituent as in 3i (R = OMe and R′ = 5-Br, 2-OH) resulted in a decrease in potency by nearly half with the MIC values of 77 and 65 µg/ml against the two Gram −ve bacteria. In pyrazoline derivatives (4a–h), compound 4f with 4 and 4′ dimethoxy substitution has exhibited the highest potency with 68 and 64 µg/ml against two Gram +ve bacteria (S. aureus and B. subtilis). Interestingly, the least potent compound 4e (R = 4-OMe & R′ = 4-Br) against Gram +ve bacteria (MIC = 218 & 248 µg/ml) exhibited highest potency against the Gram −ve bacteria (E. coli and K. pneumoniae) with MIC values of 16 and 14 µg/ml respectively. In general, chalcone showed better activity against Gram +ve bacteria than the pyrazoline showed greater potency against Gram −ve organisms. For instance, compound 3k (R = 4-OMe & R′ = 4-Br) showed MIC of 72 & 64 µg/ml, against the two Gram −ve bacteria and its pyrazoline analog, 4e exhibited grater potency with MIC of 16 and 14 µg/ml against the same organisms.
In vitro antimycobacterial activity
The title compounds were also screened for in vitro antimycobacterial activity against H37Rv strain, and results were presented in Table 3. The results indicated that the compounds 3a–p and 4a–h possessed significant antimycobacterial activity with MIC in the range of 6.25–100 µM. Among the series, compound 4b with 5-Bromo-2-hydroxy substitution on phenyl ring possessed significant activity with MIC of 6.25 µM, comparable to the standard inh with MIC of 5.86 µM.
In vitro cytotoxicity study
Cytotoxicity studies were performed against MDA-MB-231 & SKOV3 cell lines. All the compounds (3a–4h) exhibited low cytotoxicity with the IC50 values in the range of 8.52–164.56 µM, and values were presented in Table 4. The most active compound against Mtb H37R 4b was found to be nontoxic to MDA-MB-231 (breast cancer cells) and less toxic to SKOV3 (ovarian cancer cells) with IC50 of 37.5 µM.
In silico prediction of physicochemical and drug-likeness parameters
Physicochemical properties of a compound play a vital role in drug absorption, distribution metabolism, and excretion. All the synthesized molecules were further evaluated for their drug-likeness behavior through ADME (absorption, distribution, metabolism, and excretion) properties. Their molecular weights were <725 Daltons with <6 HBDs and <20 HBAs (Table 5) fulfilling the criteria of Lipinski’s rule of five and was found to be in the satisfactory range for evaluation of the drug-likeness behavior of the molecules. All other parameters, like octanol/water partition coefficient and solubility are in between the acceptable range only.
Apparent Caco-2 permeability of all the compounds except compound 3p was found to be in the acceptable range. However, compound 3p showed moderate permeability with P-CACO-2 permeability of 440.98 (nm/s). The rotatable bonds for the compounds were in the reasonable range only. Overall, the predicted physicochemical values indicated that the maximum number of titled analogs possessed the values that are well-matched with the optimum range values, and overall the compounds possessed the drug-likeness behavior.
Molecular docking studies
In order to find out the putative binding mode of significantly active and weakly active compounds, a molecular docking study was performed, and the results were given in Table 6. Before the docking studies, validation of the protein was done by checking the RMSD and found to be 0.5 Å indicating that the docking protocol was justified.
The co-crystallized ligand showed a significant docking score (−11.20) and binding energy (−67.00) (Table 6). By keen inspection of 2D and 3D representation of the co-crystallized ligand (Figs. 5, 6), a hydrogen bond found with the amino-acid residue TYR-158 (distance 2.78 Å) of protein. Apart from hydrogen bond interaction, the co-crystallized ligand also exhibited one aromatic bond interaction with the amino-acid residue PRO-156 (distance 2.56 Å) and one water hydrogen-bonding interaction. All these interactions made the co-crystallized ligand more stable in the active site of the target protein with a significant docking score. Significantly active molecule 4b displayed a docking score of −10.50 and energy of −44.50. The molecule showed a significant docking score because pyrazole hydrogen offered a hydrogen bond interaction with the surrounded water molecule (Figs. 7, 8). This hydrogen makes the compound more significant, and in the in vitro analysis, also the compound exhibited significant MIC with 6.25 µM when compared to 5.86 µM shown by the standard inh.

3D representation of the co-crystal ligand in the active site of the target 4TZK

2D representation of the co-crystal ligand in the active site of the target 4TXK

3D representation of the significantly active compound 4b in the active site of the target 4TZK

2D representation of the significantly active compound 4b in the active site of the target 4TZK
Weakly active compound 3h did not show any crucial hydrogen bond interaction with the surrounded amino-acid residues. However, it revealed an aromatic bond interaction with the amino-acid residue PRO-156 and displayed the docking score of −6.7 and docking energy of −42.50 (Figs. 9, 10). Due to a lack of hydrogen bond interaction with the target protein, the compound 3h might have shown less in vitro activity against M. tuberculosis with MIC of 100 µM.

3D representation of the weakly active compound 3 h in the active site of the target 4TZK

2D representation of the weakly active compound 3 h in the active site of the target 4TZK
Conclusion
A series of naphthyl chalcones and their pyrazoline derivatives were synthesized by standard protocol. All the synthesized compounds have shown significant antibacterial activity. Among the series, compound 4b found to exhibit potent activity against M. tuberculosis with the MIC of 6.28 µM, and in docking studies, it revealed good hydrogen bond interaction with surrounded amino-acid residues. All the compounds in the series have shown minimal to moderate cytotoxicity; however, compound 4b was found to be nontoxic to MDA-MB-231 cell lines. In summary, our results on the biological screening and in silico studies of the synthesized compounds offered an ethical framework that may lead to the discovery of new potent antitubercular agents.
References
Ahmad A, Husain A (2016) Synthesis, antimicrobial, and antitubercular activities of some novel pyrazoline derivatives. J Saudi Chem Soc 20:577–584. https://doi.org/10.1016/j.jscs.2014.12.004
Ahmad I, Prakash J, Chanda D et al. (2013) Syntheses of lipophilic chalcones and their conformationally restricted analogs as antitubercular agents. Bioorg Med Chem Lett 23:1322–1325. https://doi.org/10.1016/j.bmcl.2012.12.096
Burmaoglu S, Algul O, Gobek et al. (2017) Design of potent fluoro-substituted chalcones as antimicrobial agents. J Enzym Inhib Med Chem 32:490–495. https://doi.org/10.1080/14756366.2016.1265517
Casey JM, GJ. Carlson, JW. Ford, TE. Strecker, E Hamel, Mary Lynn Trawick KGP (2019) Synthesis and biological evaluation of structurally diverse α-conformationally restricted chalcones and related analogues. MedChemComm 1–26. https://doi.org/10.1039/C9MD00127A.
Castaño LF, Cuartas V, Bernal A et al. (2019) New chalcone-sulfonamide hybrids exhibiting anticancer and antituberculosis activity. Eur J Med Chem 176:50–60. https://doi.org/10.1016/j.ejmech.2019.05.013
Chiaradia LD, Graziela P, Martins A et al. (2012) Synthesis, biological evaluation, and molecular modeling of chalcone derivatives as potent inhibitors of Mycobacterium tuberculosis protein tyrosine phosphatases (PtpA and PtpB). J Med Chem 55:390–402
Farah SI, Abdelrahman AA, North EJ, Chauhan H (2015) Opportunities and challenges for natural products as novel antituberculosis agents. Mary Ann Liebert, Inc. XX:14–16. https://doi.org/10.1089/adt.2015.673.
Figueroa-valverde L, Hau-heredia L, García-cervera E et al. (2017) Activity exerted by a naphthalene-oxirane derivative on the ischemia/reperfusion injury. Biomed Res 28:16–22
Gomes MN, Braga RC, Grzelak EM et al. (2017) QSAR-driven design, synthesis and discovery of potent chalcone derivatives with antitubercular activity. Eur J Med Chem 137:126–138. https://doi.org/10.1016/j.ejmech.2017.05.026
Hans RH, Guantai EM, Lategan C et al. (2010) Synthesis, antimalarial and antitubercular activity of acetylenic chalcones. Bioorg Med Chem Lett 20:942–944. https://doi.org/10.1016/j.bmcl.2009.12.062
He X, Alian A, Stroud R, Montellano PRO De (2006) Pyrrolidine carboxamides as a novel class of inhibitors of enoyl acyl carrier protein reductase from Mycobacterium tuberculosis. 6308–6323. https://doi.org/10.1021/jm060715y.
Khan SA, Asiri AM (2017) Green synthesis, characterization and biological evaluation of novel chalcones as anti bacterial agents. Arab J Chem 10:S2890–S2895. https://doi.org/10.1016/j.arabjc.2013.11.018
Kumar D, Ahmad I, Shukla A, Khan F (2014) QSAR and docking studies on chalcone derivatives for antitubercular activity against M. tuberculosis H 37 Rv. J Chemom 1–24. https://doi.org/10.1002/cem.2606.
Kumar RK, Sharma RSK, Kumar DR et al. (2016) ISSN 0975-413X CODEN (USA): PCHHAX characterization, synthesis and biological evaluation of naphthalene based piperazines as anti bacterial agents. Der Pharma Chem 8:374–379
Lin Y, Zhou Y, Flavin MT et al. (2002) Chalcones and flavonoids as anti-tuberculosis agents. Bioorg Med Chem 10:2795–2802
Lopes T, Ventura B, Calixto SD et al. (2015) Antimycobacterial and anti-inflammatory activities of substituted chalcones focusing on an anti-tuberculosis dual treatment approach. Molecules 20:8072–8093. https://doi.org/10.3390/molecules20058072
Malothu N, Bhandaru JS, Kulandaivelu U et al. (2015) Synthesis, in vitro antimycobacterial evaluation and docking studies of some new 5,6,7,8-tetrahydropyrido[4′,3′:4,5]thieno[2,3-d]pyrimidin-4(3H)-one Schiff Bases. Bioorg Med Chem Lett 4. https://doi.org/10.1016/j.bmcl.2015.12.083.
Marrapu VK, Chaturvedi V, Singh S et al. (2011) Novel aryloxy azolyl chalcones with potent activity against Mycobacterium tuberculosis H37Rv. Eur J Med Chem 46:4302–4310. https://doi.org/10.1016/j.ejmech.2011.06.037
Nayyar A, Malde A, Jain R (2006) Synthesis, anti-tuberculosis activity, and 3D-QSAR study of ring-substituted-2/4-quinolinecarbaldehyde derivatives. Bioorg Med Chem 14:7302–7310. https://doi.org/10.1016/j.bmc.2006.06.049
Patole J, Shingnapurkar D, Ratledge C (2006) Schiff base conjugates of p-aminosalicylic acid as antimycobacterial agents. Bioorg Med Chem Lett 16:1514–1517. https://doi.org/10.1016/j.bmcl.2005.12.035
Rachakonda V, Alla M, Sravanti S, Ummani R (2013) Design, diversity-oriented synthesis and structure activity relationship studies of quinolinyl heterocycles as antimycobacterial agents. Eur J Med Chem 70:536–547. https://doi.org/10.1016/j.ejmech.2013.10.034
Rokade S, Agrawal S, Shastri J (2010) Antimicrobial susceptibility testing of rapidly growing mycobacteria by microdilution—experience of a tertiary care centre. Indian J Med Microbiol 28:48–51. https://doi.org/10.4103/0255-0857.58729
Sharma M, Chaturvedi V, Manju YK et al. (2009) Substituted quinolinyl chalcones and quinolinyl pyrimidines as a new class of anti-infective agents. Eur J Med Chem 44:2081–2091. https://doi.org/10.1016/j.ejmech.2008.10.011
Sivakumar PM, Seenivasan SP, Doble M (2007) Synthesis, antimycobacterial activity evaluation, and QSAR studies of chalcone derivatives. Bioorg Med Chem Lett 17:1695–1700. https://doi.org/10.1016/j.bmcl.2006.12.112
Zhou B, He Y, Zhang X et al. (2010) Targeting mycobacterium protein tyrosine phosphatase B for antituberculosis agents. PNAS 107:4573–4578. https://doi.org/10.1073/pnas.0909133107
Acknowledgements
One of the authors thankful to the Central Mass Division, IICT, Hyderabad for providing mass spectrometry, Molecular Biology Department for providing the antimycobacterial activity. BKK and SM thankful to the Department of Biotechnology, Indo-Spain, New Delhi (Ref. No: BT/IN/Spain/39/SM/2017-2018).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Pola, S., Banoth, K.K., Sankaranarayanan, M. et al. Design, synthesis, in silico studies, and evaluation of novel chalcones and their pyrazoline derivatives for antibacterial and antitubercular activities. Med Chem Res 29, 1819–1835 (2020). https://doi.org/10.1007/s00044-020-02602-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-020-02602-8