
Abstract
Purpose of Review
Dietary obesity is primarily attributed to an imbalance between food intake and energy expenditure. Adherence to lifestyle interventions reducing weight is typically low. As a result, obesity becomes a chronic state with increased co-morbidities such as insulin resistance and diabetes. We review the effects of brain insulin action and dopaminergic signal transmission on food intake, reward, and mood as well as potential modulations of these systems to counteract the obesity epidemic.
Recent Findings
Central insulin and dopamine action are interlinked and impact on food intake, reward, and mood. Brain insulin resistance causes hyperphagia, anxiety, and depressive-like behavior and compromises the dopaminergic system. Such effects can induce reduced compliance to medical treatment. Insulin receptor sensitization and dopamine receptor agonists show attenuation of obesity and improvement of mental health in rodents and humans.
Summary
Modulating brain insulin and dopamine signaling in obese patients can potentially improve therapeutic outcomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The world is facing a global obesity epidemic, with an estimated 1.9 billion people categorized as being overweight including 650 million obese people [1]. Obesity arises as the result of increased energy intake and decreased energy expenditure and greatly increases the risk of developing type 2 diabetes [2, 3]. Obesity and diabetes are characterized by insulin resistance, a condition where the body is unable to properly respond to insulin. Insulin resistance can be induced by multiple factors including glucotoxicity, lipotoxicity, low-grade inflammation or organelle stress. Importantly, insulin resistance impacts peripheral tissues as well as the brain. Food consumption is so far understood to be regulated by the homeostatic system, which apparently resides in the hypothalamus, and the reward system, which is thought to include the mesolimbic dopaminergic pathways from the ventral tegmental area to the nucleus accumbens and the prefrontal cortex [4]. The homeostatic system is regulated by peripheral hormones such as insulin and leptin, senses the current energy state of the body and controls food intake through hypothalamic neurons [5]. The reward system is regulated by physiological stimuli such as hunger [6], taste, cue-induced, and palatable food intake, thereby altering food liking and wanting [4, 7, 8]. Interestingly, homeostatic and reward systems interact and their dysregulation is linked to obesity [9, 10]. Conversely, the consumption of a diet enriched with long-chain saturated fatty acids impairs central insulin action and inhibits dopamine function in the brain [8, 11]. Dysregulation of both systems further promotes preference for high-calorie diets and results in hyperphagia, establishing a vicious cycle of over-eating (hyperphagia) that causes long-term obesity and the development of type 2 diabetes.
In addition, both systems play a crucial role in regulating emotional behavior. Thus, diabetes, insulin resistance, and reduced dopamine function are associated with behavioral abnormalities and mood disorders such as anxiety and depression. Depressive disorders, a severe category of mood disorders, are accompanied by lack of motivation and, in some cases, can lead to poor compliance to follow a therapy regime or even suicide [12, 13]. Obese and diabetic patients are prone to develop depressive disorders [14], whereas patients with depression have an increased prevalence for type 2 diabetes [15, 16]. This correlation is poorly understood but point to an interaction of insulin signaling and dopamine function. Here we will review the current understanding of this interaction. We will investigate for evidence that the dysregulation of both systems is responsible for the poor adherence for long-term weight loss after dietary interventions due to the inability to properly regulate the hedonic system and negatively impacts on mood and motivation.
Brain Insulin Signaling Affects Food Intake and Reward
The brain is an insulin-responsive organ and brain insulin action has a crucial effect on food intake and reward. Mechanistically, insulin binds to the insulin and IGF-1 receptor (IR and IGF-1R) causing the autophosphorylation of the receptors, followed by the recruitment of insulin receptor substrate (IRS) proteins and their subsequent phosphorylation. Phosphorylated IRS proteins act as a critical node activating the PI3-kinase-AKT and the MAP-kinase-ERK pathway [17]. On the one hand, the PI3-kinase-AKT pathway regulates neuronal protein content, autophagy, synaptic function, plasticity, and proliferation of neuronal progenitors via activation of downstream proteins such as mTOR complex 1 (mTORC1), glycogen synthase kinase 3β (GSK3β), or Forkhead box O (FoxO). On the other hand, activation of the MAP-kinase-ERK pathway controls mitochondrial function, proliferation, and differentiation [18, 19]. This regulation can be influenced by the activation of stress-activated protein kinases (SAPK) such as c-Jun kinase (JNK), p38 kinase, or IκB kinase (IKK) in the brain. The activities of these kinases are induced by cytokines, long-chain saturated fatty acids, or oxidative stress. They are elevated in type 2 diabetes, cause inhibitory serine phosphorylation of IRS proteins and inhibit the interaction of insulin signaling proteins which results in the abrogation of the insulin signal [18].
Insulin receptors are expressed throughout the brain including the hypothalamus which controls energy homeostasis and the striatum as part of the dopamine system, revealing overlapping expression patterns of the insulin and dopamine systems [20]. In the late 1970s Woods et al. have already demonstrated that insulin infusion into the brain of baboons reduced food intake, which has been further confirmed and refined in various other models. Insulin reduces food intake by regulating the activation of POMC and Agrp neurons in the hypothalamus engaging anorexigenic signals in the hypothalamus [5]. Thus, diabetic and insulin-resistant mice as well as mice with a knockout of the insulin receptor in the brain exhibit hyperphagia and diet-induced obesity [21,22,23]. But central insulin action affects not only the homeostatic but also the reward system. The reward or hedonic system is mainly controlled by dopamine signaling. Although it is assumed that hypothalamic mechanisms controlling food intake and energy expenditure are important in modulating energy balance, the explosive prevalence of dietary obesity clearly implicates non-homeostatic mechanisms as significant contributors. In the midbrain, food and drug reward is mediated by dopaminergic projections from the ventral tegmental area (VTA) to the nucleus accumbens (ventral striatum), known as the mesoaccumbens pathway, from the substantia nigra pars compacta (SNpc) to the dorsal striatum, known as the nigrostriatal pathway, and from the VTA to the medial prefrontal cortex, known as the mesocorticolimbic pathway. In light of dopamine signaling and food intake, it is important to mention that dopamine signaling impacts the food consumption in a region-specific manner [24]. Dopamine action in the ventrolateral neostriatum and dorsal striatum affects food intake and food preference, whereas in the nucleus accumbens dopamine signaling controls food seeking [25,26,27]. Moreover, dopaminergic neurons in the VTA modulate reward-related and goal-directed behaviors and exhibit numerous interactions with different brain regions [28]. The dopaminergic system is regulated via insulin in at least three molecular ways: (i) insulin regulates the uptake of released dopamine by induction of dopamine reuptake transporter (DAT) expression, (ii) insulin alters dopamine half-life or action by regulating the protein expression of the dopamine-degrading enzymes monoamine oxidases (MAO) and DAT, and (iii) insulin affects the spike frequency of cholinergic interneurons and dopaminergic neurons [29••, 30, 31••, 32, 33•]. In addition, diet-induced insulin resistance decreases the rate-limiting enzyme for dopamine synthesis, tyrosine hydroxylase, suggesting that insulin might affect TH synthesis in the brain [34] (Fig. 1). A causal role of brain insulin and dopamine signaling on weight regulation was shown in mice deficient for the insulin receptor in dopaminergic, tyrosine hydroxylase (TH)-positive neurons. These animals exhibit increased body weight due to increased food intake, showing that reduced dopaminergic insulin sensitivity is crucial for the development and manifestation of obesity [32]. Intra-ventral tegmental area (VTA) injection of insulin reduces food anticipatory behavior in mice by suppressing excitatory synaptic transmission onto dopamine neurons, which is reduced in the presence of hyperinsulinemia [35•, 36, 37]. This indicates that insulin alters neuronal plasticity within the dopaminergic system and this function is disrupted in hyperinsulinemic conditions such as obesity and insulin resistance. Insulin also reduces the intake of high-fat food in sated mice in the VTA [38], preventing over-eating in this state, a feature also present in obese subjects [39•, 40]. Intranasal insulin treatment in lean women modulates intrinsic reward circuitry [41], supporting that insulin in humans affects the dopamine system. In line, insulin not only reduces the response to food pictures in brain regions affected by dopamine of healthy, sated subjects but also reduces ratings of food palatability via mesolimbic pathways in insulin sensitive patients [42, 43]. Consistently, the response to food images is enhanced in type 2 diabetic patients compared to healthy controls [44]. Insulin is also able to regulate striatal function in lean men, while overweight men do not respond to insulin, indicating brain insulin resistance in this dopaminergic brain area [45•]. Indeed, aberrant dopamine signaling is present in obese humans and animals. In several rodent models of obesity, central dopamine neurotransmission is altered before, during, and after obesity develop. We have previously reported that this dopamine deficit is already in place as early as postnatal day 1 in rats inbred to become obese (obesity-prone (OP)) when compared to rats inbred to become lean (obesity-resistant (OR)) [46]. Therefore, the dopamine deficit predates the interaction of the dam with the pups during weaning and any effects of the offspring’s dietary history. This finding effectively implicates the prenatal environment and potentially maternal hormonal levels, including insulin, in central dopamine deficits observed in OP rats and confirms transgenerational aspects of obesity. The response of insulin receptors in the offspring may be a crucial determinant of dopamine aberrations observed in OP and diabetes-prone animals. Obese patients exhibit decreased striatal D2 receptor density, which negatively correlated with BMI [47]. Although decreased D2 receptor availability in the striatum can be affected by genetic predisposition, environmental factors might also influence this phenomenon [48,49,50,51]. While dopamine antagonism does not result in a major alteration of food consumption, it alters food-related motivation [52]. Haloperidol, a D2 receptor antagonist, reduces lever presses for preferred food but increases consumption of freely available less preferred food in healthy mice [53]. Dopamine injection into the lateral hypothalamus reduces food intake via reduced meal size in obese Zucker rats [54]. Bromocriptine, a dopamine agonist, has been shown to reduce obesity and improves glycemia in obese rodents and humans [55,56,57], indicating the therapeutic potential in counteracting obesity by enhancing the dopamine system. In summary, altered insulin and dopamine signaling is present in a variety of obese mouse models and humans [46, 58,59,60,61,62,63,64], highlighting insulin and dopamine action as a therapeutic target to regulate food intake and motivation-related behavior.

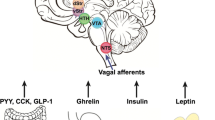
Brain insulin resistance impacts dopamine function on multiple molecular levels. Obesity-induced insulin resistance causes a reduction in TH expression [24], the rate-limiting enzyme in dopamine production. Further insulin receptor deficiency in brain causes unrestraint MAO expression and with it decreased dopamine half-life in the dopaminergic brain regions [21]. Insulin receptor deficiency on astrocytes reduces ATP exocytosis, leading to decreased purinergic signaling on dopaminergic neurons [19]. Besides these effects, insulin regulates DAT activity and thus dopamine action in the synaptic cleft [20], while also increasing spike frequency in cholinergic interneurons [23] and TH-dopaminergic neurons [22], highlighting the profound effect of insulin action on dopamine function. TH tyrosine hydroxylase, MAO monoamine oxidase. Lightning symbol highlights impact of insulin on dopamine function
Insulin and Dopamine Signaling Controls Emotional Behavior
Depressive disorders, diabetes, and insulin resistance associate [65, 66], yet the molecular mechanisms for this linkage are not well understood. Some epidemiological studies suggest an interaction of depression with type 2 diabetes and postulate inflammatory responses as a common mediator [67, 68]. Indeed, inflammation is one of many inducers of insulin resistance [18, 69]. In addition, psychological stress is linked to depression syndromes, affects the dopaminergic system, and has been shown to cause insulin resistance [70,71,72,73]. Children with increased depressive syndromes exhibit higher insulin resistance and the occurrence of depressive syndromes can predict the deterioration of insulin resistance [74]. Further, children with highest insulin resistance showed an association between altered brain morphology and depressive syndromes [75]. Thus, it seems that insulin resistance and depressive behavior might be functionally interconnected. Consistently, a variety of mouse models of insulin resistance exhibit signs of depressive-like behavior, suggesting that insulin resistance can influence this behavior. Mice fed a high-fat diet and db/db mice (mouse model of type 2 diabetes) exhibit central insulin resistance, depressive-like behaviors, and increased anxiety [29••, 31, 76, 77]. Depressive disorders are associated with impairment in neural circuits related to emotion and cognition and altered synaptic plasticity. This occurs in regions with high IR expression and insulin sensitivity such as the prefrontal cortex or hippocampus. Insulin improves synaptic plasticity and neuronal transmission, and exerts neuroprotective functions [19, 20], features which are impaired in depressive disorders. Further, treating obese animals with the insulin sensitizer rosiglitazone or pioglitazone ameliorated depressive-like behavior indicating that insulin action improves mood [78, 79]. Adding to this, prenatal stress reduces signaling of the closely related IGF-1R in hippocampus and frontal cortex and causes depression, which can be rescued by intracerebroventricular injection of IGF-1, which is able to activate the insulin receptor cascade [80, 81].
The monoamine deficiency hypothesis postulates that a reduction in serotonin, dopamine, and/or norepinephrine can be causal for the development of depressive disorders. Altered dopamine action has been shown to modulate depressive-like behavior [82,83,84]. A key region regulating dopaminergic VTA function represents the insulin-sensitive habenula (Hb) [28, 85, 86]. Structural and functional alterations of the Hb in humans are linked to depressive disorders [65, 87]. Altering the firing pattern of dopaminergic midbrain neurons or selective inhibition of VTA dopamine neurons induces depressive-like behavior [83, 84]. We were able to show that a neuronal and glial knockout of IR caused depressive-like behavior and anxiety with altered dopamine signaling. In neurons, insulin was able to suppress MAO A and B expression, enzymes crucial for monoamine degradation, and enhances dopamine half-life after electrically evoked dopamine release [31••]. IR deficiency in GFAP-positive glia cells caused reduced ATP exocytosis, resulting in decreased purinergic signaling on dopaminergic neurons and subsequently anxiety- and depressive-like behavior [29••]. Recent data show that knockout of the insulin receptor in dopaminergic neurons using the DAT-Cre mouse model does not affect food consumption or emotional behavior early in life, suggesting that loss of IR in distinct dopaminergic cell populations differentially affect metabolism and behavior [32, 88,89,90]. The combined lack of IR and IGF-1R only in the hippocampus or in the amygdala can induce increased anxiety-related behavior, supporting the hypothesis that insulin action can influence dopamine-dependent behavior [91]. Clearly, more research is needed to decipher the precise effect of insulin action in different brain cell populations and regions and on dopamine function regulating food intake and anxiety- and depressive-like behavior.
Neuroinflammation Impacts Insulin and Dopamine Function
Increased food intake and especially the increased consumption of saturated, long-chain fatty acids cause a low-grade inflammation in peripheral tissues and the brain. There is a strong association between inflammation and depression in humans and rodents [92•]. Major contributors of the inflammatory response are activated macrophages. Macrophages have been proposed to play a pivotal role in the pathogenesis of depression [93]. Macrophages secrete cytokines, such as tumor necrosis factor (TNF) α, which does affect not only the inflammatory response but also the insulin signaling cascade [94]. Increased pro-inflammatory cytokines affect synaptic plasticity and neuronal transmission and are implicated in the development of depressive disorders [95]. In line, adipose tissue secretes vast amounts of TNFα in obesity, which induces insulin resistance and is upregulated in depressive states [96]. Treating mice with the x TNFα monoclonal antibody infliximab results in protection against depressive-like behavior [97]. In addition, the widely used antidepressant bupropion reduces TNFα and improves metabolism in stressed rodents [98]. A prominent effector of TNFα represents the stress-activated kinase JNK [99]. Type 2 diabetic mice exhibit increased activation of JNK in brain, which induces insulin resistance due to serine phosphorylation of insulin receptor substrates [100, 101]. Conversely, JNK1 deficiency improves brain insulin sensitivity and reduces anxiety- and depressive-like behavior [102, 103]. In addition, inhibition of JNK protects dopaminergic neurons and improves behavior in a mouse model of neurodegeneration [104, 105].
Further, there seems to be an association between microbial-associated molecular patterns (MAMPs) generated by the gut microbiome, inflammation, insulin resistance, and dopamine function [106]. Obesity is associated with insulin resistance, altered dopamine function, and an altered microbiome [107]. Microbes produce MAMPS such as lipopolysaccharide (LPS), which can cause neuroinflammation, insulin resistance and depressive-like behavior [107,108,109]. Supporting this, healthy C57BL/6 mice which received an adoptive transfer of microbiota of high-fat diet (HFD) donor animals exhibited increased anxiety-like behavior compared to mice given the fecal transplants of normal chow diet-fed mice [110]. HFD feeding impairs membrane integrity, and with this there is an increase of endotoxin release, including LPS, into circulation [107, 111]. LPS per se induces insulin resistance and affects dopamine function representing a potential mechanism of how inflammation, altered insulin, and dopamine function are interlinked [108, 112]. Further, some microbes can generate short-chain fatty acids and precursors of neurotransmitters, such as dopamine [113], which affect insulin and dopamine function. Yet, whether actually these gut-derived metabolites penetrate the brain and affect dopamine-dependent behavior warrants further investigation.
The hypothalamus-pituitary-adrenal (HPA) axis regulates responses to stress and affect metabolism and emotional behavior. A dysregulated HPA axis is present in obesity and depression and inflammation can activate the HPA axis [114, 115]. Pro-inflammatory cytokines can induce a heightened response of the HPA axis and has been observed in depression syndromes [116]. Increased levels of glucocorticoid levels can induce insulin resistance but also affect dopamine function pointing to an additional mechanism of how inflammation might impact brain insulin signaling and dopamine function [117, 118]. Here, time and strength of the HPA axis activation differentially affects dopamine function, further complicating this crosstalk [118]. Overall these data highlight the close relationship between inflammation, insulin resistance, and depression.
Conclusion
Dietary obesity other metabolic disorders, and diabetes all share significant central monoamine neurotransmitter aberrations with psychoactive disorders like depression. In this article, we attempted to highlight some of recent advances in understanding how such deficits may be linked to central insulin receptor signaling. Beyond the role of peripheral insulin resistance in obesity and diabetes, there is substantial promise in the study of the role of brain insulin in behavioral and mood disorders that may open new pathways in novel drug target discovery and in drug development for the treatment of such disorders.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Donaldson L, Rutter P. Healthier, fairer, safer: the global health journey, 2007-2017. Geneva: World Health Organization; 2017.
Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the global burden of disease study 2010. Lancet. 2012;380(9859):2224–60.
da Rocha Fernandes J, Ogurtsova K, Linnenkamp U, Guariguata L, Seuring T, Zhang P, et al. IDF diabetes atlas estimates of 2014 global health expenditures on diabetes. Diabetes Res Clin Pract. 2016;117:48–54.
Lutter M, Nestler EJ. Homeostatic and hedonic signals interact in the regulation of food intake. J Nutr. 2009;139(3):629–32.
Belgardt BF, Bruning JC. CNS leptin and insulin action in the control of energy homeostasis. Ann N Y Acad Sci. 2010;1212:97–113.
Volkow ND, Wang GJ, Fowler JS, Logan J, Jayne M, Franceschi D, et al. “Nonhedonic” food motivation in humans involves dopamine in the dorsal striatum and methylphenidate amplifies this effect. Synapse. 2002;44(3):175–80.
Drewnowski A, Greenwood MR. Cream and sugar: human preferences for high-fat foods. Physiol Behav. 1983;30(4):629–33.
Teegarden SL, Scott AN, Bale TL. Early life exposure to a high fat diet promotes long-term changes in dietary preferences and central reward signaling. Neuroscience. 2009;162(4):924–32.
Saper CB, Chou TC, Elmquist JK. The need to feed: homeostatic and hedonic control of eating. Neuron. 2002;36(2):199–211.
Volkow ND, Wang GJ, Baler RD. Reward, dopamine and the control of food intake: implications for obesity. Trends Cogn Sci. 2011;15(1):37–46.
Cone JJ, Chartoff EH, Potter DN, Ebner SR, Roitman MF. Prolonged high fat diet reduces dopamine reuptake without altering DAT gene expression. PLoS One. 2013;8(3):e58251.
DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160(14):2101–7.
Osterberg L, Blaschke T. Adherence to medication. N Engl J Med. 2005;353(5):487–97.
Luppino FS, de Wit LM, Bouvy PF, Stijnen T, Cuijpers P, Penninx BW, et al. Overweight, obesity, and depression: a systematic review and meta-analysis of longitudinal studies. Arch Gen Psychiatry. 2010;67(3):220–9.
Geoffroy MC, Li L, Power C. Depressive symptoms and body mass index: co-morbidity and direction of association in a British birth cohort followed over 50 years. Psychol Med. 2014;44(12):2641–52.
Roy T, Lloyd CE. Epidemiology of depression and diabetes: a systematic review. J Affect Disord. 2012;142 Suppl:S8–21.
Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7(2):85–96.
Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol. 2014;6(1). https://doi.org/10.1101/cshperspect.a009191.
Wardelmann K, Blumel S, Rath M, Alfine E, Chudoba C, Schell M, et al. Insulin action in the brain regulates mitochondrial stress responses and reduces diet-induced weight gain. Mol Metab. 2019;21:68–81.
Kleinridders A, Ferris HA, Cai W, Kahn CR. Insulin action in brain regulates systemic metabolism and brain function. Diabetes. 2014;63(7):2232–43.
Bruning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, et al. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000;289(5487):2122–5.
Coleman DL. Obese and diabetes: two mutant genes causing diabetes-obesity syndromes in mice. Diabetologia. 1978;14(3):141–8.
Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci. 2002;5(6):566–72.
Salamone JD, Correa M. Dopamine and food addiction: lexicon badly needed. Biol Psychiatry. 2013;73(9):e15–24.
Dunnett SB, Iversen SD. Regulatory impairments following selective 6-OHDA lesions of the neostriatum. Behav Brain Res. 1982;4(2):195–202.
Salamone JD, Correa M. The mysterious motivational functions of mesolimbic dopamine. Neuron. 2012;76(3):470–85.
Vucetic Z, Reyes TM. Central dopaminergic circuitry controlling food intake and reward: implications for the regulation of obesity. Wiley Interdiscip Rev Syst Biol Med. 2010;2(5):577–93.
Morales M, Margolis EB. Ventral tegmental area: cellular heterogeneity, connectivity and behaviour. Nat Rev Neurosci. 2017;18(2):73–85.
•• Cai W, Xue C, Sakaguchi M, Konishi M, Shirazian A, Ferris HA, et al. Insulin regulates astrocyte gliotransmission and modulates behavior. J Clin Invest. 2018;128(7):2914–26. This manuscript provided the first evidence that astrocytic insulin signaling plays an important role in mesolimbic dopaminergic signaling, providing a potential mechanism by which insulin receptors in the CNS play a role in depression in patients with insulin resistance.
Figlewicz DP, Szot P, Chavez M, Woods SC, Veith RC. Intraventricular insulin increases dopamine transporter mRNA in rat VTA/substantia nigra. Brain Res. 1994;644(2):331–4.
•• Kleinridders A, Cai W, Cappellucci L, Ghazarian A, Collins WR, Vienberg SG, et al. Insulin resistance in brain alters dopamine turnover and causes behavioral disorders. Proc Natl Acad Sci U S A. 2015;112(11):3463–8 This article demonstrated that brain insulin resistance alters central dopamine exocytosis and induces anxiety and depressive-like behaviors .
Konner AC, Hess S, Tovar S, Mesaros A, Sanchez-Lasheras C, Evers N, et al. Role for insulin signaling in catecholaminergic neurons in control of energy homeostasis. Cell Metab. 2011;13(6):720–8.
• Stouffer MA, Woods CA, Patel JC, Lee CR, Witkovsky P, Bao L, et al. Insulin enhances striatal dopamine release by activating cholinergic interneurons and thereby signals reward. Nat Commun. 2015;6:8543 This study reported that insulin can upregulate synaptic dopamine release in the nucleus accumbens and the dorsal striatum through a 2-step mechanism that involves striatal cholinergic interneurons expressing insulin receptors.
Li Y, South T, Han M, Chen J, Wang R, Huang XF. High-fat diet decreases tyrosine hydroxylase mRNA expression irrespective of obesity susceptibility in mice. Brain Res. 2009;1268:181–9.
• Liu S, Globa AK, Mills F, Naef L, Qiao M, Bamji SX, et al. Consumption of palatable food primes food approach behavior by rapidly increasing synaptic density in the VTA. Proc Natl Acad Sci U S A. 2016;113(9):2520–5 This manuscript provided evidence that exposure to palatable foods induces long-lasting synaptic plasticity in mesolimbic dopamine neurons and suggested that targeting this pathway with brain-delivered insulin may suppress food cravings .
Liu S, Labouebe G, Karunakaran S, Clee SM, Borgland SL. Effect of insulin on excitatory synaptic transmission onto dopamine neurons of the ventral tegmental area in a mouse model of hyperinsulinemia. Nutr Diabetes. 2013;3:e97.
Mebel DM, Wong JC, Dong YJ, Borgland SL. Insulin in the ventral tegmental area reduces hedonic feeding and suppresses dopamine concentration via increased reuptake. Eur J Neurosci. 2012;36(3):2336–46.
Labouebe G, Liu S, Dias C, Zou H, Wong JC, Karunakaran S, et al. Insulin induces long-term depression of ventral tegmental area dopamine neurons via endocannabinoids. Nat Neurosci. 2013;16(3):300–8.
• Kullmann S, Heni M, Veit R, Scheffler K, Machann J, Haring HU, et al. Intranasal insulin enhances brain functional connectivity mediating the relationship between adiposity and subjective feeling of hunger. Sci Rep. 2017;7(1):1627 This article evaluated the effects of intranasal insulin on brain functional connectivity in healthy young adults. The authors concluded that brain insulin action may regulate feeding behavior and induce weight loss by altering brain functional connectivity within and between cognitive and homeostatic brain regions.
Dalton M, Blundell J, Finlayson G. Effect of BMI and binge eating on food reward and energy intake: further evidence for a binge eating subtype of obesity. Obes Facts. 2013;6(4):348–59.
Kullmann S, Frank S, Heni M, Ketterer C, Veit R, Haring HU, et al. Intranasal insulin modulates intrinsic reward and prefrontal circuitry of the human brain in lean women. Neuroendocrinology. 2013;97(2):176–82.
Guthoff M, Grichisch Y, Canova C, Tschritter O, Veit R, Hallschmid M, et al. Insulin modulates food-related activity in the central nervous system. J Clin Endocrinol Metab. 2010;95(2):748–55.
Tiedemann LJ, Schmid SM, Hettel J, Giesen K, Francke P, Buchel C, et al. Central insulin modulates food valuation via mesolimbic pathways. Nat Commun. 2017;8:16052.
Chechlacz M, Rotshtein P, Klamer S, Porubska K, Higgs S, Booth D, et al. Diabetes dietary management alters responses to food pictures in brain regions associated with motivation and emotion: a functional magnetic resonance imaging study. Diabetologia. 2009;52(3):524–33.
• Heni M, Wagner R, Kullmann S, Gancheva S, Roden M, Peter A, et al. Hypothalamic and striatal insulin action suppresses endogenous glucose production and may stimulate glucose uptake during hyperinsulinemia in lean but not in overweight men. Diabetes. 2017;66(7):1797–806 This manuscript focused on increasing insulin-mediated suppression of endogenous glucose production and stimulating peripheral glucose uptake in obese patients. The conclusion was that brain insulin may improve glucose metabolism during systemic hyperinsulinemia and that central insulin resistance in obesity may have an adverse impact on whole-body glucose homeostasis.
Geiger BM, Behr GG, Frank LE, Caldera-Siu AD, Beinfeld MC, Kokkotou EG, et al. Evidence for defective mesolimbic dopamine exocytosis in obesity-prone rats. FASEB J. 2008;22(8):2740–6.
Wang GJ, Volkow ND, Logan J, Pappas NR, Wong CT, Zhu W, et al. Brain dopamine and obesity. Lancet. 2001;357(9253):354–7.
Gluskin BS, Mickey BJ. Genetic variation and dopamine D2 receptor availability: a systematic review and meta-analysis of human in vivo molecular imaging studies. Transl Psychiatry. 2016;6:e747.
Hayden EP, Klein DN, Dougherty LR, Olino TM, Laptook RS, Dyson MW, et al. The dopamine D2 receptor gene and depressive and anxious symptoms in childhood: associations and evidence for gene-environment correlation and gene-environment interaction. Psychiatr Genet. 2010;20(6):304–10.
Pohjalainen T, Rinne JO, Nagren K, Lehikoinen P, Anttila K, Syvalahti EK, et al. The A1 allele of the human D2 dopamine receptor gene predicts low D2 receptor availability in healthy volunteers. Mol Psychiatry. 1998;3(3):256–60.
Thompson J, Thomas N, Singleton A, Piggott M, Lloyd S, Perry EK, et al. D2 dopamine receptor gene (DRD2) Taq1 a polymorphism: reduced dopamine D2 receptor binding in the human striatum associated with the A1 allele. Pharmacogenetics. 1997;7(6):479–84.
Fibiger HC, Carter DA, Phillips AG. Decreased intracranial self-stimulation after neuroleptics or 6-hydroxydopamine: evidence for mediation by motor deficits rather than by reduced reward. Psychopharmacology. 1976;47(1):21–7.
Salamone JD, Steinpreis RE, McCullough LD, Smith P, Grebel D, Mahan K. Haloperidol and nucleus accumbens dopamine depletion suppress lever pressing for food but increase free food consumption in a novel food choice procedure. Psychopharmacology. 1991;104(4):515–21.
Yang ZJ, Meguid MM, Chai JK, Chen C, Oler A. Bilateral hypothalamic dopamine infusion in male Zucker rat suppresses feeding due to reduced meal size. Pharmacol Biochem Behav. 1997;58(3):631–5.
Cincotta AH, Meier AH. Reductions of body fat stores and total plasma cholesterol and triglyceride concentrations in several species by bromocriptine treatment. Life Sci. 1989;45(23):2247–54.
Cincotta AH, Meier AH. Bromocriptine (Ergoset) reduces body weight and improves glucose tolerance in obese subjects. Diabetes Care. 1996;19(6):667–70.
Meier AH, Cincotta AH, Lovell WC. Timed bromocriptine administration reduces body fat stores in obese subjects and hyperglycemia in type II diabetics. Experientia. 1992;48(3):248–53.
Baik JH. Dopamine signaling in food addiction: role of dopamine D2 receptors. BMB Rep. 2013;46(11):519–26.
Blum K, Thanos PK, Gold MS. Dopamine and glucose, obesity, and reward deficiency syndrome. Front Psychol. 2014;5:919.
Hendrick OM, Luo X, Zhang S, Li CS. Saliency processing and obesity: a preliminary imaging study of the stop signal task. Obesity (Silver Spring). 2012;20(9):1796–802.
Kim B, Feldman EL. Insulin resistance as a key link for the increased risk of cognitive impairment in the metabolic syndrome. Exp Mol Med. 2015;47:e149.
Kok P, Roelfsema F, Frolich M, van Pelt J, Stokkel MP, Meinders AE, et al. Activation of dopamine D2 receptors simultaneously ameliorates various metabolic features of obese women. Am J Physiol Endocrinol Metab. 2006;291(5):E1038–43.
Pijl H. Reduced dopaminergic tone in hypothalamic neural circuits: expression of a “thrifty” genotype underlying the metabolic syndrome? Eur J Pharmacol. 2003;480(1–3):125–31.
Zhang Z, Hao CJ, Li CG, Zang DJ, Zhao J, Li XN, et al. Mutation of SLC35D3 causes metabolic syndrome by impairing dopamine signaling in striatal D1 neurons. PLoS Genet. 2014;10(2):e1004124.
Pearson S, Schmidt M, Patton G, Dwyer T, Blizzard L, Otahal P, et al. Depression and insulin resistance: cross-sectional associations in young adults. Diabetes Care. 2010;33(5):1128–33.
Pan A, Ye X, Franco OH, Li H, Yu Z, Zou S, et al. Insulin resistance and depressive symptoms in middle-aged and elderly Chinese: findings from the nutrition and health of aging population in China study. J Affect Disord. 2008;109(1–2):75–82.
Mezuk B, Eaton WW, Albrecht S, Golden SH. Depression and type 2 diabetes over the lifespan: a meta-analysis. Diabetes Care. 2008;31(12):2383–90.
Stuart MJ, Baune BT. Depression and type 2 diabetes: inflammatory mechanisms of a psychoneuroendocrine co-morbidity. Neurosci Biobehav Rev. 2012;36(1):658–76.
Dandona P, Aljada A, Bandyopadhyay A. Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol. 2004;25(1):4–7.
Kaneyuki H, Yokoo H, Tsuda A, Yoshida M, Mizuki Y, Yamada M, et al. Psychological stress increases dopamine turnover selectively in mesoprefrontal dopamine neurons of rats: reversal by diazepam. Brain Res. 1991;557(1–2):154–61.
Li L, Li X, Zhou W, Messina JL. Acute psychological stress results in the rapid development of insulin resistance. J Endocrinol. 2013;217(2):175–84.
Pruessner JC, Champagne F, Meaney MJ, Dagher A. Dopamine release in response to a psychological stress in humans and its relationship to early life maternal care: a positron emission tomography study using [11C]raclopride. J Neurosci. 2004;24(11):2825–31.
Yang L, Zhao Y, Wang Y, Liu L, Zhang X, Li B, et al. The effects of psychological stress on depression. Curr Neuropharmacol. 2015;13(4):494–504.
Shomaker LB, Tanofsky-Kraff M, Stern EA, Miller R, Zocca JM, Field SE, et al. Longitudinal study of depressive symptoms and progression of insulin resistance in youth at risk for adult obesity. Diabetes Care. 2011;34(11):2458–63.
Singh MK, Leslie SM, Packer MM, Zaiko YV, Phillips OR, Weisman EF, et al. Brain and behavioral correlates of insulin resistance in youth with depression and obesity. Horm Behav. 2019;108:73–83.
Sharma AN, Elased KM, Garrett TL, Lucot JB. Neurobehavioral deficits in db/db diabetic mice. Physiol Behav. 2010;101(3):381–8.
Sharma S, Fulton S. Diet-induced obesity promotes depressive-like behaviour that is associated with neural adaptations in brain reward circuitry. Int J Obes. 2013;37(3):382–9.
Kurhe Y, Mahesh R. Pioglitazone, a PPARgamma agonist rescues depression associated with obesity using chronic unpredictable mild stress model in experimental mice. Neurobiol Stress. 2016;3:114–21.
Sharma AN, Elased KM, Lucot JB. Rosiglitazone treatment reversed depression- but not psychosis-like behavior of db/db diabetic mice. J Psychopharmacol. 2012;26(5):724–32.
Basta-Kaim A, Szczesny E, Glombik K, Stachowicz K, Slusarczyk J, Nalepa I, et al. Prenatal stress affects insulin-like growth factor-1 (IGF-1) level and IGF-1 receptor phosphorylation in the brain of adult rats. Eur Neuropsychopharmacol. 2014;24(9):1546–56.
Kleinridders A. Deciphering brain insulin receptor and insulin-like growth factor 1 receptor signalling. J Neuroendocrinol. 2016;28(11). https://doi.org/10.1111/jne.12433.
Belujon P, Grace AA. Dopamine system dysregulation in major depressive disorders. Int J Neuropsychopharmacol. 2017;20(12):1036–46.
Chaudhury D, Walsh JJ, Friedman AK, Juarez B, Ku SM, Koo JW, et al. Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature. 2013;493(7433):532–6.
Tye KM, Mirzabekov JJ, Warden MR, Ferenczi EA, Tsai HC, Finkelstein J, et al. Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature. 2013;493(7433):537–41.
Shumake J, Ilango A, Scheich H, Wetzel W, Ohl FW. Differential neuromodulation of acquisition and retrieval of avoidance learning by the lateral habenula and ventral tegmental area. J Neurosci. 2010;30(17):5876–83.
Unger J, McNeill TH, Moxley RT 3rd, White M, Moss A, Livingston JN. Distribution of insulin receptor-like immunoreactivity in the rat forebrain. Neuroscience. 1989;31(1):143–57.
Boulos LJ, Darcq E, Kieffer BL. Translating the habenula—from rodents to humans. Biol Psychiatry. 2017;81(4):296–305.
Evans MC, Kumar NS, Inglis MA, Anderson GM. Leptin and insulin do not exert redundant control of metabolic or emotive function via dopamine neurons. Horm Behav. 2018;106:93–104.
Lammel S, Steinberg EE, Foldy C, Wall NR, Beier K, Luo L, et al. Diversity of transgenic mouse models for selective targeting of midbrain dopamine neurons. Neuron. 2015;85(2):429–38.
Stuber GD, Stamatakis AM, Kantak PA. Considerations when using cre-driver rodent lines for studying ventral tegmental area circuitry. Neuron. 2015;85(2):439–45.
Soto M, Cai W, Konishi M, Kahn CR. Insulin signaling in the hippocampus and amygdala regulates metabolism and neurobehavior. Proc Natl Acad Sci U S A. 2019;116(13):6379–84.
• Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol. 2016;16(1):22–34 This review article discussed evidence identifying the mechanisms through which inflammatory pathways interact with neurotransmitters and neurocircuits to influence the risk for depression .
Smith RS. The macrophage theory of depression. Med Hypotheses. 1991;35(4):298–306.
Hotamisligil GS. Mechanisms of TNF-alpha-induced insulin resistance. Exp Clin Endocrinol Diabetes. 1999;107(2):119–25.
Felger JC, Lotrich FE. Inflammatory cytokines in depression: neurobiological mechanisms and therapeutic implications. Neuroscience. 2013;246:199–229.
Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91.
Liu YN, Peng YL, Liu L, Wu TY, Zhang Y, Lian YJ, et al. TNFalpha mediates stress-induced depression by upregulating indoleamine 2,3-dioxygenase in a mouse model of unpredictable chronic mild stress. Eur Cytokine Netw. 2015;26(1):15–25.
Ahmed M, El-Bakly WM, Zaki AM. Abd Alzez LF, El serafi O. Bupropion effects on high-fat diet-induced steatohepatitis and endothelial dysfunction in rats: role of tumour necrosis factor-alpha. J Pharm Pharmacol. 2014;66(6):793–801.
Westwick JK, Weitzel C, Minden A, Karin M, Brenner DA. Tumor necrosis factor alpha stimulates AP-1 activity through prolonged activation of the c-Jun kinase. J Biol Chem. 1994;269(42):26396–401.
Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J Biol Chem. 2000;275(12):9047–54.
Kleinridders A, Lauritzen HP, Ussar S, Christensen JH, Mori MA, Bross P, et al. Leptin regulation of Hsp60 impacts hypothalamic insulin signaling. J Clin Invest. 2013;123(11):4667–80.
Belgardt BF, Mauer J, Wunderlich FT, Ernst MB, Pal M, Spohn G, et al. Hypothalamic and pituitary c-Jun N-terminal kinase 1 signaling coordinately regulates glucose metabolism. Proc Natl Acad Sci U S A. 2010;107(13):6028–33.
Mohammad H, Marchisella F, Ortega-Martinez S, Hollos P, Eerola K, Komulainen E, et al. JNK1 controls adult hippocampal neurogenesis and imposes cell-autonomous control of anxiety behaviour from the neurogenic niche. Mol Psychiatry. 2018;23(2):362–74.
Wang W, Shi L, Xie Y, Ma C, Li W, Su X, et al. SP600125, a new JNK inhibitor, protects dopaminergic neurons in the MPTP model of Parkinson’s disease. Neurosci Res. 2004;48(2):195–202.
Crocker CE, Khan S, Cameron MD, Robertson HA, Robertson GS, Lograsso P. JNK inhibition protects dopamine neurons and provides behavioral improvement in a rat 6-hydroxydopamine model of Parkinson’s disease. ACS Chem Neurosci. 2011;2(4):207–12.
Boulange CL, Neves AL, Chilloux J, Nicholson JK, Dumas ME. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. 2016;8(1):42.
Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761–72.
Rorato R, Borges BC, Uchoa ET, Antunes-Rodrigues J, Elias CF, Elias LLK. LPS-Induced low-grade inflammation increases hypothalamic JNK expression and causes central insulin resistance irrespective of body weight changes. Int J Mol Sci. 2017;18(7). https://doi.org/10.3390/ijms18071431.
Sampson TR, Mazmanian SK. Control of brain development, function, and behavior by the microbiome. Cell Host Microbe. 2015;17(5):565–76.
Bruce-Keller AJ, Salbaum JM, Luo M, Blanchard E, Taylor CM, Welsh DA, et al. Obese-type gut microbiota induce neurobehavioral changes in the absence of obesity. Biol Psychiatry. 2015;77(7):607–15.
Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57(6):1470–81.
Ling Z, Zhu Y, Tong C, Snyder JA, Lipton JW, Carvey PM. Progressive dopamine neuron loss following supra-nigral lipopolysaccharide (LPS) infusion into rats exposed to LPS prenatally. Exp Neurol. 2006;199(2):499–512.
Galland L. The gut microbiome and the brain. J Med Food. 2014;17(12):1261–72.
Bose M, Olivan B, Laferrere B. Stress and obesity: the role of the hypothalamic-pituitary-adrenal axis in metabolic disease. Curr Opin Endocrinol Diabetes Obes. 2009;16(5):340–6.
Lamers F, Vogelzangs N, Merikangas KR, de Jonge P, Beekman AT, Penninx BW. Evidence for a differential role of HPA-axis function, inflammation and metabolic syndrome in melancholic versus atypical depression. Mol Psychiatry. 2013;18(6):692–9.
Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9(1):46–56.
Geer EB, Islam J, Buettner C. Mechanisms of glucocorticoid-induced insulin resistance: focus on adipose tissue function and lipid metabolism. Endocrinol Metab Clin N Am. 2014;43(1):75–102.
Sinclair D, Purves-Tyson TD, Allen KM, Weickert CS. Impacts of stress and sex hormones on dopamine neurotransmission in the adolescent brain. Psychopharmacology. 2014;231(8):1581–99.
Acknowledgements
This work was in part supported by the Deutsche Forschungsgemeinschaft (DFG) grant project KL 2399/4-1 (to A.K.), by a grant from the German Ministry of Education and Research (BMBF) and the State of Brandenburg (DZD grant 82DZD00302 to A.K.), by NIH/NIDDK (R01 DK065872 and ARRA 3R01DK065872-04S1; ENP) and the Tufts Center for Neuroscience Research (P30 NS047243), and an Award of Excellence in Biomedical Research by the Smith Family Foundation (ENP) and the Graduate Program in Pharmacology and Drug Development, Sackler School of Graduate Biomedical Sciences, Tufts University School of Medicine (ENP).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
André Kleinridders and Emmanuel N. Pothos declare they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Nutrition and the Brain
Rights and permissions
About this article

Cite this article
Kleinridders, A., Pothos, E.N. Impact of Brain Insulin Signaling on Dopamine Function, Food Intake, Reward, and Emotional Behavior. Curr Nutr Rep 8, 83–91 (2019). https://doi.org/10.1007/s13668-019-0276-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13668-019-0276-z