
Abstract
Merkel cell carcinoma (MCC) is an aggressive cutaneous neuroendocrine carcinoma. Incidence of MCC continues to rise, and risk factors include advanced age, pale skin, chronic sun exposure, and immune suppression. Diagnosing MCC utilizes a combination of morphology and immunohistochemistry. Merkel cell polyomavirus (MCPyV) is present in approximately 70–80% of MCCs and represents a key pathogenic driver in those MCCs. In contrast, MCPyV-negative MCCs arise through progressive accumulation of ultraviolet-light induced somatic mutations. Staging of MCC proceeds according to the American Joint Commission on Cancer (AJCC) 8th Edition, which utilizes features of the primary tumor together with regional lymph node(s) (clinically and/or pathologically detected) and/or distant metastases. Many potentially useful biomarkers have been studied to refine risk stratification in MCC. In recent years, the host immune infiltrate has been leveraged as immune checkpoint blockade has emerged as an efficacious mode of treatment for patients with advanced MCC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Merkel cell carcinoma (MCC) is an aggressive primary cutaneous neuroendocrine carcinoma with frequent metastasis. Death due to disease is common. MCC develops in older patients with pale skin with chronic exposure to ultraviolet light and/or immune suppression [1,2,3,4,5,6,7]. The diagnosis of MCC relies on a combination of morphologic recognition of a neuroendocrine phenotype in cutaneous tumors consisting of monomorphous round basaloid cells together with judicious application of immunohistochemical studies. A pivotal development that significantly advanced the understanding of MCC was the identification of Merkel cell polyomavirus (MCPyV) as a pivotal driver of carcinogenesis in approximately 70% of MCCs [8]. MCPyV can be detected by polymerase chain reaction amplification of MCPyV-specific sequences or immunohistochemical detection of MCPyV-specific proteins. In contrast, MCPyV-negative MCCs appear to arise through progressive accumulation of ultraviolet light (UV)-induced somatic mutations. Staging in MCC proceeds according to the American Joint Commission on Cancer (AJCC) 8th Edition, which utilizes features of the primary tumor (size and extent of invasion) together with the involvement of regional lymph nodes (clinically and/or pathologically detected) and distant metastases to predict prognosis and stratify patient risk in MCC. A number of potentially useful biomarkers have been proffered including p63 and the density, composition and distribution of the tumor-associated immune infiltrate. The latter has been recently leveraged therapeutically as immune checkpoint blockade has emerged as a robust and efficacious mode of treatment for patients with advanced MCC.
Incidence and Epidemiology
A review of demographic information from 14,414 patients with MCC from the National Cancer Data Base (spanning 1998–2012) highlighted several important themes regarding the pathobiology of MCC [9]. First, MCC occurs almost twice as commonly in men (62.1%) as in women (37.9%). Second, MCC affects older patients: approximately 81.7% of patients with MCC are in their 7th–9th decade of life, including 18.6% between 60 and 69 years, 33.1% between 70 and 79 years, and 30% between 80 and 89 years. Third, MCC most commonly arises on the head and neck (42.6%), followed by the upper limb and shoulder (23.6%). Finally, MCC overwhelmingly affects Caucasians (96.4%) and is exceedingly rare among individuals of African American (1.2%) and Asian (0.8%) descent. Taken together, MCC is a disease commonly affecting elderly, Caucasian men on sites of chronic sun exposure.
In the United States, the incidence of MCC has increased roughly fivefold over the past 30 years: from 1.5 cases per million in 1986 to 4.4 cases per million in 2001 to 7.9 cases per million in 2011 [4, 10, 11]. Similar trends have been reported in older, fair skinned individuals living in other parts of the world, with highest incidence rates occurring in regions with highest UV exposure (including the United States, Australia and New Zealand).
Risk Factors for MCC
Risk factors for the development of MCC in part reflect the epidemiology of the disease and ultimately, manifest the divergent pathways driving MCC development: (1) infection by MCPyV (see below) or (2) progressive accumulation of UV-induced somatic mutations. As MCC is related to cumulative UV-induced mutations, risk factors include advanced patient age, fair-skin and chronic sun exposure, and these in turn directly reflect the disease demographics: older Caucasian men on the head and neck.
A parallel risk factor for the development of MCC is immunosuppression. Patients with immune suppression due to hematologic malignancy (most commonly chronic lymphocytic leukemia) [12, 13] or HIV/AIDS [3], and iatrogenic immune suppression in the setting of solid organ transplantation [14] and autoimmune disease treatment [15] are at increased risk for the development of MCC compared to immune competent patients.
Cell of Origin in MCC
As of yet, the precise cell of origin for MCC has not been identified. Intraepidermal Merkel cells are post-mitotic and terminally differentiated and reside in the basal layer of epidermis, while the vast majority of MCCs are dermal-based proliferations lacking an intraepithelial component. Endogenous intraepidermal Merkel cells, therefore, are unlikely to be the cells giving rise to MCC. Possible candidates include epidermal stem cells, B-cells and fibroblasts. Some studies implicate epidermal/dermal stem cells as the origin of Merkel cells [16]. Merkel cells have been shown to derive from epidermal progenitors during development and are replenished during adulthood by epidermal stem cells. The Merkel cell lineage is dependent on the Atoh1/Math1 transcription factor [16], as its deletion results in loss of Merkel cells.
Because MCCs have been shown to express B-cell markers, including PAX-5 and TdT, some authors have suggested they arise from primitive B-cells [17]. In a series of studies, ~ 90% (128/143) MCCs express PAX-5 [18,19,20,21,22], whereas ~ 65% (117/181) express TdT [19, 21,22,23,24,25,26]. Effectively all lesions tested showed variable expression of immunoglobulins. Further, a subset of MCCs additionally showed monoclonal immunoglobulin gene rearrangement, although this was not demonstrated in the tumor cells themselves [22]. Together, these findings implicated the cell of origin of MCCs to be an early B cell. However, these findings may also represent aberrant expression of B-cell markers as other malignancies (including small cell carcinoma of the lung) also express PAX-5 and TdT at variable frequencies. Finally, when considering the potential cell of origin for MCC, it is possible that different MCCs arise from distinct subsets of progenitor cells [27,28,29].
Merkel Cell Carcinoma Polyomavirus
The frequent association between MCC and immunosuppression prompted the search to identify a relationship to an underlying pathogen. RNA sequencing from MCC samples identified sequences of a novel human polyomavirus, named Merkel Cell Polyomavirus (MCPyV). Subsequent testing of 10 MCCs revealed MCPyV in 8/10 (80%) of tumors, whereas 10/84 control tissues (11%) also tested positive for MCPyV DNA, but most of those showed comparatively lower copy numbers. Further analysis also revealed that MCPyV DNA integration occurred at a single genomic site in the majority of neoplastic cells from any given tumor, suggesting that infection and integration of MCPyV is an early event in Merkel cell carcinogenesis [8]. Depending on the patient population, MCPyV can be detected in ~ 60–80% of MCC by either polymerase chain reaction (PCR) or immunohistochemical detection of MCPyV T-antigen (see below).
The mechanisms by which MCPyV transforms cells are numerous, but rely largely on 2 MCPyV-encoded proteins: large T-antigen (LT-ag) and small T-antigen (ST-ag) [30]. The main function of LT-ag is to bind to and inactivate the retinoblastoma (RB) protein. RB serves as a critical tumor suppressor that functions normally to prevent cellular entry into S-phase by repressing E2F transcriptional activation of cyclin E [31]. Of note, tumor specific alterations/polymorphisms in the LT-ag sequence result in consistent truncation of C-terminal elements in LT-ag, which contains elements that counteract pathways leading to cellular proliferation and is necessary for MCC development [30]. ST-ag has several critical oncogenic functions. The first involves interfering with the function of FBXW7 protein, a key component of the ubiquitin ligase complex SCFFbxw7. In general, the SCF ubiquitin ligase complex attaches ubiquitin moieties to target proteins. Ubiquitination, in turn, designates those target proteins for proteolytic destruction. FBXW7 is one of a large family of proteins that provide the SCF complex with target protein specificity. Thus, ST-ag interaction with FBXW7 results in the stabilization of SCFFBXW7 targets, including LT-ag (itself a target of SCFFBXW7), c-Myc and cyclin E. Another critical function of ST-ag is to increase cellular levels of phosphorylated (and inactivated) 4E-BP1, a eukaryotic translational initiation factor, which results in widespread dysregulation of translation. In addition, ST-ag drives the concomitant expression of genes involved in glycolysis—a key requirement for rapidly dividing MCC cells [29, 32].
Detection of Merkel Cell Carcinoma Polyomavirus in Tissue
The first study to detect MCPyV in MCC demonstrated a relative frequency of 80% of MCCs to harbor MCPyV [8]. Subsequent studies (mostly relying on PCR based amplification of MCPyV sequences) have demonstrated a frequency that ranges from 40–100% in MCC [23, 33,34,35,36,37,38,39,40,41,42,43,44]. The range in sensitivity of detection reflects a myriad of factors, including geographical differences in MCPyV prevalence (lower in Australian populations compared to North American) and technical factors (tissue substrate [formalin fixed paraffin embedded versus fresh frozen], efficacy of primers used and length of DNA template to be amplified). MCPyV has additionally been detected at varying frequencies and in other cutaneous malignancies [45] (including SCCs and BCCs), in normal skin, and in hematolymphoid cells [38], although most studies agree that detection of MCPyV is a relatively specific marker of MCC [46].
The development of a monoclonal antibody against a specific region of the T-antigen—unique to the MCPyV (the CM2B4 clone)—enabled the detection and visualization of MCPyV in situ. In general, CM2B4 detects nuclear T-antigen expression with variable cytoplasmic positivity. The reported sensitivity for immunohistochemical detection using CM2B4 ranges from 39 to 90% [38, 41, 44, 47,48,49] and the sensitivity of immunohistochemistry (IHC) depends in most instances on the extent of viral copy numbers detected in the tissue [44]. Studies in which PCR and IHC detection of MCPyV in tissue are compared to one another reveal generally good agreement between the two, with PCR being consistently more sensitive than IHC. In cases where both approaches could be applied, PCR detected MCPyV in 76% (85/112) of MCCs, whereas IHC (using CM2B4 clone) detected MCPyV T-antigen in 56% (63/112) of tumors [38, 41, 44, 47,48,49].
Diagnosis of MCC: Clinical and Histopathologic Features
Merkel cell carcinoma typically presents as a rapidly growing, violaceous firm nodule on the skin that may be ulcerated. MCC exhibits a variable distribution (intraepidermal, dermal or subcutaneous) and pattern of growth in the skin. At low power, MCC may grow as an expansile, well-circumscribed nodule or as an infiltrative tumor (Fig. 1). Typically, the tumor effaces the dermal architecture and adjacent structures as it evolves with variable extension into the subcutis. The cells of MCC may be arranged as sheets, nests, trabeculae or as variable admixtures of these patterns. Although MCC is typically an intradermal tumor, intraepidermal MCC may occasionally occur—either exclusively (i.e. as MCC in situ with variable intraepidermal pagetoid spread) or in association with invasive MCC [50].

Merkel cell carcinoma a Scanning magnification reveals a diffuse proliferation of small round blue cells effacing the dermis (H&E, ×20). b The tumor consists of sheets of cells and admixed lymphocytes (H&E, ×100). c The tumor cells are tightly packed with numerous mitotic figures (solid arrowheads) and apoptotic bodies (open arrowheads). The tumor cells exhibit scant pale eosinophilic cytoplasm and enlarged oval-irregular nuclei with coarse granular chromatin (H&E, ×400; inset: H&E, ×600)
The cells comprising MCC generally exhibit scant pale eosinophilic cytoplasm and enlarged oval-irregular nuclei (often with molding among adjacent nuclei) and with finely granular (“salt and pepper”) chromatin and indistinct nucleoli. Typically, mitotic figures and apoptotic bodies are abundant. Areas of geographic necrosis may also be seen in some cases. MCC may also exhibit divergent differentiation within the tumor, notably with focal areas of squamous, sarcomatoid and rarely eccrine differentiation [51,52,53,54,55,56]. In addition, MCC may arise in association with other distinctive malignancies, most commonly cutaneous squamous cell carcinoma, which may be either in situ or invasive [53, 56]. The difference between ‘divergent differentiation’ and a ‘separate associated malignancy’ typically rests on the extent to which the second component grows distinctly from the MCC.
Diagnosis of MCC: Immunohistochemical Features
In general, MCC are positive with antibodies directed against cytokeratins. The most specific and highly sensitive (> 90%) marker of MCC is cytokeratin 20 (CK20), which is expressed in a ‘perinuclear dot-like’ pattern (Fig. 2). Additionally, cytokeratin cocktails (Cam5.2, cytokeratin AE1/AE3) are also sensitive markers and may similarly manifest with perinuclear dot-like positivity. Reflecting its neuroendocrine phenotype, MCC virtually always expresses at least one neuroendocrine marker, including CD56, chromogranin, synaptophysin, and neurofilament, and these may also exhibit a perinuclear dot-like or cytoplasmic pattern of positivity. Finally, MCPyV may be detected in ~ 70–80% of MCCs—either by immunohistochemical detection of MCPyV large T-antigen expression in the nucleus and/or cytoplasm of tumor cells.

Immunohistochemical profile of Merkel cell carcinoma Merkel cell carcinoma may show variable perinuclear dot-like positivity for a Cam 5.2 (×400), b CK20 (×400), c Synaptophysin (also shows diffuse cytoplasmic; ×400), and d Chromogranin (×400). The tumor cells are negative for TTF-1 (e; ×400) and LCA (f; ×400)
The immunohistochemical evaluation of a tumor presumed to be MCC generally includes antibodies for CK20, neuroendocrine markers (synaptophysin and chromogranin) as well as pertinent negative epitopes, including melanocytic markers (some combination of SOX-10, HMB-45, tyrosinase and MART-1), lymphoid markers (CD45, CD3, CD20) and TTF-1 to exclude cutaneous metastasis. The relative utility of these different antibodies will be discussed in the context of the various differential diagnostic considerations below.
Differential Diagnosis of MCC
Basal Cell Carcinoma
In most cases, the morphologic difference between basal cell carcinoma (BCC) and MCC is readily apparent by evaluation of H&E stained tissue sections. Like MCC, BCC consists of variably sized and shaped islands of basaloid cells. However, whereas MCC shows characteristic ‘neuroendocrine’ (speckled or finely granular) chromatin, BCC shows hyperchromatic nuclei with smooth to coarse chromatin. BCC often exhibits a characteristic palisading arrangement of the basal cells around the periphery of these islands and typically has an associated mucinous stroma. Further, between the mucinous stroma and the basaloid cells, there is often a cleft or retraction artifact present.
In some cases, however, morphologic overlap between BCC and MCC may be apparent. Ball and Tanhuanco-Kho [57] described 30 such cases where MCCs variably exhibited mucinous stroma (93%), stromal retraction (90%), and peripheral palisading of tumor cells (27%). Immunohistochemical studies are useful in such cases to inform the diagnosis. In this regard, CK20 represents the most sensitive and specific marker, demonstrating positivity (usually in a peri-nuclear dot-like pattern) in 212/241 MCCs (88% sensitivity), whereas CK20 was reportedly negative in the tumor cells of all 72 cases of BCC tested to date (100% specificity) [25, 26, 58,59,60,61,62,63,64,65,66,67,68,69,70]. Additionally, McPyV T-antigen by IHC can only be detected in exceptionally rare cases of BCC (2/88; 2%); thus, the specificity of detecting MCPyV T-ag is high for MCC [45].
Melanoma
Melanoma can exhibit overlapping morphology with MCC. In particular, cases with an intraepithelial pattern of growth and prominent pagetoid scatter may mimic melanoma [50, 71, 72]. For such cases, immunohistochemical studies for melanocytic antigens are useful. S100 has only rarely been reported positive in MCC (3/146; 2%) [73,74,75], while other melanocytic antigens (MART-1/Melan-A; HMB-45; SOX-10; MiTF) offer additional support for the diagnosis of melanoma, although the prevalence of their expression in MCC has been largely understudied. The proclivity of melanoma to exhibit reactivity for cytokeratins poses additional caveats, although positivity for CK20 has not been reported in melanoma.
Lymphoma/Leukemia
As MCCs often grow as sheets of monotonous mononuclear cells, their differential diagnosis often also includes lymphoma. Immunohistochemical studies for MCC-specific antigens, including CK20, Cam5.2, chromogranin, and synaptophysin are generally negative in hematolymphoid proliferations. However, recognition that up to 70% of MCCs (72/103) express TdT [19, 22, 25, 26] and 94% MCCs (45/48) express the B-cell marker PAX-5 [19, 22] is a critical pitfall in the differential diagnosis of MCC and lymphoma. An additional possible pitfall is the co-expression of CD56 in MCC, which may also be expressed in cutaneous lymphomas, including extranodal NK/T-cell lymphoma and blastic plasmacytoid dendritic cell neoplasm. Application of a broad panel of antibodies is important when the differential diagnosis includes a neoplasm of hematolymphoid lineage, and it is important to recognize the MCC may arise in association with CLL.
Metastatic Small Cell Carcinoma of the Lung
An additional challenging differential diagnostic challenge in the skin is differentiating between primary cutaneous MCC and small cell lung carcinoma (SCLC) metastatic to the skin. Whereas CK20 is positive in 212/241 MCCs (~ 88%), [25, 26, 58,59,60,61,62,63,64,65,66,67,68,69,70] only 15/203 (~ 7%) of SCLC show CK20-positivity [25, 58, 59, 61, 64, 69]. Furthermore, MCC has only rarely been reported to express TTF-1 (2/172), [26, 64, 66,67,68,69, 76] whereas TTF-1 highlights ~ 80% of SCLC (115/145) [25, 64, 66, 69, 76]. Additional informative markers include MCPyV T-antigen. Of note, B-cell markers PAX-5 (55%; 22/40) and TdT (28%; 11/40) are expressed at variable frequencies in SCLC. Detection of MCPyV sequences by PCR has only rarely been reported in SCLC (2/62) [46, 77]. Thus far, the relative frequency of immunohistochemical detection of MCPyV T-antigen in SCLC has not been reported, but would be expected to be low. Other useful markers include MASH1, which reportedly exhibited nuclear positivity in 49/59 SCLC (83%) compared to 0/30 MCCs [76].
Staging MCC
Historically, as many as five different staging systems have been utilized to stratify risk and prognosis for patients with MCC [78]. As for most other solid cancers, staging and prognosis of MCC depend largely on the extent of disease burden. At presentation, 65% of patients present with local disease only, 26% of patients present with regional lymph node metastases, and 8% present with distant metastases [9]. Prognosis in these patients directly reflects the extent of their disease at presentation. Five year overall survival (OS) rates are 50.6% for those with disease localized to the primary site, 35.4% for those with regional lymph node metastases, and 13.5% for those with distant metastases [9]. These differences form the foundation for the currently used TNM based staging system.
When considering the primary tumor, the two most important variables that determine patient survival are (1) tumor size and (2) the extent of anatomic invasion. The cut-offs for these are applied as follows: pT1 (≤ 2 cm), pT2 (> 2 cm, but ≤ 5 cm), pT3 (> 5 cm) and pT4 (primary tumor invades the underlying fascia, cartilage, muscle or bone). Five year OS rates accurately and robustly reflect categorizing primary MCC according to these criteria: 55.8% (pT1); 41.1% (pT2/pT3) and 31.8% (pT4) [9]. A number of additional histopathologic features of the primary tumor have also been studied and shown to correlate with patient survival and are therefore also reported in pathologic descriptions of the primary tumor. In a study of 156 patients with MCC, features of the primary tumor that associated with patient survival in univariate analyses included tumor thickness, tumor size, deepest anatomic level of involvement, tumor growth pattern, presence of lymphovascular invasion (LVI), presence of tumor infiltrating lymphocytes and presence of solar elastosis. In multivariate models, only stage, tumor thickness, tumor growth pattern and LVI independently associated with patient survival. When patients with negative lymph nodes (confirmed by pathologic evaluation) were considered in isolation, histopathologic features that associated with survival included the deepest anatomic compartment of involvement, growth pattern of the tumor, and tumor associated lymphocytic infiltrate [2].
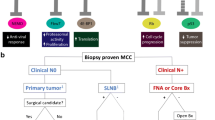
Regional metastases in MCC are described first according to whether the lymph node disease (or lack thereof) was determined by pathologic or clinical evaluation of the lymph node basin. In general, outcome is worse based on the extent of regional nodal disease burden. Some patients (most often due to other comorbidities) are only staged by clinical modalities (imaging studies and/or physical exam). Those with clinically evident lymph node metastases have the worst prognosis. Patients with clinically negative lymph nodes may either have clinically occult metastases (i.e. microscopic metastases requiring pathologic confirmation but which evade clinical detection) or pathologically confirmed negative lymph nodes. Therefore, patients with clinically negative lymph nodes collectively have an intermediate prognosis. Among patients who do undergo pathologic evaluation of their regional lymph nodes, those with pathologically confirmed lymph node negative disease have the best prognosis, while survival for patients with pathologically confirmed lymph node positive (i.e. clinically occult) disease is slightly worse. Given these findings, sentinel lymph node biopsy is recommended for staging of patients with MCC who are “…reasonably healthy…” [9]. In general, both morphologic assessment together with immunohistochemical studies are recommended to maximize sensitivity in the appraisal of SLNs in MCC [79]. An additional important variable captured in the updated AJCC staging system is the distinction of patients with regional lymph node disease without a known primary MCC (Stage IIIA) from patients with clinically evident regional lymph node disease with a known primary MCC (Stage IIIB). A number of studies have shown improved prognosis for the former compared to the latter set of patients, although the reasons underlying this difference remain unclear [80,81,82].
Prognostic Biomarkers in Merkel Cell Carcinomas
In order to improve risk modeling in MCC and to improve stratification of patient outcomes, a number of studies have explored various biomarkers as surrogates of patient survival. High levels of expression of the receptor tyrosine kinase CKIT in primary MCC showed a trend towards worse survival compared to patients with low CKIT expression (p = 0.07); however, activating mutations in CKIT have not been identified in MCC [83, 84]. Additional biomarkers previously explored in MCC include: nuclear survivin expression [85]; activating mutations in PIK3CA, a key kinase in the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) oncogenic pathway MCC [86]; expression of hedgehog pathway molecules [87, 88], although hedgehog inhibitors have not shown efficacy against MCC [89]; and markers of cell cycle and cell proliferation [90, 91]. In general, MCPyV + MCCs have been shown to have a better prognosis compared to MCPyV- MCCs [39, 86, 92, 93]—both when primary and metastatic lesions are tested [94], although not all studies agree [95, 96].
p63 as a Prognostic Marker in MCC
However, probably the best studied biomarkers predictive of patient outcome in MCC is p63 [93, 97,98,99]. In a series of 47 primary MCCs, p63 positivity (25/47; 53%) associated with significantly worse survival compared to patients whose tumors lack p63 expression (22/47; 47%; p < 0.0001) [97]. Positivity for p63 does not appear to correlate with squamous differentiation in the tumor cells. The same authors explored p63 in a larger cohort of 70 patients [98] and found: (1) patients with p63-positive MCC had a significantly reduced OS and disease-free survival compared to patients with p63-negative MCCs; (2) in multivariate analyses, only p63 and disease stage were independent prognostic indicators; and (3) among patients presenting with early stage MCC (stage I–II), patients whose tumors were p63-positive exhibited worse survival compared to those with p63-negative tumors (p < 0.0001) [98]. However, not all studies have confirmed this, as Higaki-Mori et al. showed no differences in survival in their cohort of MCC patients according to p63 expression [93]. In the largest single institution study to date, [100] Stetsenko et al. showed that among 128 patients with MCC that p63-positivity (together with stage) was indeed an independent predictor of adverse MCC-specific survival. However, when patients were grouped according to their stage at presentation, p63 expression did not distinguish patient outcomes within the distinct stage groups, and p63 expression itself did not appear to differ among the clinical stage groups. Thus, to the extent that stage information is known at the time of diagnosis, the prognostic utility of a standardized p63 assessment remains controversial [100].
The Immune System as a Biomarker in MCC
The relationship between the integrity of the immune system and the propensity to develop MCC is well established [3, 13,14,15]. The impact of chronic immune suppression on MCC patient survival was demonstrated in a landmark study from Paulson et al. [101] who showed that (1) among 471 patients with MCC those with immune suppression (n = 41; 40% at 3 years) showed shorter MCC-specific survival compared to immune competent patients (n = 430; 74% at 3 years) and (2) immune status was a stage-independent predictor of MCC specific survival as immune suppressed patients had reduced MCC specific survival compared to immune competent patients regardless of whether they presented with localized disease, regional lymph node metastases or distant metastases.
The components of the immune system driving prognosis was first investigated by Paulson et al. [102] who performed gene expression studies on a series of 35 MCCs and showed that CD8 + T-cell associated gene expression (in particular, CD8A and granzyme) was highest among patients with better MCC outcomes. High intratumoral CD8 + T-cell infiltration was independently associated with improved outcomes in univariate and multivariate analyses in a validation cohort of 146 primary and metastatic (nodal and distant) MCCs [102]. An assessment of the immune infiltrates in 116 MCCs revealed that higher densities of tumor-associated CD3 + T-cells correlated with improved patient survival [103]. Feldmeyer et al. [104] applied automated image analysis and showed that the density of CD3 + and CD8 + T-cells at the tumor periphery robustly associated with OS in a series of 62 primary MCCs, and higher densities of CD8 + T-cell along the tumor periphery further correlated with longer DSS. The impact of the T-cell infiltrate was most pronounced among MCPyV + but not MCPyV- MCC [104]. Together, these findings establish the tumor-associated T-cell infiltrate as a powerful prognostic biomarker in MCC.
Mutational Signature of MCC Depends on MCPyV Status
Wong et al. [105] subjected 32 MCCs (including 13 MCPyV + and 21 MCPyV- tumors or cell lines) and Harms et al. [106] subjected 16 MCCs (including 7 MCPyV + and 9 MCPyV- tumors) to DNA sequencing to determine differences and/or similarities between the mutational signature of MCPyV + and MCPyV- MCCs (Fig. 3). Several fundamental observations emerged from these seminal studies. First, MCPyV- MCC carry a significantly higher mutational burden compared to MCPyV + MCC. Second, the vast majority of those mutations are UV-signature mutations. Third, the most common mutations observed in MCPyV- MCCs included mutations in tumor suppressor genes including TP53, RB, and NOTCH family members. Together, these studies establish a dichotomous molecular genetic paradigm for MCC development: MCPyV- tumors driven by the progressive accumulation of UV-induced somatically acquired mutations and MCPyV + tumors driven by infection and integration of the MCPyV and expression of oncogenic LT-ag and ST-ag proteins [29, 105, 106]. It is worth emphasizing that in geographic regions with lower UV exposure, most MCCs are driven by MCPyV. In contrast, countries with high UV exposure have a higher incidence of MCPyV-negative MCCs, which are typified by a high burden of UV-induced DNA mutations [107]. To the extent that combined SCC/Merkel cell carcinomas are negative for MCPyV [41, 108,109,110], their mutational background generally resembles histopathologically ‘pure’ MCPyV-MCC [111]. Furthermore, cytokeratin 20-negative MCCs exhibit mutational profiles that ultimately reflect their MCPyV status (similar to the above studies): namely, an enrichment in UV-signature mutations impacting tumor suppressors (TP53, RB) and oncogenes (PIK3CA) in MCPyV- compared to MCPyV + tumors. In addition, CK20-negative MCCs additionally harbor mutations in TET2, APC, and BAP1—not previously described in CK20-positive MCCs [112].

Models for Merkel cell carcinomagenesis. Left: Merkel cell carcinoma polyomavirus (MCPyV) infection and expression of long T-antigen and short T-antigen abrogate RB functions directly and interfere with TP53-dependent pathways. Immunohistochemical studies for MCPyV T-antigen highlight T-antigen in the tumor cells (×100). Right: In MCPyV-negative MCCs, the accumulation of UV-induced mutations (most commonly affecting TP53 and RB) contribute to tumor development. Immunohistochemical studies for MCPyV T-antigen are negative in the tumor cells (×100)
These observations are further supported by analyses of the MCC transcriptome [113]. Harms et al. applied DNA microarrays to determine the gene expression profiles of 30 MCC (including 12 MCPyV + and 14 MCPyV- tumors) from 27 patients and showed (1) distinct patterns of gene expression in MCC compared to other primary cutaneous carcinomas and (2) distinct patterns of gene expression in MCPyV + compared to MCPyV- MCCs. In particular, MCPyV- MCCs showed upregulation of Notch and receptor tyrosine kinase signaling pathways compared to MCPyV + MCCs [113].
Therapies for Merkel Cell Carcinoma
Surgical excision of the primary tumor remains the mainstay of treatment for patients with local/regional MCC [114]. Additional front line management strategies depend on the presence of absence of clinically detectable lymph node disease. For patients with clinically detectable regional lymph node metastases, options include completion lymphadenectomy and/or adjuvant radiation therapy. For patients without clinically evident regional lymph node disease, sentinel lymph node (SLN) biopsy (SLNB) performed at the time of surgical excision of the primary MCC is recommended for staging and to direct subsequent management. Patients with negative SLN typically receive radiation to the primary site and the regional lymph node basin. Patients with positive SLN typically proceed to completion lymphadenectomy and/or radiation therapy [27, 29], as MCC is responsive to radiation therapy. The benefit of radiation therapy in early stage patients was established in a study of 4843 patients with localized MCC (stage I/II disease). For those patients, surgical excision followed by adjuvant radiation therapy independently correlated with improved OS compared to surgery alone, whereas in patients with regional nodal metastases (n = 2065), adjuvant radiation therapy or chemotherapy did not correlate with improved OS [115]. Radiation therapy is also reserved for treatment of patients who are not good surgical candidates or as a palliative measure in patients with inoperable tumors [116].
Patients with systemic involvement by MCC historically have had relatively few efficacious options. Although MCC is sensitive to many chemotherapeutic agents including varying combinations of cisplatin, carboplatin, etoposide, taxols (including paclitaxel/docetaxel) and anthracylcines (including doxorubicin), responses have not been shown to be durable, and a clear survival benefit has not been demonstrated [27, 29]. Most of these are applied in MCC based on its morphologic similarity to small cell lung carcinoma (and the susceptibility of the latter to these agents). Based on mutational studies, somatically acquired mutations occasionally activate the phosphatidylinositol-3 kinase (PI3K) pathway, [105, 106] thus indicating the potential therapeutic utility of PI3K pathway inhibitors in MCC [28].
The abundance of neoantigens in MCC (either in the form of virally encoded proteins in MCPyV-positive cases or in the form of numerous UV-driven mutations in MCPyV-negative tumors) [27, 29, 105, 106] the increased susceptibility to develop MCC in immunosuppressed populations and the close relationship between the density and composition of the tumor associated immune infiltrate and patient survival together strongly implicate the susceptibility of MCC to immune checkpoint blockade therapy. In the first clinical trial, 26 previously untreated patients with advanced MCC were given the PD-1 inhibitor pembrolizumab [117]. The objective response rate was 56%, including 4 patients who had a complete response and 10 who had a partial response. In a second trial, 88 patients with stage IV chemotherapy-refractory MCC (i.e. they had failed prior chemotherapy) were given the PD-L1 inhibitor avelumab with an objective response rate of 31.8% (28 of 88 patients—including 8 complete responders and 20 partial responders) [118]. Given the efficacy of avelumab in chemotherapy-resistant metastatic MCC resulted in its FDA approval for MCC. Additional reports of isolated patients with MCC who had clinical responses to checkpoint blockade have been described [119, 120]. Of note, thus far, responses to immune checkpoint blockade were not associated with either MCPyV status or PD-L1 expression in the tumor cells, and there is a critical unmet need to identify biomarkers predictive of response in MCC.
Summary and Future Directions
Merkel cell carcinoma is an aggressive, high grade neuroendocrine carcinoma of the skin that continues to present formidable diagnostic and clinical challenges. Recognition of its distinctive histopathologic and immunophenotypic features facilitates accurate diagnosis. Refinements to the staging system have improved patient risk stratification. The identification of molecular-genetic drivers has greatly improved understanding of pathogenesis. Together with the recognition of the intricate relationship between MCC and the host immune system, these observations have culminated in potentially efficacious therapies, and the identification of informative biomarkers predictive of response will facilitate tailored management strategies accordingly.
References
Albores-Saavedra J, Batich K, Chable-Montero F, Sagy N, Schwartz AM, Henson DE. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol. 2010;37(1):20 – 7.
Andea AA, Coit DG, Amin B, Busam KJ. Merkel cell carcinoma: histologic features and prognosis. Cancer 2008;113(9):2549–58.
Engels EA, Frisch M, Goedert JJ, Biggar RJ, Miller RW. Merkel cell carcinoma and HIV infection. Lancet 2002;359(9305):497–8.
Fitzgerald TL, Dennis S, Kachare SD, Vohra NA, Wong JH, Zervos EE. Dramatic Increase in the Incidence and Mortality from Merkel Cell Carcinoma in the United States. Am Surg 2015;81(8):802–6.
Guler-Nizam E, Leiter U, Metzler G, Breuninger H, Garbe C, Eigentler TK. Clinical course and prognostic factors of Merkel cell carcinoma of the skin. Br J Dermatol. 2009;161(1):90–4.
Tarantola TI, Vallow LA, Halyard MY, et al. Prognostic factors in Merkel cell carcinoma: analysis of 240 cases. J Am Acad Dermatol. 2013;68(3):425–32.
Warner CL, Cockerell CJ. The new seventh edition American Joint Committee on Cancer staging of cutaneous non-melanoma skin cancer: a critical review. Am J Clin Dermatol. 2011;12(3):147–54.
Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008;319(5866):1096–100.
Harms KL, Healy MA, Nghiem P, et al. Analysis of prognostic factors from 9387 Merkel cell carcinoma cases forms the basis for the new 8th edition AJCC staging system. Ann Surg Oncol. 2016;23(11):3564–71.
Hodgson NC. Merkel cell carcinoma: changing incidence trends. J Surg Oncol. 2005;89(1):1–4.
Paulson KG, Park SY, Vandeven NA, et al. Merkel cell carcinoma: current United States incidence and projected Increases Based on changing demographics. J Am Acad Dermatol. 2017. https://doi.org/10.1016/j.jaad.2017.10.028
Tadmor T, Liphshitz I, Aviv A, Landgren O, Barchana M, Polliack A. Increased incidence of chronic lymphocytic leukaemia and lymphomas in patients with Merkel cell carcinoma - a population based study of 335 cases with neuroendocrine skin tumour. Br J Haematol. 2012;157(4):457–62.
Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;58(3):375–81.
Penn I, First MR. Merkel’s cell carcinoma in organ recipients: report of 41 cases. Transplantation 1999;68(11):1717–21.
Hemminki K, Liu X, Ji J, Sundquist J, Sundquist K. Kaposi sarcoma and Merkel cell carcinoma after autoimmune disease. Int J Cancer 2012;131(3):E326-E8.
Van Keymeulen A, Mascre G, Youseff KK, et al. Epidermal progenitors give rise to Merkel cells during embryonic development and adult homeostasis. J Cell Biol. 2009;187(1):91–100.
Sauer CM, Haugg AM, Chteinberg E, et al. Reviewing the current evidence supporting early B-cells as the cellular origin of Merkel cell carcinoma. Crit Rev Oncol/Hematol. 2017;116:99–105.
Dong HY, Liu W, Cohen P, Mahle CE, Zhang W. B-cell specific activation protein encoded by the PAX-5 gene is commonly expressed in merkel cell carcinoma and small cell carcinomas. Am J Surg Pathol. 2005;29(5):687–92.
Kolhe R, Reid MD, Lee JR, Cohen C, Ramalingam P. Immunohistochemical expression of PAX5 and TdT by Merkel cell carcinoma and pulmonary small cell carcinoma: a potential diagnostic pitfall but useful discriminatory marker. Int J Clin Exp Pathol. 2013;6(2):142–7.
Mhawech-Fauceglia P, Saxena R, Zhang S, et al. Pax-5 immunoexpression in various types of benign and malignant tumours: a high-throughput tissue microarray analysis. J Clin Pathol. 2007;60(6):709 – 14.
Murakami I, Takata K, Matsushita M, et al. Immunoglobulin expressions are only associated with MCPyV-positive Merkel cell carcinomas but not with MCPyV-negative ones: comparison of prognosis. Am J Surg Pathol. 2014;38(12):1627–35.
Zur Hausen A, Rennspiess D, Winnepenninckx V, Speel EJ, Kurz AK. Early B-cell differentiation in Merkel cell carcinomas: clues to cellular ancestry. Cancer Res. 2013;73(16):4982–7.
Bhatia K, Goedert JJ, Modali R, Preiss L, Ayers LW. Merkel cell carcinoma subgroups by Merkel cell polyomavirus DNA relative abundance and oncogene expression. Int J Cancer 2010;126(9):2240–6.
Buresh CJ, Oliai BR, Miller RT. Reactivity with TdT in Merkel cell carcinoma: a potential diagnostic pitfall. Am J Clin Pathol. 2008;129(6):894–8.
Sidiropoulos M, Hanna W, Raphael SJ, Ghorab Z. Expression of TdT in Merkel cell carcinoma and small cell lung carcinoma. Am J Clin Pathol. 2011;135(6):831–8.
Sur M, AlArdati H, Ross C, Alowami S. TdT expression in Merkel cell carcinoma: potential diagnostic pitfall with blastic hematological malignancies and expanded immunohistochemical analysis. Mod Pathol. 2007;20(11):1113–20.
Schadendorf D, Lebbe C, Zur Hausen A, et al. Merkel cell carcinoma: epidemiology, prognosis, therapy and unmet medical needs. Eur J Cancer 2017;71:53–69.
Harms PW. Update on Merkel cell carcinoma. Clin Lab Med. 2017;37(3):485–501.
Becker JC, Stang A, DeCaprio JA, et al. Merkel cell carcinoma. Nat Rev Dis Primers. 2017;3: 170–77.
Wendzicki JA, Moore PS, Chang Y. Large T and small T antigens of Merkel cell polyomavirus. Curr Opin Virol. 2015;11:38–43.
Sherr CJ. Cancer cell cycles. Science 1996;274(5293):1672–7.
Berrios C, Padi M, Keibler MA, et al. Merkel cell polyomavirus small T antigen promotes pro-glycolytic metabolic perturbations required for transformation. PLoS Pathog. 2016;12(11):e1006020.
Kassem A, Schopflin A, Diaz C, et al. Frequent detection of Merkel cell polyomavirus in human Merkel cell carcinomas and identification of a unique deletion in the VP1 gene. Cancer Res. 2008;68(13):5009–13.
Becker JC, Houben R, Ugurel S, Trefzer U, Pfohler C, Schrama D. MC polyomavirus is frequently present in Merkel cell carcinoma of European patients. J Invest Dermatol. 2009;129(1):248–50.
Garneski KM, Warcola AH, Feng Q, Kiviat NB, Leonard JH, Nghiem P. Merkel cell polyomavirus is more frequently present in North American than Australian Merkel cell carcinoma tumors. J Invest Dermatol. 2009;129(1):246–8.
Duncavage EJ, Zehnbauer BA, Pfeifer JD. Prevalence of Merkel cell polyomavirus in Merkel cell carcinoma. Mod Pathol. 2009;22(4):516 – 21.
Sastre-Garau X, Peter M, Avril MF, et al. Merkel cell carcinoma of the skin: pathological and molecular evidence for a causative role of MCV in oncogenesis. J Pathol. 2009;218(1):48–56.
Shuda M, Arora R, Kwun HJ, et al. Human Merkel cell polyomavirus infection I. MCV T antigen expression in Merkel cell carcinoma, lymphoid tissues and lymphoid tumors. Int J Cancer 2009;125(6):1243–9.
Sihto H, Kukko H, Koljonen V, Sankila R, Bohling T, Joensuu H. Clinical factors associated with Merkel cell polyomavirus infection in Merkel cell carcinoma. J Natl Cancer Inst. 2009;101(13):938–45.
Loyo M, Guerrero-Preston R, Brait M, et al. Quantitative detection of Merkel cell virus in human tissues and possible mode of transmission. Int J Cancer 2010;126(12):2991–6.
Busam KJ, Jungbluth AA, Rekthman N, et al. Merkel cell polyomavirus expression in merkel cell carcinomas and its absence in combined tumors and pulmonary neuroendocrine carcinomas. Am J Surg Pathol. 2009;33(9):1378–85.
Andres C, Belloni B, Puchta U, Sander CA, Flaig MJ. Prevalence of MCPyV in Merkel cell carcinoma and non-MCC tumors. J Cutan Pathol. 2010;37(1):28–34.
Houben R, Schrama D, Alb M, et al. Comparable expression and phosphorylation of the retinoblastoma protein in Merkel cell polyoma virus-positive and negative Merkel cell carcinoma. Int J Cancer 2010;126(3):796–8.
Bhatia K, Goedert JJ, Modali R, Preiss L, Ayers LW. Immunological detection of viral large T antigen identifies a subset of Merkel cell carcinoma tumors with higher viral abundance and better clinical outcome. Int J Cancer 2010;127(6):1493–6.
Mertz KD, Paasinen A, Arnold A, et al. Merkel cell polyomavirus large T antigen is detected in rare cases of nonmelanoma skin cancer. J Cutan Pathol. 2013;40(6):543–9.
Duncavage EJ, Le BM, Wang D, Pfeifer JD. Merkel cell polyomavirus: a specific marker for Merkel cell carcinoma in histologically similar tumors. Am J Surg Pathol. 2009;33(12):1771–7.
Jung HS, Choi YL, Choi JS, et al. Detection of Merkel cell polyomavirus in Merkel cell carcinomas and small cell carcinomas by PCR and immunohistochemistry. Histol Histopathol. 2011;26(10):1231–41.
Leitz M, Stieler K, Grundhoff A, Moll I, Brandner JM, Fischer N. Merkel cell polyomavirus detection in Merkel cell cancer tumors in Northern Germany using PCR and protein expression. J Med Virol. 2014;86(10):1813–9.
Leroux-Kozal V, Leveque N, Brodard V, et al. Merkel cell carcinoma: histopathologic and prognostic features according to the immunohistochemical expression of Merkel cell polyomavirus large T antigen correlated with viral load. Hum Pathol. 2015;46(3):443–53.
D’Agostino M, Cinelli C, Willard R, Hofmann J, Jellinek N, Robinson-Bostom L. Epidermotropic Merkel cell carcinoma: a case series with histopathologic examination. J Am Acad Dermatol. 2010;62(3):463–8.
Gomez LG, DiMaio S, Silva EG, Mackay B. Association between neuroendocrine (Merkel cell) carcinoma and squamous carcinoma of the skin. Am J Surg Pathol. 1983;7(2):171–7.
Cerroni L, Kerl H. Primary cutaneous neuroendocrine (Merkel cell) carcinoma in association with squamous- and basal-cell carcinoma. Am J Dermatopathol. 1997;19(6):610–3.
Iacocca MV, Abernethy JL, Stefanato CM, Allan AE, Bhawan J. Mixed Merkel cell carcinoma and squamous cell carcinoma of the skin. J Am Acad Dermatol. 1998;39(5 Pt 2):882–7.
Boutilier R, Desormeau L, Cragg F, Roberts P, Walsh N. Merkel cell carcinoma: squamous and atypical fibroxanthoma-like differentiation in successive local tumor recurrences. Am J Dermatopathol. 2001;23(1):46 – 9.
Hwang JH, Alanen K, Dabbs KD, Danyluk J, Silverman S. Merkel cell carcinoma with squamous and sarcomatous differentiation. J Cutan Pathol. 2008;35(10):955–9.
Sirikanjanapong S, Melamed J, Patel RR. Intraepidermal and dermal Merkel cell carcinoma with squamous cell carcinoma in situ: a case report with review of literature. J Cutan Pathol. 2010;37(8):881–5.
Ball NJ, Tanhuanco-Kho G. Merkel cell carcinoma frequently shows histologic features of basal cell carcinoma: a study of 30 cases. J Cutan Pathol. 2007;34(8):612–9.
Moll R, Lowe A, Laufer J, Franke WW. Cytokeratin 20 in human carcinomas. A new histodiagnostic marker detected by monoclonal antibodies. Am J Pathol. 1992;140(2):427–47.
Chan JK, Suster S, Wenig BM, Tsang WY, Chan JB, Lau AL. Cytokeratin 20 immunoreactivity distinguishes Merkel cell (primary cutaneous neuroendocrine) carcinomas and salivary gland small cell carcinomas from small cell carcinomas of various sites. Am J Surg Pathol. 1997;21(2):226–34.
Schirren CG, Rutten A, Kaudewitz P, Diaz C, McClain S, Burgdorf WH. Trichoblastoma and basal cell carcinoma are neoplasms with follicular differentiation sharing the same profile of cytokeratin intermediate filaments. Am J Dermatopathol. 1997;19(4):341–50.
Scott MP, Helm KF. Cytokeratin 20: a marker for diagnosing Merkel cell carcinoma. Am J Dermatopathol. 1999;21(1):16–20.
McNiff JM, Eisen RN, Glusac EJ. Immunohistochemical comparison of cutaneous lymphadenoma, trichoblastoma, and basal cell carcinoma: support for classification of lymphadenoma as a variant of trichoblastoma. J Cutan Pathol. 1999;26(3):119 – 24.
Poniecka AW, Alexis JB. An immunohistochemical study of basal cell carcinoma and trichoepithelioma. Am J Dermatopathol. 1999;21(4):332–6.
Hanly AJ, Elgart GW, Jorda M, Smith J, Nadji M. Analysis of thyroid transcription factor-1 and cytokeratin 20 separates merkel cell carcinoma from small cell carcinoma of lung. J Cutan Pathol. 2000;27(3):118–20.
Nicholson SA, McDermott MB, Swanson PE, Wick MR. CD99 and cytokeratin-20 in small-cell and basaloid tumors of the skin. Appl Immunohistochem Mol Morphol. 2000;8(1):37–41.
Leech SN, Kolar AJ, Barrett PD, Sinclair SA, Leonard N. Merkel cell carcinoma can be distinguished from metastatic small cell carcinoma using antibodies to cytokeratin 20 and thyroid transcription factor 1. J Clin Pathol. 2001;54(9):727–9.
Yang DT, Holden JA, Florell SR. CD117, CK20, TTF-1, and DNA topoisomerase II-alpha antigen expression in small cell tumors. J Cutan Pathol. 2004;31(3):254 – 61.
Llombart B, Monteagudo C, Lopez-Guerrero JA, et al. Clinicopathological and immunohistochemical analysis of 20 cases of Merkel cell carcinoma in search of prognostic markers. Histopathology 2005;46(6):622–34.
Bobos M, Hytiroglou P, Kostopoulos I, Karkavelas G, Papadimitriou CS. Immunohistochemical distinction between merkel cell carcinoma and small cell carcinoma of the lung. Am J Dermatopathol. 2006;28(2):99–104.
Rekhi B, Kane SV, Jambhekar NA. Clinicopathological spectrum of a series of Merkel cell carcinomas diagnosed at a tertiary cancer referral center in India, with current concepts. Ann Diagn Pathol. 2015;19(5):341–6.
Hashimoto K, Lee MW, D’Annunzio DR, Balle MR, Narisawa Y. Pagetoid Merkel cell carcinoma: epidermal origin of the tumor. J Cutan Pathol. 1998;25(10):572–9.
Miraflor AP, LeBoit PE, Hirschman SA. Intraepidermal Merkel cell carcinoma with pagetoid Bowen’s disease. J Cutan Pathol. 2016;43(11):921–6.
Skelton HG, Smith KJ, Hitchcock CL, McCarthy WF, Lupton GP, Graham JH. Merkel cell carcinoma: analysis of clinical, histologic, and immunohistologic features of 132 cases with relation to survival. J Am Acad Dermatol. 1997;37(5 Pt 1):734–9.
Kontochristopoulos GJ, Stavropoulos PG, Krasagakis K, Goerdt S, Zouboulis CC. Differentiation between merkel cell carcinoma and malignant melanoma: an immunohistochemical study. Dermatology 2000;201(2):123–6.
Acebo E, Vidaurrazaga N, Varas C, Burgos-Bretones JJ, Diaz-Perez JL. Merkel cell carcinoma: a clinicopathological study of 11 cases. J Eur Acad Dermatol Venereol. 2005;19(5):546 – 51.
Ralston J, Chiriboga L, Nonaka D. MASH1: a useful marker in differentiating pulmonary small cell carcinoma from Merkel cell carcinoma. Mod Pathol. 2008;21(11):1357–62.
Andres C, Ihrler S, Puchta U, Flaig MJ. Merkel cell polyomavirus is prevalent in a subset of small cell lung cancer: a study of 31 patients. Thorax. 2009;64(11):1007–8.
Moshiri AS, Nghiem P. Milestones in the staging, classification, and biology of Merkel cell carcinoma. J Natl Compr Canc Netw. 2014;12(9):1255–62.
Su LD, Lowe L, Bradford CR, Yahanda AI, Johnson TM, Sondak VK. Immunostaining for cytokeratin 20 improves detection of micrometastatic Merkel cell carcinoma in sentinel lymph nodes. J Am Acad Dermatol. 2002;46(5):661–80.
Foote M, Veness M, Zarate D, Poulsen M. Merkel cell carcinoma: the prognostic implications of an occult primary in stage IIIB (nodal) disease. J Am Acad Dermatol. 2012;67(3):395–9.
Tarantola TI, Vallow LA, Halyard MY, et al. Unknown primary Merkel cell carcinoma: 23 new cases and a review. J Am Acad Dermatol. 2013;68(3):433–40.
Chen KT, Papavasiliou P, Edwards K, et al. A better prognosis for Merkel cell carcinoma of unknown primary origin. Am J Surg. 2013;206(5):752–7.
Andea AA, Patel R, Ponnazhagan S, et al. Merkel cell carcinoma: correlation of KIT expression with survival and evaluation of KIT gene mutational status. Hum Pathol. 2010;41(10):1405–12.
Swick BL, Srikantha R, Messingham KN. Specific analysis of KIT and PDGFR-alpha expression and mutational status in Merkel cell carcinoma. J Cutan Pathol. 2013;40(7):623–30.
Kim J, McNiff JM. Nuclear expression of survivin portends a poor prognosis in Merkel cell carcinoma. Mod Pathol. 2008;21(6):764–9.
Nardi V, Song Y, Santamaria-Barria JA, et al. Activation of PI3K signaling in Merkel cell carcinoma. Clin Cancer Res. 2012;18(5):1227–36.
Brunner M, Thurnher D, Pammer J, et al. Expression of hedgehog signaling molecules in Merkel cell carcinoma. Head Neck 2010;32(3):333–40.
Kuromi T, Matsushita M, Iwasaki T, et al. Association of expression of the hedgehog signal with Merkel cell polyomavirus infection and prognosis of Merkel cell carcinoma. Hum Pathol. 2017;69:8–14.
Carroll TM, Williams JS, Daily K, et al. Hedgehog signaling inhibitors fail to reduce Merkel cell carcinoma viability. J Invest Dermatol. 2017;137(5):1187–90.
Fernandez-Figueras MT, Puig L, Musulen E, et al. Prognostic significance of p27Kip1, p45Skp2 and Ki67 expression profiles in Merkel cell carcinoma, extracutaneous small cell carcinoma, and cutaneous squamous cell carcinoma. Histopathology 2005;46(6):614–21.
Henderson SA, Tetzlaff MT, Pattanaprichakul P, et al. Detection of mitotic figures and G2 + tumor nuclei with histone markers correlates with worse overall survival in patients with Merkel cell carcinoma. J Cutan Pathol. 2014;41(11):846 – 52.
Sihto H, Kukko H, Koljonen V, Sankila R, Bohling T, Joensuu H. Merkel cell polyomavirus infection, large T antigen, retinoblastoma protein and outcome in Merkel cell carcinoma. Clin Cancer Res. 2011;17(14):4806–13.
Higaki-Mori H, Kuwamoto S, Iwasaki T, et al. Association of Merkel cell polyomavirus infection with clinicopathological differences in Merkel cell carcinoma. Hum Pathol. 2012;43(12):2282–91.
Haymerle G, Janik S, Fochtmann A, et al. Expression of Merkelcell polyomavirus (MCPyV) large T-antigen in Merkel cell carcinoma lymph node metastases predicts poor outcome. PLoS ONE. 2017;12(8):e0180426.
Schrama D, Peitsch WK, Zapatka M, et al. Merkel cell polyomavirus status is not associated with clinical course of Merkel cell carcinoma. J Invest Dermatol. 2011;131(8):1631–8.
Hall BJ, Pincus LB, Yu SS, Oh DH, Wilson AR, McCalmont TH. Immunohistochemical prognostication of Merkel cell carcinoma: p63 expression but not polyomavirus status correlates with outcome. J Cutan Pathol. 2012;39(10):911–7.
Asioli S, Righi A, Volante M, Eusebi V, Bussolati G. p63 expression as a new prognostic marker in Merkel cell carcinoma. Cancer 2007;110(3):640–7.
Asioli S, Righi A, de Biase D, et al. Expression of p63 is the sole independent marker of aggressiveness in localised (stage I-II) Merkel cell carcinomas. Mod Pathol. 2011;24(11):1451–61.
Fleming KE, Ly TY, Pasternak S, Godlewski M, Doucette S, Walsh NM. Support for p63 expression as an adverse prognostic marker in Merkel cell carcinoma: report on a Canadian cohort. Hum Pathol. 2014;45(5):952 – 60.
Stetsenko GY, Malekirad J, Paulson KG, et al. p63 expression in Merkel cell carcinoma predicts poorer survival yet may have limited clinical utility. Am J Clin Pathol. 2013;140(6):838 – 44.
Paulson KG, Iyer JG, Blom A, et al. Systemic immune suppression predicts diminished Merkel cell carcinoma-specific survival independent of stage. J Invest Dermatol. 2013;133(3):642–6.
Paulson KG, Iyer JG, Tegeder AR, et al. Transcriptome-wide studies of merkel cell carcinoma and validation of intratumoral CD8 + lymphocyte invasion as an independent predictor of survival. J Clin Oncol. 2011;29(12):1539–46.
Sihto H, Bohling T, Kavola H, et al. Tumor infiltrating immune cells and outcome of Merkel cell carcinoma: a population-based study. Clin Cancer Res. 2012;18(10):2872–81.
Feldmeyer L, Hudgens CW, Ray-Lyons G, et al. Density, distribution, and composition of immune infiltrates correlate with survival in Merkel cell carcinoma. Clin Cancer Res. 2016;22(22):5553–63.
Wong SQ, Waldeck K, Vergara IA, et al. UV-Associated mutations underlie the etiology of MCV-negative Merkel cell carcinomas. Cancer Res. 2015;75(24):5228–34.
Harms PW, Vats P, Verhaegen ME, et al. The distinctive mutational spectra of polyomavirus-negative Merkel CELL carcinoma. Cancer Res. 2015;75(18):3720–7.
Bloom R, Amber KT, Nouri K. An increased risk of non-Hodgkin lymphoma and chronic lymphocytic leukemia in US patients with Merkel cell carcinoma versus Australian patients: a clinical clue to a different mechanism of pathogenesis? Australas J Dermatol. 2016;57(3):e114-6.
Paik JY, Hall G, Clarkson A, et al. Immunohistochemistry for Merkel cell polyomavirus is highly specific but not sensitive for the diagnosis of Merkel cell carcinoma in the Australian population. Hum Pathol. 2011;42(10):1385–90.
Martin B, Poblet E, Rios JJ, et al. Merkel cell carcinoma with divergent differentiation: histopathological and immunohistochemical study of 15 cases with PCR analysis for Merkel cell polyomavirus. Histopathology 2013;62(5):711 – 22.
Iwasaki T, Matsushita M, Kuwamoto S, et al. Usefulness of significant morphologic characteristics in distinguishing between Merkel cell polyomavirus-positive and Merkel cell polyomavirus-negative Merkel cell carcinomas. Hum Pathol. 2013;44(9):1912–7.
Pulitzer MP, Brannon AR, Berger MF, et al. Cutaneous squamous and neuroendocrine carcinoma: genetically and immunohistochemically different from Merkel cell carcinoma. Mod Pathol. 2015;28(8):1023–32.
Harms PW, Collie AM, Hovelson DH, et al. Next generation sequencing of Cytokeratin 20-negative Merkel cell carcinoma reveals ultraviolet-signature mutations and recurrent TP53 and RB1 inactivation. Mod Pathol. 2016;29(3):240–8.
Harms PW, Patel RM, Verhaegen ME, et al. Distinct gene expression profiles of viral- and paxnonviral-associated merkel cell carcinoma revealed by transcriptome analysis. J Invest Dermatol. 2013;133(4):936–45.
Bichakjian CK, National Comprehensive Cancer Network Guidelines Version 1.2017 Merkel cell carcinoma http://merkelcell.org/wp-content/uploads/2015/10/MccNccn.pdf (2017). Accessed 4 June 2017.
Bhatia S, Storer BE, Iyer JG, et al. Adjuvant radiation therapy and chemotherapy in Merkel cell carcinoma: survival analyses of 6908 cases from the national cancer data base. J Natl Cancer Inst. 2016;108(9).
Mortier L, Mirabel X, Fournier C, Piette F, Lartigau E. Radiotherapy alone for primary Merkel cell carcinoma. Arch Dermatol. 2003;139(12):1587–90.
Nghiem PT, Bhatia S, Lipson EJ, et al. PD-1 blockade with Pembrolizumab in advanced Merkel-Cell carcinoma. N Engl J Med. 2016;374(26):2542–52.
Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17(10):1374–85.
Winkler JK, Bender C, Kratochwil C, Enk A, Hassel JC. PD-1 blockade: a therapeutic option for treatment of metastatic Merkel cell carcinoma. Br J Dermatol. 2017;176(1):216–9.
Walocko FM, Scheier BY, Harms PW, Fecher LA, Lao CD. Metastatic Merkel cell carcinoma response to nivolumab. J Immunother Cancer 2016;4:79.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
MTT has received an honorarium from Myriad Genetics, Seattle Genetics and Novartis for Advisory Boards. MTT declares no relevant conflicts of interest to the material presented herein.
Ethical Approval
This article does not contain any studies with human participants performed by any of the authors. This article does not contain any studies with animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Tetzlaff, M.T., Nagarajan, P. Update on Merkel Cell Carcinoma. Head and Neck Pathol 12, 31–43 (2018). https://doi.org/10.1007/s12105-018-0898-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12105-018-0898-2