
Abstract
p-Sulphonic-N-propionyl aniline 3 was produced from sulphanilic acid with propionic acid under reflux for 2 h, then the compound 3 reacted with thionyl chloride and the corresponding imidoyl chloride 4 was obtained. When imidoyl chloride 4 reacted with potassium isothiocyanate and aniline, the corresponding thiourea derivatives 6 was produced. Compound 6 was reacted with tricyanovinylamine to produce the thiazole derivative 7 after cyclization. The microwave-assisted condensation of 2,3,4.6,-tetra-O-acetyl-β-D-glucopyranosylisothiocynate 8 with thiazole derivative 7 afforded the thiourea derivatives 9. Compound 10 a, b has been synthesized by reacting thiourea derivatives 9 with chloroacetone. The prepared compounds were screened for antibacterial properties against Staphylococcus aureus and Escherichia coli, and antifungal activity against Candida sp. The structure assignments of the new synthesized compounds are based on spectroscopic data.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Many spectra of biological activities appeared from different natural products and these were subjected to medicinal applications, and it is seen that carbohydrates are a very important constituent of these products. N- and S-linked sugar derivatives of these compounds showed antileukemic [1] antimalarial [2], antifungal [3], antiviral [4], and anticancer activities [5]. In medicine, carbonyl glycosides were used as an anticancer agent using the boron neutron capture therapy [3]. Chemotherapy is considered an important choice between the strategies utilized for cancer treatment [3, 6]. The important objective of most chemotherapeutic agents is the induction of apoptosis of cancer cells [7].
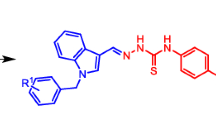
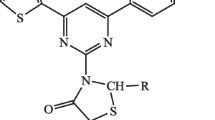
Thiazoles have a prominent position between heterocycles. An enormous number of thiazoles taken from microbial and marine origins show important biological activity such as antifungal, antiviral, antibiotic and antitumor activities [8, 9]. On the other hand, sugar isothiocyanates are among the most versatile synthetic intermediates found in carbohydrate chemistry [10]. They play a fundamental role in the synthesis of many series of functional groups such as isonitrile, carbodiimide, amide, and N-thiocarbonyl derivatives. So, isothiocyanates have an important role to play in heterocyclic compounds and the process of glucosylate is important for the synthesis of N-glucosylated thiadiazole through the C–N bond, aided by compounds such as 1a–d and 2 [11,12,13] as in Fig. 1. In this research, peracetylated glycopyranosyl thiourea derivatives were synthesized and converted to an iminothiazole ring.

Examples of glucosylimino derivatives
Results and discussion
Treatment of sulphanilic acid with propionic acid using zinc dust under reflux for 2 h resulted in p-sulphonic-N-propionyl aniline 3. The structure of 3 was deduced from its analytical and spectral data, where IR spectrum showed absorption bands ranging from 3324 to 3265 cm−1 assignable for (NH), 1648 cm−1 for (C=O) and 3502 cm−1 for (OH). The 1HNMR spectrum showed singlet signal at 11.13 for NH (D2O exchangeable) and singlet signal at δ 9.27 for OH. The reaction of 3 with thionyl chloride in benzene with stirring at room temperature produced imidoyl chloride 4 (Scheme 1). The products were elucidated using 1HNMR and MS spectra and elemental analysis. Treatment of 4 with potassium isothiocyanate yielded the intermediate imidoylisothiocyanates 5 (Scheme 1).

Synthesized of compound 7
A reaction of 5 with aniline in the presence of acetone as a solvent at room temperature with stirring for 3 h resulted in the thiourea derivative 6 in 72% yield. IR spectrum showed strong absorption bands at 3184−3106, 1643 and 1375 cm−1 attributed to NH, C=N and C=S, respectively. The 1H NMR spectrum showed a band for OH and NH (exchange with D2O) at 9.53, 8.53 and 7.23 ppm, respectively. Compound 6 was reacted with tricyanovinylamine (prepared from tetracyanoethylene with ammonium acetate) [14], which produced thiazole derivatives 7. The IR spectrum showed the absence of NH group and the presence of CN and NH2 at 2210 and 3225 cm−1 The 1H NMR showed a signal for NH2 at 8.53 ppm (Scheme 1). The suggested reaction mechanism for compound 7 is the nucleophilic attack of thione of the sugar at olefin followed by elimination of CN, formation of NH2 and formation of thiazole derivative as shown in Fig. 2.

The suggested reaction mechanism for compound 7
N-(2,3,4.6,-tetra-O-acetyl-β-D-glucopyranosyl)-N′-(-4-amino-5-cyano-2-(phenylimino)thiazol-3(2H)-yl)propylideneamino)benzenesulfonic acid) thiourea 9 was synthesized by nucleophilic addition of (4E)-4-(1-((2E)-4-amino-5-cyano-2-(phenylimino)thiazol-3(2H)-yl)propylideneamino)benzenesulfonic acid 7 on 2,3,4.6,-tetra-O-acetyl-β-D-glucopyranosylisothiocynate 8( scheme 2). The reaction proceeded in the microwave oven for 25–30 min using dioxane as a solvent. IR spectrum of compound 9 showed stretching band of C=S bond at 1374 cm−1, another band at 3489–3165 cm−1 due to N–H bands and C=O bonds of glucopyranosyl at 1745 cm−1 Fig. 4. The mechanism for the formation of derivative 9 is proceeded by the nucleophilic attack of the lone pair of electrons on NH2 group at the carbon of the isothiocynate group followed by H transfer to form the thiourea derivative as shown in Fig. 3.

Synthesized of compound 9

The suggested reaction mechanism for compound 9

Tatutomerism of compound 9
The reaction of compound 9 with chloroacetone in the presence of chloroform gave two products as an isomer, namely 10a and 10b( scheme 3). The product yield was 46–69%. Compounds 10a and 10b have structural similarity Therefore, it is impossible to separate these isomers (by recrystallization or column chromatography). IR spectra showed the absence of NH. 1H NMR spectrum confirmed the presence of two isomers 10a and 10b. The isomer 10a showed chemical shifts of proton β-anomeric H-′1 at δ = 6.59 ppm and another proton β- anomeric H-′1 of 10b appeared at δ = 5.64 ppm. Another evidence is the absence of NH of thiourea at 9–10 ppm.

Synthesized of compounds 10a and 10b
The mechanism for the formation of 10a,b is proceeded by the nucleophilic attack of thiourea derivative on CH2 anion of chloroacetone followed by cyclization and loss of water as shown in Fig. 5.

The suggested reaction mechanism for compound 10a,b
Antimicrobial activity
All the synthesized compounds were screened for their antifungal activity against Candida sp., using the agar well diffusion method [15, 16] and then ketoconazole was used as a standard for antifungal activity. The synthesized compounds were also screened for antibacterial activity with Escherichia coli representing Gram-negative bacteria, and Staphylococcus aureus representing Gram-positive bacteria using Ampicillin as a standard drug for antibacterial activity (Table 1). Antimicrobial activity had been tested by measuring the diameter of the inhibition zone in millimeters (Table 1). First, the medium was sterilized using autoclaving at 120 °C (15 lb/in2). 30 ml of the agar medium with the relative strains of bacteria and fungi were transferred into each sterile petri plate. The plates were standing at room temperature for solidification. The sterile cork is used to make a well of 6 diameter. The standard drug and extracts were placed in the 6 mm diameter well. Antibacterial activity plates were then incubated at 37 °C for 24 h for antibacterial activity assay and 28 °C for 48 h for antifungal activity assay. All the synthesized compounds were dissolved in DMSO which was used as a negative control. The tested compounds exhibited antifungal activity against Candida sp. The activity of the synthesized compounds is arranged in the following order 10a,b > 7 > 9. On the other hand, the compound 7 was the least active, while the compound 10a,b was the most active ones.
Conclusion
We have synthesized an efficient method for the reaction of (2,3,4,6-tetra-O-acetyl–D-glucopyranosyl)thioureas containing an iminothiazole ring with chloroacetone under refluxing conditions which produced two isomers. The presence of two iminothiazole isomers was determined by 1HNMR spectroscopy.
Experimental
General procedures
Melting points were determined on Electrothermal IA 9,100 series digital melting point apparatus in capillaries and are uncorrected. IR spectra were obtained in the solid state as potassium bromide discs using a Perkin-Elmer model 1430 spectrometer. 1HNMR spectra were recorded on a Varian/Gemini 400 MHz spectrometer in DMSO-d6 as a solvent and TMS as an internal standard (chemical shifts in δ, ppm). Mass spectra were measured on an instrument VG-7035 at 70 or 15 eV. Elemental analyses were performed at the Microanalytical Centre, Cairo University, and Giza, Egypt.
4-(Propionamido)benzenesulfonic acid (3)
A mixture of sulphanilic acid (1 mmol) and zinc dust (1immol) in propionic acid (10 ml) was heated under reflux for 2 h, and then the reaction mixture was poured into ice water (30 ml) with stirring until the precipitate formed and the solid was collected by filtration. The residue was washed with water, filtrated and dried are crystallized by water. m.p. 136–138 °C. Yield: (92%). FT-IR (υ cm− 1): 3324–3265 (NH), 1648 (C=O), 3502 (OH). 1HNMR (400 MHz, DMSO): δ 1.16 (t, 3H, CH3), 3.21 (q, 2H, CH2), 7.32–7.39 (dd, 2H, J = 7.9, C6H4), 7.64–7.36 (dd, 2H, J = 7.9, C6H4), 9.27 (s, 1H, OH), 11.13 (s, 1H, NH), anal. Calcd for (C9H11NO4S, 229.25): C, 47.15; H, 4.84; N, 6.11; S, 13.99. Found: C, 47.12; H, 4.81; N, 6.09; S, 13.97.
4-(1-Chloropropylideneamino)benzenesulfonic acid (4)
Thionyl chloride (2.5 mL) was added dropwise to a cold solution of 4-propionamido)benzenesulfonic acid 3 (3 mmol) in benzene (10 mL). The reaction mixture was stirred at room temperature for 2 h. The solvent was then evaporated and the solid was recrystallized from aqueous ethanol to afford the pure products 4. Yellow solid; mp 165–166 °C; yield 70%. FT-IR (υ cm−1): 1489 (C=N), 3562 (OH) 1HNMR (400 MHz, DMSO): δ 9.23 (s, 1H, OH), 7.52–7.63 (dd, 2H, J = 7.8 C6H4), 7.91–7.93 (dd, 2H, J = 7.8, C6H4), 1.18 (t, 3H, CH3), 3.35 (q, 2H, CH2). Anal. Calcd for (C9H10ClNO3S, 247.7): C, 43.64; H, 4.07; Cl, 14.31; N, 5.65; S, 12.95. Found: C, 43.61; H, 4.27; Cl, 14.33; N, 5.65; S, 12.92.
4-(AminoN-(1-(phenylimino)propyl)methanethioamino)benzenesulfonic acid (6)
4-(1-chloropropylideneamino) benzenesulfonic (1 mmol) 4 acid was dissolved in acetone (3 ml) at 0 °C and a solution of potassium isothiocyanate (1 mmol) in acetone (2 ml) was added drop wise. The mixture was stirred in the cooling bath at 0 °C. The precipitate potassium chloride was removed by filtration. The filtrate was stirred on an ice bath, and aniline (1 mmol) was added. The reaction mixture was allowed to react at room temperature with stirring, then the solvent was removed by evaporation, and the residue was triturated with diethyl ether, washed once more, and dried in vacuo to give the thiourea derivatives 6. mp 205–207 °C yield 72%; FT-IR (υ cm− 1): 3184 − 3015 (NH), 1643 (C=N), 1375 (C=S). 1HNMR (400 MHz, DMSO): δ 9.53 (s,1H, OH),7.23 (s,1H, NH), 8.52 (s, 1H, NH), 6.95–6.73 (dd, 2H, J = 8.5, C6H4), 7.23-7.54-7.63 (dd, 2H, J = 8.5, C6H4), 7.87(m, 5H, C6H5) 3.37 (q, 2H, CH2), 2.18 (t, 3H, CH3). Anal. Calcd for: (C16H17N3O3S2, 363.45): C, 52.87; H, 4.71; N, 11.56; S, 17.64. Found: C, 52.84; H, 4.73; N, 11.58; S, 17.67.
(4Z)-4-(1-((2E)-5-amino-4-cyano-2-(phenylimino)thiazol-3(2H)-yl)propylideneamino)benzenesulfonic acid (7)
Compound 6 (1 mmol) and tricyanovinylamine (1 mmol) dissolved in ethyl acetate was refluxed for 10 h and then the precipitate formed to which a mixture of ethyl acetate and diethyl ether was added to yield compound 7, recrystallization from toluene. mp 263–265 °C. FT-IR (υ cm− 1): 3235, 3124 (NH2), 2210 (CN) and 1647 (C=N). 1HNMR (400 MHz, D2O): δ 9.79 (s, 1H, OH), 8.53 (s,1H, NH2), 7.13–7.33 (dd, 2H, J = 8.3 Hz, C6H4), 7.49 (m, 5H, C6H5), 3.21 (q, 2H, CH2), 2.13 (t, 3H, CH3). 13C NMR (DMSO-d6): δ 7.38 (CH3), 19.0 (CH2), 57.3 (C=C–NH2), 114.8 (CN), 122.8 (C-2 C-6), 123.4 (C-2′, C-6′), 128.1 (C-4), 129.9 (C-3′,C-5′), 132.9 (C-3, C-5), 147.5 (C-4′), 149.5 (C-1), 153.4 (C-1′), 163.1 (C=N), 165.3 (C=N), 189.3(C=C–NH2). EIMS (m/z) 427 (M+. 100%), 255 (80%), 171 (73%), 91(53%). Anal. Calcd for: (C19H17N5O3S2): C, 53.38; H, 4.01; N, 16.38; S, 15.00. Found. C, 53.41; H, 4.04; N, 16.40; S, 15.12.
N-(2,3,4.6,-Tetra-O-acetyl-β-D-glucopyranosylimino)-N′-(-5-amino-4-cyano-2-(phenyl)thiazol-3(2H)-yl)propylideneamino)benzenesulfonic acid) thiourea (9)
A mixture of (4E)-4-(1-((2E)-4-amino-5-cyano-2-(phenylimino)thiazol-3(2H)-yl)propylideneamino)benzenesulfonic acid 7 (1 mmol) and 2,3,4.6,-tetra-O-acetyl-β-D-glucopyranosylisothiocyanate 8 (1 mmol) was heated for 25–30 min in a microwave at 750 watts in 1,4-dioxane. This mixture was cooled to room temperature, recrystallized from a mixture of ethanol and toluene by ratio (1:1 in volume) which yielded a pale yellow color, 45%, mp. 215–217 °C. FT-IR (υ cm− 1):3489–3165 (NH), 2106 (CN), 1745 (C=O), 1374 (C=S). 1HNMR (400 MHz, DMSO): δ 9.27 (s, 1H, OH), 10.53 (s,1H, NH), 8.68 (s, 1H, NH), 7.43–7.63 (dd, 2H, J = 8.2 Hz, C6H4), 7.49 (m, 5H, C6H5), 6.93–6.75 (dd, 2H, J = 8.2 Hz, C6H4), 6.37 (s, 1H, H′1), 2.29 (4 s, 12H, 4COCH3), 4.52–5.46 (m, 6H, glucosyl ring), 3.39 (q, 2H, CH2), 2.38 (t, 3H, CH3). 13C NMR (DMSO-d6) : δ 9.38 (CH3), 21.0–20.1 (4 × COCH3), 23.2 (CH2), 57.3 (C=C–NH2), 61.6 (C-6′), 66.3 (C-4′), 66.7(C-2′), 70.1 (C-3′), 72.4 (C-5′), 81.2 (C-1′), 115.9 (CN), 122.9 (C-2 C-6), 123.5 (C-2′, C-6′), 128.1 (C-4), 128.9 (C-3′,C-5′), 131.6 (C-3, C-5), 147.5 (C-4′), 149.5 (C-1), 153.4 (C-1′), 163.1 (C=N), 165.3 (C=N), 169.3–169.5 (4 × COCH3), 187.4 (C=S). EIMS (m/z) 816 (M+., 29%), 426 (100%), 331 (56%), 43 (19%). Anal. Calcd for:(C34H36N6O12S3): C, 49.99; H, 4.44; N, 10.29; S, 11.78. Found. C, 50.03; H, 4.47; N, 10.31; S, 11.80.
(4Z)-4-(1-((2E,4E)-5-(4-Methyl-3-(2,3,4.6,-tetra-O-acetyl-β-D-glucopyranosylimino)thiazol-2(3H)-ylideneamino)-4-cyano-2-(phenylimino)thiazol-3(2H)-yl)propylideneamino)benzenesulfonic acid (10a, 10b)
Compound 9 (3 mmol) was dissolved in dry chloroform (25 ml) and chloroacetone (3 mmol) was added drop by drop with stirring. The reaction mixture was refluxed for 17 h and the solvent was concentrated under vacuum and left overnight. The solid was separated and filtrated, and then purified by recrystallization from ethanol and toluene (1:1). mp. 221–226 °C. FT-IR (υ cm− 1): 2123 (CN), 1745 (C=O),1573,1565 (C=C), 1615(C=N). EIMS (m/z) 855 (M + 1, 33%). Anal. Calcd: (C37H38N6O12S3): C, 51.98; H, 4.48; N, 9.83; S, 11.25. Found. C, 52.00; H, 4.45; N, 9.85; S, 11.28.
(10a). 1HNMR (400 MHz, DMSO): δ 9.27 (s, 1H,OH), 7.51 (m, 5H, C6H5), 7.36–7.72 (dd, 2H, J = 7.8 Hz C6H4), 7.12–6.95 (dd, 2H, J = 7.6, C6H4), 6.59 (d, 1H, J 9.3 Hz, H′1),5.94 (t, 1H, J 9.3 Hz, H-2′), 2.32 (4 s, 12H, 4COCH3), 4.49–5.32 (m, 5H, glucosyl ring), 5.03 (s, 1H, C=CH), 3.25 (q, 2H, CH2), 2.91 (s, 3H, CH3), 2.07 (t, 3H, CH3).13C NMR (DMSO-d6) : δ 9.38 (CH3), 21.0–20.1 (4 × COCH3), 23.2 (CH2), 57.3 (C=C–NH2), 61.6 (C-6′), 66.3 (C-4′), 66.7(C-2′), 70.1 (C-3′), 72.4 (C-5′), 81.2 (C-1′), 115.9 (CN), 122.9 (C-2 C-6), 123.5 (C-2′, C-6′), 128.1 (C-4), 128.9 (C-3′,C-5′), 131.6 (C-3, C-5), 147.5 (C-4′), 149.5 (C-1), 153.4 (C-1′), 163.1 (C=N), 165.3 (C=N), 171.2–169.9 (4 × COCH3).
(10b). 1HNMR (400 MHz, DMSO): δ 9.63 (s, 1H,OH), 7.48–7.65 (dd, 2H, C6H4), 7.52 (m, 5H, C6H5), 7.16–6.97 (dd, 2H, J = 7.2, C6H4), 2.21 (st, 3H, CH3), 6.42 (t, 1H, J 9.15 Hz, H-2′),5.21 (s, 1H, C=CH), 5.64 (d, 1H, J 9.3 Hz, H′1), 2.39 (4 s, 12H, 4COCH3), 4.45–5.39 (m, 5H, glucosyl ring), 3.35 (q, 2H, CH2), 2.37 (t, 3H, CH3).13C NMR (DMSO-d6) : δ 9.38 (CH3), 21.0–20.1 (4 × COCH3), 23.2 (CH2), 57.3 (C=C–NH2), 61.6 (C-6′), 66.3 (C-4′), 66.7(C-2′), 70.1 (C-3′), 72.4 (C-5′), 81.2 (C-1′), 115.9 (CN), 122.9 (C-2 C-6), 123.5 (C-2′, C-6′), 128.1 (C-4), 128.9 (C-3′,C-5′), 131.6 (C-3, C-5), 147.5 (C-4′), 149.5 (C-1), 153.4 (C-1′), 163.1 (C=N), 165.3 (C=N), 169.8–168.8 (4 × COCH3).
References
S. Hout, N. Azas, A. Darque, M. Robin, C.D. Giorgio, M. Gasquet, J. Galy, P. Timon-Savid, Parasitology 129, 525 (2004)
K. Taujiihara, M. Ozeki, T. Morikawa, M. Kawamori, Akaike, Y. Arai, J. Med. Chem. 25, 441 (1982)
S.-T. Huang, I.-J. Hsei, Chen-Chinpiao, Bioorg. Med. Chem. 14, 6106 (2006)
Q.-L. Wei, S.-S. Zang, J. Gao, W. Li, L.-Z. Xu, Z.-G. Yu, Bioorg. Med. Chem. 14, 7146 (2006)
P.C. Tome Joao, G. Neves Maria, A. Tome, A.S. Gavaleiro Jose, F. Mendonca Ana, I.N. Duarte, M.L. Valdeira, Bioorg. Med. Chem. 14, 3878 (2005)
P. Kedar, S. Pande, P. Deshmukh, Der Pharma Chemica 3, 28 (2011)
L.F. Tietze, U. Bothe, U. Griesbach, Nakaichi, T. Hasegawa, Bioorg. Med. Chem. 9, 1747 (2001)
J.K. De Martino, D.L. Boger, Drug Future 33, 969 (2008)
W.J. Curran, Oncology 63, 29 (2002)
L.S. Povarov, N.P. Potapova, E.V. Bakina, M.N. Preobrazhenskaya, B.V. Rozynov, Bioorg. Khim. 18, 1117 (1992)
J.G. Fernández-Bolaños, Ó López, V. Ulgar, I. Maya, J. Fuentes, Tetrahedron 10, 3011 (1999)
B.N. Berad, S.M. Bhiwagade, A.G. Ulhe, Der. Pharm. Chem. 4, 1730 (2012)
S.A. Gharad, B.N. Berad, Phosphorus, Sulfur Silicon Relat. Elem 190, 2315 (2016)
O.V. Ershov, A.V. Eremkin, Ya.S. Kayukov, A.N. Lyshchikov, O.E. Nasakin, V.A. Tafeenko, Zh. Org. Khim. 44, 575 (2008)
P.F. Olurinola, A laboratory manual of pharmaceutical microbiology. (Idu, Abuja, 1996)
D. Srinivasan, N. Sangeetha, T. Suresh, J. Ethnopharmacol. 74, 217 (2001)
Acknowledgements
A. F. El-Farargy thanks Professor Norbert Krause, Lehrstuhl für Organische Chemie, TU Dortmund, for his repeated invitations, kind hospitality, and the facilities offered. The authors thank College of Science, Jouf University, Sakaka, Kingdom of Saudi and Chemistry Department and Faculty of Science, Zagazig University, Zagazig, Egypt for their continuous help and support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ghoneim, A.A., El-Farargy, A.F. Synthesis and antimicrobial evaluation of new glucosylimino thiazole derivatives. J IRAN CHEM SOC 16, 1391–1399 (2019). https://doi.org/10.1007/s13738-019-01617-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-019-01617-2