
Abstract
Purpose of Review
To describe updates on the pathogenesis, diagnosis, and management of Merkel cell carcinoma (MCC).
Recent Findings
Sequencing studies revealed that MCCs have either a low mutational burden and integrated Merkel cell polyomavirus (MCPyV), or they have a high number of ultraviolet-associated somatic mutations and no MCPyV. Clinically, prognosis was better for stage III MCC of unknown primary than known primary. Similarly, lack of immunosuppression conferred better prognosis. The immunogenicity of MCC was reflected in high response rates to PD-1/PD-L1 checkpoint inhibitors.
Summary
MCC is a rare but aggressive neuroendocrine skin cancer associated with advanced age and immunosuppression. Approximately 80% of MCCs are MCPyV driven, whereas MCPyV-negative tumors have mutations in genes such as p53 and RB1. MCC is highly immunogenic, and recently, the anti-PD-L1 antibody avelumab was approved to treat metastatic MCC. Here, we summarized features of the pathogenesis, diagnosis, and management of MCC.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Merkel cell carcinoma (MCC) is a rare and aggressive primary neuroendocrine skin cancer. In the USA, about 2000 new cases are diagnosed annually. The 5-year survival rate for local and metastatic disease are 70–80 and 20–30%, respectively [1]. Risk factors for MCC include ultraviolet (UV) exposure, older age (> 50 years), and immunosuppression (e.g., chronic lymphocytic leukemia) [2].
The discovery of the Merkel cell polyomavirus (MCPyV) in 2008 prompted further investigation of the biology and pathogenesis of MCC. Furthermore, as we gain experience with treating MCC patients, protocols for diagnosis and management are being refined. In this article, we provide updates on the pathogenesis, diagnosis, and management of MCC.
Pathogenesis
Merkel Cell Polyomavirus-Positive MCC Pathogenesis
Approximately 80% of MCC tumors in the USA are virus positive (VP-MCC) and have clonal integration of MCPyV into the host genome [3]. Like other polyomaviruses, MCPyV is a small (5.4 kb), double-stranded, non-enveloped DNA virus in the Orthopolyomavirus genus of the Polyomaviridae family [3]. The early region of the MCPyV virome encodes putative oncogenes including large T (LT) antigen, small T (sT) antigen, 57kT antigen, and a protein called alternative LT ORF (ALTO) [3,4,5,6,7]. The late region encodes the capsid proteins, VP1 and VP2 [3,4,5,6]. The LT protein contains multiple domains including conserved region 1 (CR1), DnaJ domain, RB binding site, origin binding domain (OBD), and a Helicase domain [4,5,6]. The sT protein contains CR1, DnaJ, the LT-stabilization domain (LSD), and the PP2A binding domain [8]. VP-MCC carries a low somatic mutation burden, suggesting that tumorigenesis is driven by viral oncogenes [9•]. The integration of MCPyV into the host genome occurs early in tumorigenesis (Fig. 1a). Interestingly, VP-MCC tumor development requires a mutation or deletion that disrupts LT helicase activity [5, 10]. Despite the demonstrated importance of MCPyV in MCC oncogenesis, little is known about the normal host cell of the virus, what drives viral integration, and the cell of origin for VP-MCC tumors.

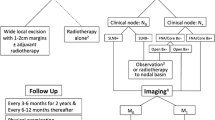
Pathogenesis and management of Merkel cell carcinoma (MCC). a Schematic model of virus-positive (VP) and virus-negative (VN) MCC pathogenesis illustrating targets of viral small T antigen (sT) and large T antigen (LT) in VP-MCC tumors, as well as mutations impacting Rb and p53 function commonly found in VN-MCC. Antitumor T cells are disinhibited by targeting PD-1 or PD-L1 with monoclonal antibody immune checkpoint inhibitors in the treatment of MCC. TCR T cell receptor, MHC major histocompatibility complex, MCPyV Merkel cell polyomavirus. b Management flowchart for MCC. 1Excision and/or radiation to primary tumor should not occur prior to SLNB [2].Adjuvant radiation to the primary tumor bed is generally recommended. Consider observation without adjuvant radiation if the primary tumor is small (<1 cm), widely excised, and without high-risk features [3].Palliative option; recurrence is not uncommon [4].PET-CT is the preferred modality. MRI and CT are alternatives [5].Anti-PD-1/PD-L1 immune checkpoint inhibitors or referral to a clinical trial is preferred over chemotherapy for metastatic disease. Avelumab is FDA-approved for treating metastatic MCC [6].Consider radiation to nodal basin in high-risk patients (e.g., immunosuppression) or if false-negative SLNB is suspected [7].Consider routine imaging in high-risk patients [8].Serology testing is only useful in patents with detectable baseline titers assessed soon after treatment. SLNB sentinel lymph node biopsy, FNA fine needle aspiration, CLND complete lymph node dissection
sT Antigen
The primary MCPyV oncogenes are thought to be the sT and LT antigens. The sT antigen is expressed from two of four alternative spliced mRNA of the MCPyV virome [11• ]. While the MCPyV LT and sT share exon 1 of the T antigen locus, the DnaJ, CR1, and Hsc70 domains found in exon 1 appear to be dispensable for sT-driven tumorigenesis [8]. In most polyomaviruses, sT antigen binds protein phosphatase 2A (PP2A) to promote AKT activation and thereby increases cell survival [12]. Like other polyomaviruses, the sT antigen of MCPyV binds PP2A, but unlike other polyomaviruses, this binding has no observed effect on in vitro or in vivo tumorigenesis [12]. Interestingly, MCPyV sT binds protein phosphatase 4C (PP4C) and protein phosphatase 4 regulatory subunit 1 (PP4R1) to form a complex targeting the NF-κB regulator NEMO, leading to reduced NF-κB translocation and transcriptional activity [13• ]. Regulation of PP4C and PP4R1 by the sT could be a mechanism by which MCPyV modulates host anti-viral response or autoimmunity [13•].
The transformative activity of MCPyV sT is due, in part, to the large T stabilization domain (LSD) located between amino acids 91–95 [14]. The LSD binds and inhibits E3 ubiquitin ligase (SCFFbw7) [14]. Because MCPyV LT is a target of Fbw7-mediated ubiquitination to induce proteasomal degradation, sT increases the half-life of LT protein [14]. Furthermore, inhibition of Fbw7 by MCPyV sT also increases the levels of other Fbw7 target proteins such as c-Myc and cyclin E which contribute to increased cell proliferation [14].
The MCPyV sT LSD also increases hyperphosphorylated 4E-binding protein 1 (4E-BP1), a regulator of cap-dependent translation. Active 4E-BP1 inhibits eukaryotic translation initiation factor 4E (eFI4E) function, thereby inhibiting translation [12]. 4E-BP1 is inactivated through phosphorylation by Mammalian target of complex 1 (mTOR1) [12]. Shuda et al. showed that, independent of mTOR1, MCPyV sT promotes hyperphosphorylation of 4E-BP1, thereby increasing protein translation [12]. Thus, MCPyV sT increases protein translation, reduces proteasomal degradation, and thereby promotes tumor cell survival via increased levels of c-Myc, cyclin E, and MCPyV LT antigen.
LT Antigen
MCPyV LT also plays specific fundamental roles in promoting VP-MCC tumorigenesis. The C-terminal domain of LT contains a helicase region important for initiating viral DNA replication [6, 8]. Importantly, loss of viral replication is necessary for tumorigenesis because initiating replication within integrated virus promotes DNA damage responses and host cell death [5, 6, 8, 10, 14, 15]. Therefore, C-terminal mutations disrupting LT helicase function are essential for VP-MCC tumor development [15]. The preservation of N-terminal LT in tumors suggests that it is necessary for host cell transformation.
MCPyV LT antigen can inhibit tumor suppressors and activate oncoproteins in MCC. First, the Rb binding domain (amino acids 211–217) of LT is never mutated in MCC [16]. This domain binds and inhibits Rb, thereby preventing Rb-mediated suppression of E2F activity [16, 17]. Release of E2F, by LT sequestering Rb, allows transcription of cell cycle genes involved in G1 to S-phase transitions, thus promoting tumor growth [16, 17]. The LT Rb-binding domain is also required for upregulation of the anti-apoptotic oncoprotein, survivin, which can be targeted pharmacologically to delay MCC xenograft tumor growth in mice [18]. Another target of the MCPyV LT is the tumor suppressor p53 [19]. Although MCPyV LT is not known to bind p53 directly, Borchert et al. showed that the LT expression leads to reduced p53 transactivation activity [19]. Taken together, integrated MCPyV employs multiple sT and LT-mediated mechanisms to promote tumor development and growth.
Merkel Cell Polyomavirus-Negative MCC Pathogenesis
The 20% of MCC that are polyomavirus negative (VN-MCC) [3] contain an exceptionally high somatic mutational burden, enriched for ultraviolet signature C > T transitions throughout the genome (Fig. 1a) [9•, 20•, 21•, 22•]. Interestingly, the two most commonly mutated tumor suppressor proteins in VN-MCC, Rb and p53, are both targets of MCPyV LT antigen [18, 19, 21•]. In VN-MCC, Rb is generally lost through genome deletion or epigenetic hypermethylation [20•, 21•, 23, 24•]. Loss of Rb in VN-MCC leads to increased E2F activity and thus increased tumor growth [17]. The majority of p53 mutations occurring in MCC are in VN-MCC tumors, which often show high p53 immunostaining [20•, 21•, 24•]. The inactivating mutations in p53 result in downregulation of p53 targets, thereby preventing tumor cell senescence, cell cycle arrest, DNA damage repair, and apoptosis [18, 20•, 21•].
Combined cutaneous squamous and Merkel cell carcinoma is an established variant of MCC that are consistently MCPyV-negative [25]. Pulitzer et al. found that, like pure VN-MCC, combined cutaneous squamous and Merkel cell carcinoma tumors have a high mutational burden including frequent mutations in both p53 and RB1, leading to increased expression of p53 and decreased expression of Rb [22•]. Cutaneous squamous and Merkel cell carcinoma also show high expression of p63 in the squamous component of the tumor [22•]. Interestingly, the ΔNp63α isoform of p63 is known to be overexpressed in squamous cell carcinoma and is thought to be a hallmark of both squamous cell and basal cell carcinoma tumorigenesis [26,27,28,29,30,31,32,33, 34•].
Taken together, loss of Rb and p53 tumor suppressors function seems to be a consistent feature of both VP-MCC and VN-MCC tumors. Whereas a number of additional oncogenic processes are consistently activated by viral oncogenes in VP-MCC, there is little commonality among the numerous additional UV-induced driver mutations found in VN-MCC tumors. A few oncogenic pathways such as MYCL and phosphoinositide 3-kinase (PI3K) activation have been implicated in VN-MCC tumors; nonetheless, only subsets of VN-MCC show activation or upregulation of these oncogenic pathways [20•, 21•]. Thus, most MCCs are MCPyV-driven tumors with low mutational burdens, and the remainder are a heterogeneous set of VN-MCC tumors driven by UV-signature mutations.
Diagnosis and Staging
MCC typically presents as a rapidly growing, asymptomatic, erythematous nodule (Fig. 2a). Polymorphous vessels and milky-red structureless areas are often seen on dermoscopy (Fig. 2b). Because MCC is rare and clinically nondescript, it is seldom suspected on clinical exam. Diagnosis is best done by an experienced dermatopathologist. Because of its aggressive nature, management of MCC requires multidisciplinary care. Referral to a cancer center with a cutaneous oncology tumor board should be considered. The general staging and management of MCC are summarized in Fig. 1b.

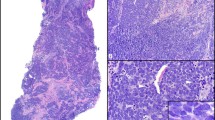
Clinical and histopathological appearance of Merkel cell carcinoma. The clinical (a) and dermatoscopic (b) presentation of a MCC tumor on the leg of a patient. The central crust is a healing punch biopsy site. c Hematoxylin and eosin (H&E) staining of a tissue section from a MCC tumor biopsy from another patient showing sheets of small blue cells with neuroendocrine features. d Immunostaining for cytokeratin 20 (CK20) shows staining in the cytoplasm of the cells with frequent paranuclear dots. Original magnification ×200
Diagnosis
Primary Tumor
MCC diagnosis requires biopsy and tissue examination with immunohistochemistry (IHC). Histologically, MCC is comprised of small round blue cells with neuroendocrine features arranged in sheets or narrow cords (Fig. 2c). IHC markers are needed to distinguish MCC from other tumor types. MCC expresses nonspecific neuroendocrine (e.g., synaptophysin, chromogranin, neuron-specific enolase) and epithelial (e.g., pancytokeratin, epithelial membrane antigen) markers. CK20 is currently the most sensitive and specific marker for MCC and is often expressed in a paranuclear dot-like pattern (Fig. 2d). Excluding cutaneous metastases from other neuroendocrine tumors is important. Negative TTF-1 staining generally excludes metastatic small cell lung cancer [35]. Pathology reports for MCC should include comments on tumor size and depth, margin status, lymphovascular invasion, and extracutaneous extension. Other factors to report include mitotic index, tumor-infiltrating lymphocytes, tumor growth pattern, and presence of a second malignancy [36].
Lymph Nodes
As regional involvement is common, sentinel lymph node biopsy (SLNB) should be considered for all MCC tumors. Immunostaining with CK20 or pancytokeratin should be used to evaluate lymph nodes. IHC with CK20 increases the sensitivity for detecting micrometastatic disease [37, 38]. The pattern of lymph node involvement may have prognostic implications, with worse overall survival when sheet-like patterns of tumor cells infiltrate the lymph node [39].
Staging
MCC can be clinically and pathologically staged using the American Joint Cancer Committee (AJCC) Cancer Staging Manual. Updates to the upcoming eighth edition of AJCC guidelines will reflect the observation that nodal disease (stage III) of unknown primary has a better prognosis than nodal disease with known primary [40]. In addition to AJCC staging, the patient’s immune status significantly impacts prognosis, with worse survival in those with impaired immunity [41]. A relatively new adjunct test uses serology for antibodies against the MCPyV sT antigen at diagnosis as an independent predictor of decreased MCC recurrence [42].
Management
Surgery
Wide local excision is the treatment approach for primary MCC. No studies have correlated margin size and recurrence risk, but 1–2-cm margins and excision down to fascia are recommended [36]. Moh’s micrographic surgery (MMS) can be considered if tissue sparing is of priority [36]. A small retrospective study of 22 patients treated with MMS showed a 5% recurrence rate after a median follow-up of 37.5 months [43]. If MMS is performed, it should be done so as to not interfere with SLNB. Extensive reconstructive procedures that involve undermining tissue should be delayed until histologic margins are confirmed negative by IHC on permanent sections and SLNB is performed [36].
Lymph Nodes
Clinical Node Negative Disease
All patients with clinically node negative, primary MCC should be offered a SLNB. A study of 240 cases suggested that tumor size does not reliably predict nodal involvement [44]. In addition, clinically occult lymph node disease is not uncommon; a study of 403 cases showed that about one third have nodal involvement on SLNB [45••]. Patients with lymph node disease have increased risk of recurrence, but the impact on overall survival is unclear [36, 46]. Nonetheless, lymph node status determines adjuvant treatment options for regional control.
Adjuvant therapy is recommended in patients with positive SLNB. A study of 29 patients with positive SLNB showed that those who received adjuvant treatment (radiation therapy [RT], chemotherapy, or complete lymph node dissection [CLND]) had a 3-year relapse-free survival of 51 versus 0% in those who did not [46]. Patients with positive SLNB should undergo CLND if they are appropriate surgical candidates. If CLND cannot be performed, RT to the nodal basin is an alternative option [36]. In a study of 171 patients with nodal disease, RT to the nodal bed was associated with higher 3-year disease-specific survival (76.2 versus 48.1%) [47••]. Patients with nodal disease should also receive baseline imaging studies.
Those with negative SLNB can be managed with regional observation alone. Recurrence in the same nodal basin is low (4–14%) after negative SLNB [48, 49]. Multiple studies have suggested that compared to observation alone, RT does not decrease regional recurrence if the SLNB is negative [48, 49]. In addition, RT to the nodal bed in SLNB negative patients does not impact 3-year disease-specific survival [47••]. In high-risk patients (e.g., immunosuppression) or if a false-negative SLNB is suspected, nodal RT can be considered.
Clinical Node Positive Disease
Clinically evident lymph node disease should be biopsied for confirmation of metastatic disease. Fine needle aspiration (FNA) or core biopsy with immunostaining is recommended and, if positive, should be followed by CLND and imaging studies. Adjuvant RT should also be considered if there is extracapsular extension or if multiple nodes are involved [36]. If the FNA or core needle biopsy is negative, an open biopsy should be considered.
Radiation
Adjuvant radiation is an option for all stages of MCC [36]. Radiation to the primary tumor bed should be initiated within weeks of surgical excision, with higher doses if resection margins are positive [36, 50]. Several studies suggest that RT decreases local and regional recurrence and may have an overall survival benefit [47••, 51]. A recent retrospective study of 171 MCC patients with known lymph node status and without distant metastases showed that RT to the primary tumor bed and/or lymph node basin improved 3-year local and regional control, disease-free survival, and overall survival, but had no effect on disease-specific survival [47••]. Another retrospective study of 1254 patients showed that local RT post-resection decreases local and regional recurrence compared to surgery alone [51]. Two large studies based on the Surveillance, Epidemiology, and End Results database suggest that adjuvant radiation improves overall survival, but not disease-specific survival [52, 53]. The only prospective randomized controlled trial investigating RT in MCC involved 83 patients with stage I disease who all underwent WLE and RT to the tumor bed. This study evaluated the effects of additional RT to the regional lymph nodes compared to observation. Prophylactic radiation to the regional nodes decreased regional recurrence compared to observation, but there was no survival benefit. However, this study dropped in recruitment and terminated prematurely after SLNB became a standard of care [54].
Observation without RT to the tumor bed may be considered if the primary tumor is small (< 1 cm), widely excised, and without other high-risk features [55]. When appropriate, patients with positive SLNB should receive RT to the regional lymph node basin, while those with negative SLNB can be observed [46, 48, 49].
In surgically unresectable cases, RT can be used as monotherapy, but recurrence is not uncommon [56]. For palliative purposes, hypofractionated or single-fraction RT can be used [36].
It is worth noting that most studies of RT in MCC are retrospective in design and are limited by variability in cohorts, with inconsistent specification of lymph node status, additional treatments (e.g., chemotherapy), and RT parameters (e.g., dose, timing, site). Many studies were performed prior to the routine use of the SLNB as a standard of care. Currently, there is a lack of prospective data investigating the role of RT in MCC management.
Imaging
Baseline imaging should be performed in patients with positive SLNB or if there is clinical suspicion of metastatic disease. FDG-PET/CT is the preferred modality [57,58,59]. PET enables detection of bone and bone marrow involvement and should be combined with CT to detect liver and lung metastases. If FDG-PET/CT is unavailable, MRI or CT are alternatives.
Somatostatin receptors (SSTR) are expressed in most MCC and are experimental targets for molecular imaging. Small studies have investigated the use of somatostatin analogues (e.g., [68Ga]DOTA-D-Phe1-Tyr3-octreotide (DOTATOC) or -octreotate (DOTATATE)) combined with PET (SSTR-PET) to identify loci of metastatic MCC. SSTR-PET has high sensitivity for the bone, soft tissue, and brain metastases, but CT is still required for reliable detection of liver and lung lesions [60].
Chemotherapy
Chemotherapy is reserved for distant metastatic disease. Guidelines are not well defined, and regimens are based on MCC’s similarity to small cell lung cancer. The most common regimen includes a platinum-based agent with or without etoposide. Others may use cyclophosphamide, doxorubicin, and vincristine (CAV), or topotecan. A recent review showed that the objective response rate (ORR) with chemotherapy is higher in the first-line compared to second-line setting (53–61 versus 23–45%). Although MCC is chemosensitive, there are high toxicity and responses that are seldom durable. The duration of response is less than 8 months with most recurrences occurring within 6 months [61•]. The effect of chemotherapy on overall survival is unclear. Chemotherapy’s shortcomings and studies of MCC biology prompted the investigation of targeted therapies as well as immunotherapies for treatment of metastatic disease.
Targeted Therapy
PI3K/Akt/mTOR Pathway Inhibition
Signaling through the PI3K/Akt/mTOR pathway is upregulated in a small subset (4–10%) of MCC due to activating mutations in PIK3CA and AKT1 [62,63,64]. There is a case report of a patient with metastatic MCC with a known PIK3CA mutation that had complete response after treatment with idelalisib, a PI3Kδ inhibitor [65]. A phase I/II clinical trial investigating the mTOR inhibitor MLN0128 for metastatic MCC is ongoing (NCT02514824).
Tyrosine Kinase Inhibition
MCC tumors frequently express tyrosine kinase receptors for signaling molecules including VEGF-A, VEGF-C, VEGFR-2, PDGF-A, and c-kit. As such, tyrosine kinase inhibitors have been studied as potential therapeutics [66, 67]. Pazopanib, a multitargeted tyrosine kinase inhibitor, was used in one patient who had progressed on chemotherapy, with a partial response at 6-month follow-up [66]. Imatinib was studied in c-kit-positive metastatic MCC in a phase II trial and showed a brief partial response in 1/23 (4%) of patients [67]. A phase II trial with cabozantinib, a small molecule inhibitor of c-MET and VEGFR-2, in patients with recurrent or metastatic MCC that progressed on chemotherapy is ongoing (NCT02036476).
Bcl-2 Antisense Oligodexoyribonucleotide Therapy
Preclinical studies found that Bcl-2, an anti-apoptotic protein, is upregulated in MCC. A phase II study of olimersen, a Bcl-2 antisense oligodexoyribonucleotide, showed no objective response in 12 patients with metastatic or regionally recurrent MCC [68].
Somatostatin Analogues
Somatostatin (SST) has an anti-proliferative effect on neuroendocrine tumor cells. Because ~ 90% of MCC express somatostatin receptors (SSTR), SST analogues are being investigated [69]. There was an early report of a complete response after treatment with lanreotide in a patient with metastatic MCC [70]. Current clinical trials include a recently completed phase I trial with pasireotide for metastatic MCC (NCT01652547) and an ongoing phase II trial with lanreotide for unresectable and/or metastatic MCC (NCT02351128).
SST analogues can also be paired with radionucleotide therapy, in which a radiolabeled SST analogue is taken up by SSTR-expressing tumor cells, leading to emission of local radiation. A phase II study investigating this approach with 177Lu-DOTATATE for treatment of SSTR-expressing NETs, including MCC, recently completed with results pending (NCT01237457).
Immunotherapy
The immunogenicity of MCC has prompted multiple immunotherapy trials in MCC [71].
Intratumoral Vaccines
Intratumoral vaccines stimulate the host immune response while circumventing systemic toxicity. Talimogene laherparepvec (T-VEC) is an oncolytic virus engineered from attenuated HSV-1 that has been modified to secrete GM-CSF. T-VEC injections are FDA-approved for unresectable and injectable melanomas [72]. An ongoing phase II trial is investigating T-VEC with or without hypofractionated RT in melanoma, MCC, and other solid tumors with skin metastases (NCT02819843).
In mouse melanoma, intratumoral injection with IL-12 plasmid vaccine followed by electroporation increases IL-12 and IFN γ, and reduces tumor vascularity [73]. This same treatment regimen was subsequently studied in a phase I trial in patients with metastatic melanoma, and the majority had stable disease, partial response, or complete response [74]. A phase II trial of IL-12 plasmid vaccine followed by in vivo electroporation for MCC recently completed enrollment (NCT01440816).
Adoptive Cell Transfer
Immunotherapy with adoptive cell transfer involves harvesting, expanding, and infusing immune cells to mount an antitumor response. This process can be autologous, in which case T cells are harvested from the patient, modified or cultured in vitro, and then reinjected into the patient. Transferred cells can also be from allogenic sources.
One ongoing phase I/II trial for metastatic MCC combines transfer of autologous MCPyV T-antigen-specific CD8+ T cells with the immune checkpoint inhibitor avelumab, in addition to either localized RT or recombinant IFN-β (NCT02584829). Another phase II study involves infusions of innate natural killer (NK) NK-92 cells and the NK stimulating cytokine IL-15 (NCT02465957).
Immune Checkpoint Inhibition
Programmed Cell Death Receptor 1 (PD-1)/Programmed Cell Death Receptor Ligand 1 (PD-L1)
Immune checkpoints prevent chronic activation of immune responses. For example, signaling to the T cell receptor PD-1 by PD-L1 expressed on tumor or immune cells leads to T cell inactivation and local immune tolerance. Blocking PD-1 signaling with monoclonal antibodies targeting PD-1 or PD-L1 enhances T cell activation and antitumor activity (Fig. 1a). In MCC, PD-L1 is expressed on tumor cells and local immune cell infiltrates [75]. In addition, MCPyV-specific tumor infiltrating lymphocytes have high levels of PD-1 expression [76]. These findings rationalize using PD-1/PD-L1 blocking antibodies in MCC.
A phase II trial studied the use of pembrolizumab, a monoclonal antibody targeting PD-1, as first-line therapy for 26 patients with metastatic MCC [77••]. The ORR was 56%, with responses observed independent of tumor viral status or PD-L1 expression. Progression-free survival at 6 months was 67%. The most common side effects were fatigue and laboratory abnormalities. By Common Terminology Criteria for Adverse Events (CTCAE), grade 3 or 4 drug-related adverse events occurred in 15% of patients, including myocarditis and elevated liver enzymes.
A larger phase II trial tested avelumab, an anti-PD-L1 monoclonal antibody, as a second-line agent in 88 patients with chemotherapy-resistant metastatic MCC [78••]. The ORR was 31.8%, with several patients achieving complete response. There was no difference in response rates across subgroups (e.g., tumor PD-L1 expression, tumor MCPyV status, baseline disease burden). Response to treatment with avelumab was durable, with an estimated 6-month response durability of 92%. Treatment with avelumab was well tolerated, and the most common side effects were fatigue and infusion reactions. Five patients had grade 3 adverse events, including lymphopenia and elevated liver enzymes, creatinine phosphokinase, and blood cholesterol. There were no grade 4 adverse events. Avelumab is now the first FDA- and EMA-approved drug for metastatic MCC. A phase III trial testing adjuvant avelumab in resected stage III MCC is ongoing (NCT03271372).
Other immune checkpoint inhibitors for metastatic MCC are currently under investigation. A phase I/II trial of nivolumab (anti-PD-1) for metastatic virus-associated tumors including MCC is ongoing. Preliminary results with 22 patients with MCC revealed an ORR of 68% [79]. A phase II trial with atezolizumab (anti-PD-L1) in combination with bevacizumab (anti-VEGF) in rare solid tumors including MCC is recruiting participants (NCT03074513).
Cytotoxic T-Lymphocyte Associated Protein 4 (CTLA-4)
Signaling through CTLA-4, another immune checkpoint receptor on T cells, downregulates T cell activity and is permissive to tumor cells’ immune evasion. In a series of five cases of metastatic MCC treated with the anti-CTLA-4 antibody, ipilimumab (3 mg/kg every 3 weeks for 4 cycles), two patients had a complete response, two had stable disease, and one had disease progression [80]. There is an ongoing phase II trial that initially was comparing adjuvant ipilimumab versus observation after complete MCC resection. However, due to the higher toxicity of anti-CTLA-4, patients are now randomized to receive either nivolumab (anti PD-1) or observation (NCT02196961).
Combinations of Checkpoint Inhibitors
Ongoing trials combining multiple immune checkpoint inhibitors for metastatic MCC include a phase II trial combining nivolumab and ipilimumab, with and without stereotactic body RT (NCT03071406), and a phase I/II trial combining tremelimumab (anti-CTLA-4) and durvalumab (anti-PD-L1) with a tumor microenvironment immunostimulant, poly-ICLC (NCT02643303).
Disease Monitoring and Follow-up
All MCC patients should be monitored closely by a dermatologist. For the first 3 years following diagnosis, a full body skin and lymph node exam should be performed every 3–6 months, and every 6–12 months thereon afterwards. Close follow-up is critical, as median time to recurrence is 9 months, with 90% of recurrences occurring within 24 months [36]. Routine imaging should be considered in high-risk patients.
Circulating biomarkers for monitoring disease progression or recurrence have recently been investigated. Based on a retrospective study of 60 MCC patients, blood levels of neuron specific enolase and chromogranin A are not useful for predicting outcomes or detecting recurrence [81]. However, circulating tumor cells have potential utility in following MCC disease course [81, 82]. The only validated biomarker to monitor for MCC recurrence is the serial evaluation of circulating anti-MCPyV sT antibody titers [42]. Increasing titers have a positive predictive value of 66% for clinical recurrence, and titers may rise before recurrence is detectable on physical exam. However, there are limitations with this approach. Circulating sT antibodies are only found in some patients with MCPyV-positive tumors (52% of MCC patients), and patients must be tested soon after initial treatment because titers may fall below detection post-treatment.
Conclusions
MCC is a rare disease requiring multi-institutional collaborations to investigate its natural history and develop treatment protocols. For patients with advanced disease, first-line immunotherapy with anti-PD-1/PD-L1 checkpoint inhibitors is well tolerated and offers a durable response compared to traditional chemotherapy. Currently, the only approved drug for metastatic MCC is avelumab. However, not all patients respond to immune checkpoint inhibitors, and additional therapies are needed. As our understanding of VP-MCC and VN-MCC biology and pathogenesis develops, additional treatments can be investigated.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major Importance
Albores-Saavedra J, Batich K, Chable-Montero F, Sagy N, Schwartz AM, Henson DE. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol. 2010;37(1):20–7.
Koljonen V, Kukko H, Pukkala E, Sankila R, Böhling T, Tukiainen E, et al. Chronic lymphocytic leukaemia patients have a high risk of Merkel-cell polyomavirus DNA-positive Merkel-cell carcinoma. Br J Cancer. 2009;101(8):1444–7.
Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319(5866):1096–100.
Pipas JM. Common and unique features of T antigens encoded by the polyomavirus group. J Virol. 1992;66(7):3979–85.
Shuda M, Feng H, Kwun HJ, Rosen ST, Gjoerup O, Moore PS, et al. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc Natl Acad Sci U S A. 2008;105(42):16272–7.
Feng H, Kwun HJ, Liu X, Gjoerup O, Stolz DB, Chang Y, et al. Cellular and viral factors regulating Merkel cell polyomavirus replication. PLoS One. 2011;6(7):e22468.
Carter JJ, Daugherty MD, Qi X, Bheda-Malge A, Wipf GC, Robinson K, et al. Identification of an overprinting gene in Merkel cell polyomavirus provides evolutionary insight into the birth of viral genes. Proc Natl Acad Sci U S A. 2013;110(31):12744–9.
Kwun HJ, Guastafierro A, Shuda M, Meinke G, Bohm A, Moore PS, et al. The minimum replication origin of merkel cell polyomavirus has a unique large T-antigen loading architecture and requires small T-antigen expression for optimal replication. J Virol. 2009;83(23):12118–28.
• Goh G, Walradt T, Markarov V, Blom A, Riaz N, Doumani R, et al. Mutational landscape of MCPyV-positive and MCPyV-negative Merkel cell carcinomas with implications for immunotherapy. Oncotarget. 2016;7(3):3403–15. Defined the mutation spectra of MCPyV-negative and MCPyV-positive MCC tumors
Fischer N, Brandner J, Fuchs F, Moll I, Grundhoff A. Detection of Merkel cell polyomavirus (MCPyV) in Merkel cell carcinoma cell lines: cell morphology and growth phenotype do not reflect presence of the virus. Int J Cancer. 2010;126(9):2133–42.
• Theiss JM, Gunther T, Alawi M, et al. A comprehensive analysis of replicating Merkel cell polyomavirus genomes delineates the viral transcription program and suggests a role for mcv-miR-M1 in episomal persistence. PLoS Pathog. 2015;11(7):e1004974. Explored MCPyV transcriptional programming using RACE, RNA-seq and ChIP-seq.
Shuda M, Kwun HJ, Feng H, Chang Y, Moore PS. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J Clin Invest. 2011;121(9):3623–34.
• Abdul-Sada H, Muller M, Mehta R, et al. The PP4R1 sub-unit of protein phosphatase PP4 is essential for inhibition of NF-kappaB by merkel polyomavirus small tumour antigen. Oncotarget. 2017;8(15):25418–32. Identified the complex between PP4R1 and PP4C that bridges MCPyV sT antigen to NEMO adaptor protein.
Kwun HJ, Shuda M, Feng H, Camacho CJ, Moore PS, Chang Y. Merkel cell polyomavirus small T antigen controls viral replication and oncoprotein expression by targeting the cellular ubiquitin ligase SCFFbw7. Cell Host Microbe. 2013;14(2):125–35.
Li J, Wang X, Diaz J, Tsang SH, Buck CB, You J. Merkel cell polyomavirus large T antigen disrupts host genomic integrity and inhibits cellular proliferation. J Virol. 2013;87(16):9173–88.
Hesbacher S, Pfitzer L, Wiedorfer K, Angermeyer S, Borst A, Haferkamp S, et al. RB1 is the crucial target of the Merkel cell polyomavirus large T antigen in Merkel cell carcinoma cells. Oncotarget. 2016;7(22):32956–68.
Nakamura T, Sato Y, Watanabe D, Ito H, Shimonohara N, Tsuji T, et al. Nuclear localization of Merkel cell polyomavirus large T antigen in Merkel cell carcinoma. Virology. 2010;398(2):273–9.
Dresang LR, Guastafierro A, Arora R, Normolle D, Chang Y, Moore PS. Response of Merkel cell polyomavirus-positive merkel cell carcinoma xenografts to a survivin inhibitor. PLoS One. 2013;8(11):e80543.
Borchert S, Czech-Sioli M, Neumann F, Schmidt C, Wimmer P, Dobner T, et al. High-affinity Rb binding, p53 inhibition, subcellular localization, and transformation by wild-type or tumor-derived shortened Merkel cell polyomavirus large T antigens. J Virol. 2014;88(6):3144–60.
• Wong SQ, Waldeck K, Vergara IA, Schroder J, Madore J, Wilmott JS, et al. UV-associated mutations underlie the etiology of MCV-negative Merkel cell carcinomas. Cancer Res. 2015;75(24):5228–34. Detected tumor harboring mutations of MCPyV-negative tumors in Rb1 , p53 , PIK3CA , AKT1 , PIK3CG , HRAS , NF1 and FGFR2 . Also investigated the status of T cell infiltrating lymphocytes and PD-L1 expression in MCC tumors.
• Harms PW, Collie AM, Hovelson DH, et al. Next generation sequencing of cytokeratin 20-negative Merkel cell carcinoma reveals ultraviolet-signature mutations and recurrent TP53 and RB1 inactivation. Mod Pathol. 2016;29(3):240–8. Identified mutations in Rb1 , p53 , PIK3CA , BAP1 , AKT1 and EZH2 in MCPyV-negative MCC tumor samples.
• Pulitzer MP, Brannon AR, Berger MF, Louis P, Scott SN, Jungbluth AA, et al. Cutaneous squamous and neuroendocrine carcinoma: genetically and immunohistochemically different from Merkel cell carcinoma. Mod Pathol. 2015;28(8):1023–32. Compared gene signatures and mutational rates of combined squamous and Merkel cell carcinoma versus pure Merkel cell carcinoma
Sahi H, Savola S, Sihto H, Koljonen V, Bohling T, Knuutila S. RB1 gene in Merkel cell carcinoma: hypermethylation in all tumors and concurrent heterozygous deletions in the polyomavirus-negative subgroup. APMIS. 2014;122(12):1157–66.
• Harms PW, Vats P, Verhaegen ME, Robinson DR, Wu YM, Dhanasekaran SM, et al. The distinctive mutational spectra of polyomavirus-negative Merkel cell carcinoma. Cancer Res. 2015;75(18):3720–7. Identified mutations in Rb1 , p53 , PIK3CA , HRAS , PRUNE2 and NOTCH in MCPyV-negative MCC.
Walsh NM. Primary neuroendocrine (Merkel cell) carcinoma of the skin: morphologic diversity and implications thereof. Hum Pathol. 2001;32(7):680–9.
Sniezek JC, Matheny KE, Westfall MD, Pietenpol JA. Dominant negative p63 isoform expression in head and neck squamous cell carcinoma. Laryngoscope. 2004;114(12):2063–72.
Reis-Filho JS, Torio B, Albergaria A, Schmitt FC. p63 expression in normal skin and usual cutaneous carcinomas. J Cutan Pathol. 2002;29(9):517–23.
Lo Muzio L, Santarelli A, Caltabiano R, Rubini C, Pieramici T, Trevisiol L, et al. p63 overexpression associates with poor prognosis in head and neck squamous cell carcinoma. Hum Pathol. 2005;36(2):187–94.
Di Como CJ, Urist MJ, Babayan I, et al. p63 expression profiles in human normal and tumor tissues. Clin Cancer Res. 2002;8(2):494–501.
Choi HR, Batsakis JG, Zhan F, Sturgis E, Luna MA, El-Naggar AK. Differential expression of p53 gene family members p63 and p73 in head and neck squamous tumorigenesis. Hum Pathol. 2002;33(2):158–64.
Bircan S, Candir O, Kapucoglu N, Baspinar S. The expression of p63 in basal cell carcinomas and association with histological differentiation. J Cutan Pathol. 2006;33(4):293–8.
Carroll DK, Carroll JS, Leong CO, Cheng F, Brown M, Mills AA, et al. p63 regulates an adhesion programme and cell survival in epithelial cells. Nat Cell Biol. 2006;8(6):551–61.
Hill NT, Gracia-Maldonado GH, Leonard MK, Harper AR, Tober KL, Oberyszyn TM, et al. Role of vitamin D3 in modulation of DeltaNp63alpha expression during UVB induced tumor formation in SKH-1 mice. PLoS One. 2014;9(9):e107052.
• Hill NT, Zhang J, Leonard MK, Lee M, Shamma HN, Kadakia M. 1alpha, 25-Dihydroxyvitamin D(3) and the vitamin D receptor regulates DeltaNp63alpha levels and keratinocyte proliferation. Cell Death Dis. 2015;6:e1781. Correlated increased expression of VDR and p63 in basal cell carcinoma and squamous cell carcinoma.
Bobos M, Hytiroglou P, Kostopoulos I, Karkavelas G, Papadimitriou CS. Immunohistochemical distinction between merkel cell carcinoma and small cell carcinoma of the lung. Am J Dermatopathol. 2006;28(2):99–104.
National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology (NCCN Guidelines) Merkel Cell Carcinoma. 2017; https://www.nccn.org/professionals/physician_gls/pdf/mcc.pdf. Accessed July 21, 2017.
Su LD, Lowe L, Bradford CR, Yahanda AI, Johnson TM, Sondak VK. Immunostaining for cytokeratin 20 improves detection of micrometastatic Merkel cell carcinoma in sentinel lymph nodes. J Am Acad Dermatol. 2002;46(5):661–6.
Allen PJ, Busam K, Hill AD, Stojadinovic A, Coit DG. Immunohistochemical analysis of sentinel lymph nodes from patients with Merkel cell carcinoma. Cancer. 2001;92(6):1650–5.
Ko JS, Prieto VG, Elson PJ, Vilain RE, Pulitzer MP, Scolyer RA, et al. Histological pattern of Merkel cell carcinoma sentinel lymph node metastasis improves stratification of stage III patients. Mod Pathol. 2016;29(2):122–30.
Chen KT, Papavasiliou P, Edwards K, Zhu F, Perlis C, Wu H, et al. A better prognosis for Merkel cell carcinoma of unknown primary origin. Am J Surg. 2013;206(5):752–7.
Paulson KG, Iyer JG, Blom A, Warton EM, Sokil M, Yelistratova L, et al. Systemic immune suppression predicts diminished Merkel cell carcinoma-specific survival independent of stage. J Invest Dermatol. 2013;133(3):642–6.
Paulson KG, Lewis CW, Redman MW, Simonson WT, Lisberg A, Ritter D, et al. Viral oncoprotein antibodies as a marker for recurrence of Merkel cell carcinoma: a prospective validation study. Cancer. 2017;123(8):1464–74.
Kline L, Coldiron B. Mohs micrographic surgery for the treatment of Merkel cell carcinoma. Dermatol Surg. 2016;42(8):945–51.
Tarantola TI, Vallow LA, Halyard MY, Weenig RH, Warschaw KE, Grotz TE, et al. Prognostic factors in Merkel cell carcinoma: analysis of 240 cases. J Am Acad Dermatol. 2013;68(3):425–32.
•• Shibayama Y, Imafuku S, Takahashi A, Nakayama J. Role of sentinel lymph node biopsy in patients with Merkel cell carcinoma: statistical analysis of 403 reported cases. Int J Clin Oncol. 2015;20(1):188–93. Established that patients with clinically node negative disease frequently have positive SLNB.
Gupta SG, Wang LC, Penas PF, Gellenthin M, Lee SJ, Nghiem P. Sentinel lymph node biopsy for evaluation and treatment of patients with Merkel cell carcinoma: the Dana-Farber experience and meta-analysis of the literature. Arch Dermatol. 2006;142(6):685–90.
•• Strom T, Carr M, Zager JS, et al. Radiation therapy is associated with improved outcomes in Merkel cell carcinoma. Ann Surg Oncol. 2016;23(11):3572–8. Established that radiation therapy improves locoregional control and survival in patients with MCC.
Grotz TE, Joseph RW, Pockaj BA, Foote RL, Otley CC, Bagaria SP, et al. Negative sentinel lymph node biopsy in Merkel cell carcinoma is associated with a low risk of same-nodal-basin recurrences. Ann Surg Oncol. 2015;22(12):4060–6.
Gunaratne DA, Howle JR, Veness MJ. Sentinel lymph node biopsy in Merkel cell carcinoma: a 15-year institutional experience and statistical analysis of 721 reported cases. Br J Dermatol. 2016;174(2):273–81.
Rush Z, Fields RC, Lee N, Brownell I. Radiation therapy in the management of Merkel cell carcinoma: current perspectives. Expert Rev Dermatol. 2011;6(4):395–404.
Lewis KG, Weinstock MA, Weaver AL, Otley CC. Adjuvant local irradiation for Merkel cell carcinoma. Arch Dermatol. 2006;142(6):693–700.
Mojica P, Smith D, Ellenhorn JD. Adjuvant radiation therapy is associated with improved survival in Merkel cell carcinoma of the skin. J Clin Oncol. 2007;25(9):1043–7.
Kim JA, Choi AH. Effect of radiation therapy on survival in patients with resected Merkel cell carcinoma: a propensity score surveillance, epidemiology, and end results database analysis. JAMA Dermatol. 2013;149(7):831–8.
Jouary T, Leyral C, Dreno B, Doussau A, Sassolas B, Beylot-Barry M, et al. Adjuvant prophylactic regional radiotherapy versus observation in stage I Merkel cell carcinoma: a multicentric prospective randomized study. Ann Oncol. 2012;23(4):1074–80.
Frohm ML, Griffith KA, Harms KL, Hayman JA, Fullen DR, Nelson CC, et al. Recurrence and survival in patients with Merkel cell carcinoma undergoing surgery without adjuvant radiation therapy to the primary site. JAMA Dermatol. 2016;152(9):1001–7.
Veness M, Foote M, Gebski V, Poulsen M. The role of radiotherapy alone in patients with merkel cell carcinoma: reporting the Australian experience of 43 patients. Int J Radiat Oncol Biol Phys. 2010;78(3):703–9.
Treglia G, Kakhki VR, Giovanella L, Sadeghi R. Diagnostic performance of fluorine-18-fluorodeoxyglucose positron emission tomography in patients with Merkel cell carcinoma: a systematic review and meta-analysis. Am J Clin Dermatol. 2013;14(6):437–47.
Siva S, Byrne K, Seel M, Bressel M, Jacobs D, Callahan J, et al. 18F-FDG PET provides high-impact and powerful prognostic stratification in the staging of Merkel cell carcinoma: a 15-year institutional experience. J Nucl Med. 2013;54(8):1223–9.
Hawryluk EB, O'Regan KN, Sheehy N, Guo Y, Dorosario A, Sakellis CG, et al. Positron emission tomography/computed tomography imaging in Merkel cell carcinoma: a study of 270 scans in 97 patients at the Dana-Farber/Brigham and Women’s Cancer Center. J Am Acad Dermatol. 2013;68(4):592–9.
Buder K, Lapa C, Kreissl MC, Schirbel A, Herrmann K, Schnack A, et al. Somatostatin receptor expression in Merkel cell carcinoma as target for molecular imaging. BMC Cancer. 2014;14:268.
• Nghiem P, Kaufman HL, Bharmal M, Mahnke L, Phatak H, Becker JC. Systematic literature review of efficacy, safety and tolerability outcomes of chemotherapy regimens in patients with metastatic Merkel cell carcinoma. Future Oncol. 2017. Comprehensive review describing the efficacy and safety of chemotherapy in MCC;13:1263–79.
Hafner C, Houben R, Baeurle A, Ritter C, Schrama D, Landthaler M, et al. Activation of the PI3K/AKT pathway in Merkel cell carcinoma. PLoS One. 2012;7(2):e31255.
Nardi V, Song Y, Santamaria-Barria JA, Cosper AK, Lam Q, Faber AC, et al. Activation of PI3K signaling in Merkel cell carcinoma. Clin Cancer Res. 2012;18(5):1227–36.
Iwasaki T, Matsushita M, Nonaka D, Kuwamoto S, Kato M, Murakami I, et al. Comparison of Akt/mTOR/4E-BP1 pathway signal activation and mutations of PIK3CA in Merkel cell polyomavirus-positive and Merkel cell polyomavirus-negative carcinomas. Hum Pathol. 2015;46(2):210–6.
Shiver MB, Mahmoud F, Gao L. Response to Idelalisib in a patient with stage IV Merkel-cell carcinoma. N Engl J Med. 2015;373(16):1580–2.
Davids MS, Charlton A, Ng SS, Chong ML, Laubscher K, Dar M, et al. Response to a novel multitargeted tyrosine kinase inhibitor pazopanib in metastatic Merkel cell carcinoma. J Clin Oncol. 2009;27(26):e97–100.
Samlowski WE, Moon J, Tuthill RJ, Heinrich MC, Balzer-Haas NS, Merl SA, et al. A phase II trial of imatinib mesylate in merkel cell carcinoma (neuroendocrine carcinoma of the skin): a southwest oncology group study (S0331). Am J Clin Oncol. 2010;33(5):495–9.
Shah MH, Varker KA, Collamore M, Zwiebel JA, Coit D, Kelsen D, et al. G3139 (Genasense) in patients with advanced merkel cell carcinoma. Am J Clin Oncol. 2009;32(2):174–9.
Sestini R, Orlando C, Peri A, Tricarico C, Pazzagli M, Serio M, et al. Quantitation of somatostatin receptor type 2 gene expression in neuroblastoma cell lines and primary tumors using competitive reverse transcription-polymerase chain reaction. Clin Cancer Res. 1996;2(10):1757–65.
Fakiha M, Letertre P, Vuillez JP, Lebeau J. Remission of Merkel cell tumor after somatostatin analog treatment. J Cancer Res Ther. 2010;6(3):382–4.
Vandeven N, Nghiem P. Rationale for immune-based therapies in Merkel polyomavirus-positive and -negative Merkel cell carcinomas. Immunotherapy. 2016;8(8):907–21.
Andtbacka RH, Kaufman HL, Collichio F, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780–8.
Lucas ML, Heller L, Coppola D, Heller R. IL-12 plasmid delivery by in vivo electroporation for the successful treatment of established subcutaneous B16.F10 melanoma. Mol Ther. 2002;5(6):668–75.
Daud AI, DeConti RC, Andrews S, et al. Phase I trial of interleukin-12 plasmid electroporation in patients with metastatic melanoma. J Clin Oncol. 2008;26(36):5896–903.
Lipson EJ, Vincent JG, Loyo M, Kagohara LT, Luber BS, Wang H, et al. PD-L1 expression in the Merkel cell carcinoma microenvironment: association with inflammation, Merkel cell polyomavirus and overall survival. Cancer Immunol Res. 2013;1(1):54–63.
Afanasiev OK, Yelistratova L, Miller N, Nagase K, Paulson K, Iyer JG, et al. Merkel polyomavirus-specific T cells fluctuate with merkel cell carcinoma burden and express therapeutically targetable PD-1 and Tim-3 exhaustion markers. Clin Cancer Res. 2013;19(19):5351–60.
•• Nghiem PT, Bhatia S, Lipson EJ, et al. PD-1 blockade with Pembrolizumab in advanced Merkel-cell carcinoma. N Engl J Med. 2016;374(26):2542–52. First randomized clinical trial of immune checkpoint inhibition for treating metastatic MCC.
•• Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17(10):1374–85. Randomized clinical trial that led to avelumab being the first FDA-approved drug for metastatic MCC.
Topalian SL, Bhatia S, Hollebecque A, et al. Non-comparative, open-label, multiple cohort, phase 1/2 study to evaluate nivolumab (NIVO) in patients with virus-associated tumors (CheckMate 358): Efficacy and safety in Merkel cell carcinoma (MCC) [abstract]. In. In: Proceedings of the 107th Annual Meeting of the American Association for Cancer Res; 2017 Apr 1–5; Washington, DC. Philadelphia (PA): AACR; 2017. Abstract nr CT074 2017.
Winkler JK, Dimitrakopoulou-Strauss A, Sachpekidis C, Enk A, Hassel JC. Ipilimumab has efficacy in metastatic Merkel cell carcinoma: a case series of five patients. J Eur Acad Dermatol Venereol. 2017;31:e389–91.
Gaiser MR, Daily K, Hoffmann J, Brune M, Enk A, Brownell I. Evaluating blood levels of neuron specific enolase, chromogranin A, and circulating tumor cells as Merkel cell carcinoma biomarkers. Oncotarget. 2015;6(28):26472–82.
Blom A, Bhatia S, Pietromonaco S, Koehler K, Iyer JG, Nagase K, et al. Clinical utility of a circulating tumor cell assay in Merkel cell carcinoma. J Am Acad Dermatol. 2014;70(3):449–55.
Funding
This research was supported by the NIH Intramural Research Program, Center of Cancer Research, National Cancer Institute.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Dr. Brownell reports work prepared as part of official duties as a US government employee for Intramural Research Program, CCR, NCI, during the conduct of the study.
Jannett Nguyen and Natasha Hill declare that they have no conflict of interest.
Disclosures
Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Skin Cancer
Rights and permissions
About this article

Cite this article
Nguyen, J., Hill, N. & Brownell, I. Merkel Cell Carcinoma: Updates on Pathogenesis, Diagnosis, and Management. Curr Derm Rep 7, 158–168 (2018). https://doi.org/10.1007/s13671-018-0221-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13671-018-0221-1