
Abstract
Rapid, economical, and reliable genotyping is an important requirement for germplasm analysis and cultivar identification in crop species. Chinese cabbage (Brassica rapa L. subsp. pekinensis (Lour.) Hanelt) originated in China and is now an economically important vegetable crop worldwide, especially in East Asia. In this study, we evaluated 1167 single nucleotide polymorphisms (SNPs) among 166 representative Chinese cabbage inbred lines using a KASP genotyping assay. On the basis of polymorphisms and principal component analysis, we selected 60 core SNPs distributed on all Brassica rapa chromosomes with allele frequencies sufficiently balanced so as to provide adequate information for genetic identification. The core set of SNPs was used for construction of a neighbor-joining dendrogram, in which the 166 inbred lines were clustered into spring, summer, and autumn ecotype groups. Clustering of the ecotype groups was better resolved than that achieved with 1167 and 360 polymorphic SNP datasets. Stability and resolution of the core SNP markers were tested using 178 commercial hybrid Chinese cabbage cultivars to confirm their utility in genetic identification. The set of 60 informative and stable SNP markers showed high discriminatory power and relatively uniform genomic distribution (4–9 markers per chromosome). The SNPs represent a cost-efficient and accurate marker set for germplasm analysis and cultivar identification and are suitable for molecular marker-assisted breeding in Chinese cabbage.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Chinese cabbage (Brassica rapa L. subsp. pekinensis) originated in China and is an important vegetable crop worldwide. Two decades ago, Chinese cabbage was grown as an autumn crop, but now is grown year-round and has spring, summer, and autumn ecotypes (Ke 2010). As a diploid (2n = 20) crop, Chinese cabbage is a model plant for genetic studies owing to its high recombination rate and rich genetic diversity. In terms of breeding, the selection of diverse genetic resources with different agronomic characteristics and understanding the genetic relationships among these breeding materials are crucial for cultivar improvement. However, little is known about such genetic materials. It is imperative to understand the genetic diversity of Chinese cabbage within available breeding lines using genome-wide molecular markers. In addition, an accurate, simple, and rapid method is urgently needed to test the purity and authenticity of seeds and for protection of intellectual property rights.
Molecular detection and utilization of genetic variation in crop genomes is one of the most important tasks for plant geneticists and breeders to understand the genomic architecture and to devise crop improvement strategies. The development and widespread adoption of molecular markers in genetic studies has provided a foundation for linking the phenotype to the genotype (Langridge et al. 2005). Molecular markers have been used to characterize the distinctness of a species by analyzing the genetic diversity and constructing a DNA fingerprint, which gave rise to the distinctness, uniformity, and stability (DUS) testing method. In recent decades, several DNA marker technologies have been applied to detect genetic diversity in cultivated Chinese cabbage (Song et al. 1990; Powell et al. 1996; Das et al. 1999; He et al. 2003; Soengas et al. 2011), such as random amplified polymorphic DNA (RAPD), amplified fragment length polymorphism (AFLP), and simple sequence repeats (SSRs). However, the different data sets are hardly comparable because of the lack of a common core set of reference genotypes and the use of different marker systems.
At present, single nucleotide polymorphisms (SNPs) are the markers of choice for genome-wide analyses, owing to the high marker density across the genome and high genetic stability and because SNPs can be readily adapted to automated genotyping methods. A number of high-throughput, cost-effective SNP genotyping platforms have been developed, such as the Illumina® GoldenGate® (Fan et al. 2003) and Infinium platforms (Steemers and Gunderson 2007), TaqMan® technology (Livak et al. 1995), and the KASP™ platform (KBiosciences; https://www.lgcgroup.com/products/kasp-genotyping-chemistry/#.W2MSyygzbIU). Many of these platforms have been used for important crop species such as barley, wheat, maize, soybean, cowpea, and pea (Allen et al. 2011; Cortés et al. 2011; Hiremath et al. 2012). KASP is a user-friendly system that provides flexibility in the numbers of SNPs and genotypes to be used for assays. Given the importance of KASP assays in genotyping variable numbers of samples with variable numbers of SNPs, assays have been developed for wheat, common bean, chickpea, and cotton (Allen et al. 2011; Cortés et al. 2011; Hiremath et al. 2012; Kuang et al. 2016). The generation of a high-throughput SNP genotype identification platform will play a crucial role in genetic diversity analysis, fingerprint construction, and assessment of cultivar purity and authenticity.
The objective of this study was to validate and obtain an appropriate set of core SNP markers suitable for identification of Chinese cabbage germplasm and cultivars. Using 166 representative Chinese cabbage lines, we identified a set of 60 core SNPs from 1167 SNP markers, which are rich in polymorphisms and evenly distributed throughout the B. rapa genome. Marker stability and resolution was tested using 178 commercial hybrid cultivars to demonstrate the utility of the markers for genetic identification. The core SNPs effectively represented the genetic diversity in the Chinese cabbage germplasm collections and can be used efficiently and reliably in DUS testing, DNA fingerprinting, cultivar identification, and analysis of genetic diversity in Chinese cabbage.
2 Materials and methods
2.1 Plant materials
A total of 166 Chinese cabbage inbred lines, which were collected from different areas in China, were used for core SNP screening in this study and consisted of 32 spring Chinese cabbages, 36 summer Chinese cabbages, and 98 autumn Chinese cabbages (Supplementary Table 1). In addition, 178 Chinese cabbage hybrid cultivars (Supplementary Table 2) obtained from 68 breeding companies or institutes were used for genetic identification.
2.2 DNA extraction
Total DNA was extracted from two to three young leaves following a standard DNA isolation protocol (Li et al. 2015). The DNA quality and concentration were measured with a NanoDrop 2000 UV spectrophotometer (Thermo Scientific, Waltham, MA, USA), and working solutions were prepared at a concentration of 10 ng/μL.
2.3 SNP selection
A total of 1167 SNPs were identified using 231 resequenced B. rapa genotypes (Su et al. 2018). These SNPs were then used to select a core set from the 166 inbred lines of Chinese cabbage. The identification of SNPs was performed using GATK software (McKenna et al. 2010) using Chiifu-401-42 (v1.5) as the reference genome (Wang et al. 2011).
High-quality SNP candidates were selected for KASP assays and were comprehensively screened in the 166 inbred lines. The following strict criteria were used for selection of high-quality SNPs for KASP assays: (1) minor allele frequency (MAF) value among the 166 genotypes ≥ 0.1; (2) read depth ≥ 20; (3) potential SNP candidates evenly distributed throughout the genome; (4) only one marker selected for markers that showed the same genotypes across the 166 inbred lines; and (5) polymorphic markers useful for genotyping in both inbred lines and hybrids.
2.4 KASP genotyping
For each SNP, two allele-specific forward primers and one common reverse primer were designed by LGC (Laboratory of the Government Chemist). Using these primers, KASP assays were performed in final reaction volumes of 1 μL in 1536-plates (no. KBS-0751-001, KBioscience), containing 1 × KASP reaction mix (KBS-1016-011, KBioscience), 12 nM each allele-specific forward primer, 30 nM reverse primer, and 4 ng genomic DNA. The GenePro™ Thermal Cycler (Hydrocycler) was used for amplification with the following cycling conditions: 15 min at 94 °C; 10 touchdown cycles of 20 s at 94 °C and 60 s at 65–57 °C (the annealing temperature for each cycle was reduced by 0.8 °C per cycle); and 26–42 cycles of 20 s at 94 °C and 60 s at 57 °C. Fluorescence detection of the reactions was performed using an Omega Fluorostar scanner (BMG PHERAstar), and the data were analyzed using KlusterCaller 1.1 software (Kbiosciences).
Following completion of the KASP PCR, reaction plates were read and the data analyzed using SNPviewer (Kbiosciences). Detected signals were plotted, with samples of the same genotype clustering together. Detailed instructions can be downloaded at www.kbioscience.co.uk. The clusters were defined from the graphs according to the following criteria: (1) clear boundaries between different genotypes and (2) the minimized missing data rate. Specific primers for KASP assays are usually 18–35 bp, with high specificity and SNP call rate.
2.5 Marker polymorphism and diversity analysis
To identify high-quality, core SNP markers, principal component analysis (PCA) was performed using Tassel 4.0 (Bradbury et al. 2007). The polymorphic information content (PIC) and gene diversity values for the SNP markers in this study were calculated using PowerMarker software (https://brcwebportal.cos.ncsu.edu/powermarker/). To assess genetic diversity within different subspecies or variant clusters, we used Genalex 6.3 (Peakall and Smouse 2006) to estimate MAF, observed heterozygosity (ObsHET), and fixation index (FST) values.
A matrix was constructed using Nei’s genetic distances and a neighbor-joining (NJ) tree was created with MEGA 5 software (Tamura et al. 2011). Population and subpopulation genetic structure were further analyzed by conducting an analysis of molecular variance (AMOVA) using Arlequin 3.5 software (Excoffier et al. 1992; Peakall and Smouse 2006). The graphical genotyping software GGT 2.0 was used to represent graphically the genotyping data for all 178 hybrids using 60 SNPs.
3 Results
3.1 Development of KASP assay markers from selected SNPs
As shown in Fig. 1, SNPs were automatically called for AA, AB, and BB genotypes. If a rare AB genotype was identified or some data points were shifted to one side, the automatic SNP calling frequently produced errors; therefore, such SNP loci were of insufficient quality to be used as a KASP marker (Fig. 1a, b). For the remaining SNP loci, KASP genotyping discriminated the two homozygous alleles and heterozygous alleles in the inbred lines (Fig. 1c, d). In total, 597 KASP markers were readily amplified and clearly distinguished the 166 Chinese cabbage genotypes.

Development of SNP markers from Chinese cabbage inbred lines for KASP genotyping. SNPs were automatically called for AA, AB, and BB genotypes. Red dots are homozygous for one allele, blue dots are homozygous for a second allele, and green dots are the heterozygous allele. a, b KASP markers that were not well amplified; c, d KASP markers that were well amplified. (Color figure online)
3.2 Identification of candidate core SNPs
We screened the 597 KASP markers on the basis of the MAF, heterozygosity, and PIC values as well as physical position. There were 227 SNPs with MAF < 0.1, of which 14 SNPs were monomorphic. Ten SNPs showed heterozygosity ≥ 0.25. To identify markers representing the core SNP set, we performed PCA of 360 polymorphic SNPs using TASSEL 4.0 software based on the 166 genotypes. On the basis of the eigenvalues (Supplementary Table 3), 60 principal components were selected when a cumulative contribution rate of 80% was taken into account (Fig. 2). We identified 60 SNPs with the maximum eigenvector values, which were considered to be the most representative markers. The genomic distribution of the 60 candidate SNPs was screened for development of KASP assays. The SNPs were distributed on the 10 chromosomes of the genome with numbers of loci per individual chromosome of 5, 9, 8, 4, 7, 5, 6, 4, 8, and 4, respectively. The physical distribution of the 60 loci on the 10 chromosomes was determined from their mapped positions on the Chiifu-401-42 genome sequence (Fig. 3). The majority of the SNP loci were distributed evenly throughout the genome. The 60 SNP loci, which satisfied the five criteria described in Materials and Methods, were selected as core SNP markers for further analysis (Table 1).

Principal component analysis of 360 SNP markers. Blue dots indicate the proportion of variance accounted for by individual principal components, and orange dots indicate the cumulative percentage of contribution of principal components to the total variance

Distribution of the 60 core SNP loci on the 10 chromosomes of the Chinese cabbage genome
3.3 Evaluation of polymorphism for the core SNP markers in inbred lines
Data from the 166 Chinese cabbage inbred lines were used to calculate the PIC, MAF, heterozygosity, and gene diversity values for each core SNP marker. The PIC for the 60 markers across all 166 accessions ranged from 0.21 to 0.37 with an average of 0.35. The PIC percentage value between 0.3 and 0.4 was 86.7% (Table 2), which suggested that the markers were strongly polymorphic.
The MAF of the 166 inbred lines ranged from 0.14 to 0.50 with an average of 0.37. The ObsHET of the 166 inbred lines ranged from 0.01 to 0.22 with an average of 0.04. Given that the 166 lines included in this study had been selfed for many generations and all were predicted to be largely homozygous, low ObsHET values among these lines were expected. Indeed, only two lines (A06_2523098 and A09_28158636) showed ObsHET values > 0.1. The genetic diversity within the germplasm collection ranged from 0.24 to 0.5 with an average of 0.45 (Table 2).
3.4 Cluster analyses of genetic distance and genetic diversity among inbred lines
In general, Chinese cabbage accessions can be grouped into spring, summer, and autumn ecotypes based on the growing season (Su et al. 2018). In addition, a number of heading types are distinguished, such as flat heading, oval heading, and straight heading types. We compared three datasets to evaluate the core SNP markers in this study. First, the dataset of 1167 SNPs was used to analyze the genetic distance and diversity among the 166 Chinese cabbage inbred lines (Supplementary Fig. 1). In the unrooted NJ tree, the inbred lines were predominantly grouped into spring, summer, and autumn ecotypes at a low genetic distance. However, some mixture of spring, summer, and autumn ecotypes was apparent. Second, the dataset comprising 360 polymorphic SNPs was used to analyze the genetic distance and diversity among the Chinese cabbage accessions (Supplementary Fig. 2). Clustering of the Chinese cabbage inbred lines into distinct clusters of spring, summer, and autumn ecotypes was improved compared with that achieved with the 1167 SNP dataset. However, several different ecotypes were still mixed together.
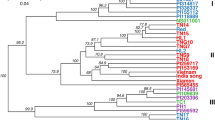
Finally, the genetic diversity among the Chinese cabbage inbred lines was analyzed using the core SNP marker dataset. In the unrooted NJ tree constructed from pairwise genetic distances, the 166 genotypes were clustered into three groups at a low genetic distance (Fig. 4). The three groups corresponded to the spring, summer, and autumn ecotypes. Thus, the clustering of Chinese cabbage accessions using the core SNP dataset was far superior to that achieved with the 1167 SNP and 360 SNP datasets. Similarly, the clustering of Chinese cabbage accessions was better than that realized using the 568 B. rapa SNPs (Su et al. 2018). Thus, our results indicated that the 60 core SNPs could effectively represent the genetic diversity among the Chinese cabbage inbred lines.

Cluster analysis of the core SNP data sets for 166 Chinese cabbage inbred lines. The unrooted dendrograms were constructed using the NJ method from distance matrices calculated from the 60 SNP dataset. The inbred lines of spring, summer, and autumn ecotypes are shown using green, red, and yellow lines, respectively. (Color figure online)
3.5 Evaluation of the efficiency of the core set of SNPs in hybrid cultivars
KASP genotyping showed that 178 hybrid cultivars of Chinese cabbage harbored two different homozygous alleles and a heterozygous allele (Fig. 5). Polymorphisms of the core SNP markers among the 178 Chinese cabbage hybrids were analyzed. The PIC values ranged from 0.12 to 0.37 with an average value of 0.33. The MAF of the 178 hybrid cultivars ranged from 0.07 to 0.49 with an average of 0.35. The ObsHET of the hybrid cultivars ranged from 0.02 to 0.97 with an average of 0.36 (Supplementary Table 4). Given that the cultivars were all seed-raised hybrids, it was expected that the heterozygosity values would be considerably higher than those among the 166 inbred lines.

Development of SNP markers from Chinese cabbage hybrid cultivars for KASP genotyping. SNPs were automatically called for AA, AB, and BB genotypes. Red dots are homozygous for one allele, blue dots are homozygous for a second allele, and green dots are the heterozygous allele. (Color figure online)
A matrix of genetic distances derived from the core SNP dataset for the 178 hybrid cultivars was used to construct an unrooted NJ tree with PowerMarker (Liu and Muse 2005). The hybrid cultivars were clustered into three groups corresponding to spring, summer, and autumn ecotypes (Fig. 6), which was consistent with the clustering of the 166 inbred lines using the core SNP markers. The NJ tree indicated that the core set of SNP markers was capable of differentiating the 178 hybrid genotypes into genetically coherent groups. In addition, DNA fingerprinting based on the SNP genotyping data for individual cultivars is feasible (Supplementary Fig. 3).

Cluster analysis of 178 Chinese cabbage hybrid cultivars using the core SNP markers. The unrooted dendrogram was constructed using the NJ method. The cultivars of spring, summer, and autumn ecotype are shown using green, red, and yellow lines, respectively. (Color figure online)
4 Discussion
4.1 Development of KASP SNP marker sets
Previously, the majority of markers used in Chinese cabbage breeding were RAPDs, AFLPs, SSRs, and InDels (Song et al. 1990; Powell et al. 1996; Das et al. 1999; He et al. 2003; Soengas et al. 2011). However, the frequency of polymorphism among Chinese cabbage accessions is reported to be limited. SNP markers have been employed in many research fields, including linkage mapping, population genetics, and comparative genomics, in a variety of crops such as rice, maize, and barley (Rafalski 2002; Varshney et al. 2008; Tian et al. 2015). Recently, SNP markers have been developed and converted for cost-effective genotyping platforms such as KASP and BeadXpress assays (Allen et al. 2011; Cortés et al. 2011; Hiremath et al. 2012; Roorkiwal et al. 2013).
KASP assays provide flexibility with respect to the number of SNPs used for genotyping. This gives KASP assays an advantage over other SNP genotyping assays. KASP assays have been shown to be suitable for estimation of genetic diversity in common bean, chickpea, and peanut (Allen et al. 2011; Cortés et al. 2011; Hiremath et al. 2012) but have not been applied previously for large-scale germplasm characterization in Chinese cabbage. In this study, candidate SNPs for KASP assays were initially selected on the basis of reproducibility, signal strength, and utility for definition of the different genotypes. Of the original 1167 SNPs, a core set of 60 SNPs was successfully screened for KASP assays. The non-utility of the remaining SNP markers is likely due to technical issues, incorrect primer design, or the need to optimize PCR conditions.
To construct an SNP array for Chinese cabbage DNA fingerprinting, a set of evaluation hybrids representing a broad genetic pool, reasonable SNP selection principles, and a reliable genotype clustering procedure is required. Polymorphism bias will be present if the genetic background of the selected materials is concentrated. In addition, Chinese cabbage DNA fingerprinting must be able to differentiate among hybrids quickly and accurately. Consequently, representative hybrids must be selected to validate the efficiency of genotype discrimination and accuracy of heterozygous base calling for candidate SNPs. Common assessment indices for selecting a set of SNPs include repeatability, discriminatory power, uniformity of distribution, and conservatism of flanking sequences. To ensure that three genotype clusters can be readily distinguished, the selected SNP should be a single-copy locus, and both inbred and hybrid lines should be used to evaluate cluster independence and stability. In addition, automatic SNP calling using KASP software is sometimes prone to error, especially when a rare AB genotype cluster is present, which needs to be improved.
4.2 Evaluation of core SNP polymorphism
Our goal in identifying core SNPs is to use the fewest SNPs to represent the most genetic diversity among Chinese cabbage germplasm. The genetic diversity of each locus was estimated by calculating the frequency of the genotype based on the PIC following the formula developed by Anderson et al. (1993). In this study, the average PIC value of Chinese cabbage was considerably higher than those reported in a recently developed KASP assay or Illumina SNP array for pigeonpea, maize, and wheat of 0.16, 0.09, and 0.33, respectively (Saxena et al. 2012; Tobias et al. 2013; Tian et al. 2015). All of these PIC values suggest a high discriminatory ability and reliable deep resolution for these SNPs. In addition, the higher PIC value of the 166 Chinese cabbage inbred lines may be indicative of higher genetic diversity in this experimental set of germplasm. The polymorphism detected in this study was assessed in accessions that are representative of the expression of different characteristics of major Chinese cabbage cultivars; thus, the core SNP markers are of importance for related studies and applications in Chinese cabbage.
Previously, SSR markers were detected within morphotypes represented by multiple accessions, and the mean PIC values reported were 0.60 (Brussels sprouts), 0.54 (broccoli), 0.57 (cauliflower), 0.65 (cabbage), and 0.31 (Pak-choi) (Federico et al. 2008; Su et al. 2017). It must be noted that for biallelic markers such as SNPs, the PIC ranges from 0 to 0.5, whereas for multiallelic markers such as SSRs, the PIC values can exceed 0.5 and approach 1. SSR markers have been used for cultivar identification for more than 10 years because of their high discriminatory power and relatively simple experimental procedures (Richard et al. 2008). Compared with SSRs, SNPs are biallelic and high-throughput and thus are easy to read, compare, and integrate between different data sources. We would also like to stress that the molecular information provided in this paper easily can be adapted and exploited in alternative technological platforms for SNP detection.
4.3 Applications of core SNP marker sets in marker-associated research and germplasm characterization
Rapid genotyping is necessary for screening a large number of DNA samples in a limited period. This is the case, for example, when a phenotypic trait is mapped at high resolution in a large population of individuals. In addition, with the development of a variety of SNP genotyping platforms, SNPs are thus ideal for DNA fingerprinting, analysis of genetic diversity, and molecular marker-assisted selection (MAS) in breeding. The identification of the 60 core SNP markers in Chinese cabbage may provide a sufficiently high marker density in many populations to allow thorough screening of the genome for discovery of quantitative trait loci, association analysis, map-based cloning, and anchoring of genome sequences with a genetic map.
Assessment of relationships within germplasm collections can assist in the selection of more distantly related lines for use in breeding programs. In this study, SNP genotyping data were used to quantify the genetic diversity and genetic distances within a Chinese cabbage germplasm collection (Fig. 3). Using cluster analysis, the relationships among a large number of genotypes were examined and the genotypes were grouped consistent with the ecotype (i.e., spring, summer, and autumn ecotypes). The clustering of the accessions using the core SNPs was much better than that achieved with the 360 and 1167 SNP datasets, which resulted in a degree of mixing of ecotypes within clusters. In this study, the majority of the branches in the dendrograms received strong support, which demonstrated the reliability of the core set. In addition, all of the inbred lines were distinguished based on polymorphism of the 60 core SNPs, which indicated that these SNPs effectively represented the genetic diversity among the Chinese cabbage germplasm collection.
4.4 Selection of SNPs for Chinese cabbage DNA fingerprinting
Protection of plant breeder’s rights is an important issue in Chinese cabbage breeding (Buanec 2010; Liu et al. 2013). Previously, for cultivar identification, a grow-out test applied in conjunction with traditional DUS technology involves growing plants to maturity and assessing several morphological characteristics that distinguish individual plants. However, environmental influences on morphological characters and time demands make it diffcult to collect morphological data (Reid et al. 2011). In recent years, some elite parents have been used frequently in breeding, which has resulted in high genetic similarity of Chinese cabbage hybrids and diffculty in distinguishing cultivars based on phenotypic traits.
Development of SNP markers in Chinese cabbage is in its infancy. Not all SNPs are suitable for DNA fingerprinting, however, and some loci do not meet array chip design requirements. Genotyping is relatively important for diploid crops such as rice, maize, and hybrid Chinese cabbage cultivars. With regard to hybrid cultivars, one SNP locus may display three genotypes, namely, AA, BB, and AB. It is extremely important to distinguish accurately the hybrid genotypes from the homozygous genotypes. Hybrid cultivars constitute the majority of the Chinese seed market, and the variety of genotypic combinations increases the complexity of genotyping. In this study, only 60 SNP markers were identified among Chinese cabbage. As a result, the ability to distinguish hybrids and the accuracy of the core marker set is more powerful. In addition, molecular markers can be used to distinguish hybrids for precise assessment of plant genotypes, but the relationship of genotype to phenotypic traits remains a crucial issue. SNP markers have the advantage over other types of molecular markers in that they can be associated with specific genes. The clustering analysis of hybrid Chinese cabbage cultivars analyzed using the core set of SNP markers differentiated all genotypes, thus indicating that the screening strategy for identification of the core SNP markers was effective.
In summary, in this study, we developed an invaluable resource of cost-effective and polymorphic KASP markers for Chinese cabbage, which are robust, simple to use, and easy to interpret and record. We identified a set of 60 representative SNPs that show a high level of polymorphism and are evenly distributed across the B. rapa genome. Genotype characteristics and genetic diversity of 166 inbred lines representative of Chinese cabbage germplasm and 178 hybrid Chinese cabbage cultivars were analyzed using a set of core SNP markers. In both germplasm collections, accessions were separated into spring, summer, and autumn ecotype groups. The core SNPs will enable breeders to genotype large numbers of accessions rapidly and economically and will assist in MAS breeding. In addition, the core SNP markers will help protect breeders’ rights through application of the markers for Chinese cabbage DNA fingerprinting in the future.
References
Allen AM, Barker GLA, Berry ST (2011) Transcript-specific, single-nucleotide polymorphism discovery and linkage analysis in hexaploid bread wheat (Triticum aestivum L.). Plant Biotechnol J 9:1086–1099. https://doi.org/10.1111/j.1467-7652.2011.00628.x
Anderson JA, Churchill GA, Autrique JE, Tanksley SD, Sorrells ME (1993) Optimizing parental selection for genetic linkage maps. Genome 36:181–186. https://doi.org/10.1139/g93-024
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635. https://doi.org/10.1093/bioinformatics/btm308
Buanec BL (2010) Protection of plant-related innovations: evolution and current discussion. World Patent Inf 28:50–62. https://doi.org/10.1016/j.wpi.2005.10.002
Cortés AJ, Chavarro MC, Blair MW (2011) SNP marker diversity in common bean (Phaseolus vulgaris L.). Theor Appl Genet 123:827–845. https://doi.org/10.1007/s00122-011-1630-8
Das S, Rajagopal J, Bhatia S, Srivastava PS, Lakshmikumaran M (1999) Assessment of genetic variation within Brassica campestris cultivars using amplified fragment length polymorphism and random amplification of polymorphic DNA markers. J Biosci 24:433–440. https://doi.org/10.1007/bf02942653
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491. https://doi.org/10.1016/1050-3862(92)90005-P
Fan JB, Oliphant A, Shen R, Kermani BG, Garcia F, Gunderson KL, Hansen M, Steemers F, Butler SL et al (2003) Highly parallel SNP genotyping. Cold Spring Harb Symp Quant Biol 68:69–78. https://doi.org/10.1101/sqb.2003.68.69
Federico L, Amy V, Thomas C (2008) Development of a set of public SSR markers derived from genomic sequence of a rapid cycling Brassica oleracea L. genotype. Theor Appl Genet 117:977–985. https://doi.org/10.1007/s00122-008-0837-9
Hiremath PJ, Kumar A, Penmetsa RV (2012) Large-scale development of cost-effective SNP marker assays for diversity assessment and genetic mapping in chickpea and comparative mapping in legumes. Plant Biotechnol J 10:716–732. https://doi.org/10.1111/j.1467-7652.2012.00710.x
Ke GL (2010) Chinese cabbage breeding. China Agricluture Press, Beijing, pp 18–24
Kuang M, Wei SJ, Wang YQ, Zhou DY, Lei MA, Fang D, Yang WH, Ma ZY (2016) Development of a core set of SNP markers for the identification of upland cotton cultivars in China. J Integr Agr 15:954–962. https://doi.org/10.1016/S2095-3119(15)61226-6
Langridge P, Chalmers K, Mohler V, Schwarz G, Mackill DJ, Mcnally KL, Scheible WR, Törjek O, Altmann T, et al (2005) Molecular marker systems in plant breeding and crop improvement. Biotechnology in agriculture & forestry Vol 55. Springer, Berlin, pp 10–12. https://doi.org/10.1038/npg.els.0002024
Li P, Zhang S, Zhang S, Li F, Zhang H, Liu X, Wu J, Wang X, Sun R (2015) Carotenoid identification and molecular analysis of carotenoid isomerase-encoding BrCRTISO, the candidate gene for inner leaf orange coloration in Chinese cabbage. Mol Breed 35:72. https://doi.org/10.1007/s11032-015-0190-z
Liu K, Muse SV (2005) POWERMARKER: integrated analysis environment for genetic marker data. Bioinformatics 21:2128–2129. https://doi.org/10.1093/bioinformatics/bti282
Liu YF, Zhang JH, Bo L, Yang XH, Yan-Gang LI, Ye W, Wang JM, Hui Z, Guan JJ (2013) Statistic analysis on quantitative characteristics for developing the DUS test guideline of Ranunculus asiaticus L. J Integr Agric 12:971–978. https://doi.org/10.1016/S2095-3119(13)60474-8
Livak KJ, Flood SJA, Marmaro J, Giusti W, Deetz K (1995) Oligonucleotides with fluorescent dyes at opposite ends provide a quenched probe system useful for detecting PCR product and nucleic acid hybridization. Genome Res 4:357–362
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M et al (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. https://doi.org/10.1101/gr.107524.110
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in excel population genetic software for teaching and research. Mol Ecol Notes 6:288–295. https://doi.org/10.1111/j.1471-8286.2005.01155.x
Powell W, Morgante M, Andre C, Hanafey M, Vogel J, Tingey S, Rafalaski A (1996) The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol Breed 3:225–238. https://doi.org/10.1007/BF00564200
Rafalski A (2002) Applications of single nucleotide polymorphisms in crop genetics. Curr Opin in Plant Biol 5:94–100. https://doi.org/10.1016/S1369-5266(02)00240-6
Reid A, Hof L, Felix G, Rücker B, Tams S, Milczynska E, Esselink D, Uenk G, Vosman B et al (2011) Construction of an integrated microsatellite and key morphological characteristic database of potato varieties on the EU common catalogue. Euphytica 182:239–249. https://doi.org/10.1007/s10681-011-0462-6
Richard GF, Kerrest A, Dujon B (2008) Comparative genomics and molecular dynamics of DNA repeats in Eukaryotes. Micriobiol Mol Biol Rev 72:686–727. https://doi.org/10.1128/MMBR.00011-08
Roorkiwal M, Sawargaonkar SL, Chitikineni A, Thudi M, Saxena RK, Upadhyaya HD, Vales MI, Riera-Lizarazu O, Varshney RK (2013) Single nucleotide polymorphism genotyping for breeding and genetics applications in Chickpea and Pigeonpea using the BeadXpress platform. Plant Genome 6:494. https://doi.org/10.3835/plantgenome2013.05.0017
Saxena RK, Varma Penmetsa R, Upadhyaya HD, Kumar A, Noelia CC, Rajeev KV (2012) Large-scale development of cost-effective single-nucleotide polymorphism marker assays for genetic mapping in Pigeonpea and comparative mapping in Legumes. DNA Res 19:449–461. https://doi.org/10.1093/dnares/dss025
Soengas P, Cartea ME, Francisco M, Lema M, Velasco P (2011) Genetic structure and diversity of a collection of Brassica rapa subsp. rapa L. revealed by simple sequence repeat markers. J Agric Sci 149(05):617–624. https://doi.org/10.1017/s002185961100013x
Song K, Osborn TC, Williams PH (1990) Brassica taxonomy based on nuclear restriction fragment length polymorphisms (RFLPs). Theor and Appl Genet 79:497–506. https://doi.org/10.1007/bf00226159
Steemers FJ, Gunderson KL (2007) Whole genome genotyping technologies on the BeadArray ™ platform. Biotechnol J 2:41–49. https://doi.org/10.1002/biot.200600213
Su T, Zhao X, Sui G, Yu S, Zhang F, Wang W, Zhang D et al (2017) Validation of a set of informative simple sequence repeats markers for variety identification in Pak-choi (Brassica rapa L.ssp. chinensis var.communis). Plant Breed 136:410–419. https://doi.org/10.1111/pbr.12483
Su T, Li P, Yang J, Sui G, Yu Y, Zhang D, Zhao X, Wang W, Wen C et al (2018) Development of cost-effective single nucleotide polymorphism marker assays for genetic diversity analysis in Brassica rapa. Mol Breed 38:42. https://doi.org/10.1007/s11032-018-0795-0
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. https://doi.org/10.1093/molbev/msr121
Tian HL, Wang FG, Zhao JR, Yi HM, Wang L, Wang R, Yang Y, Song W (2015) Development of maizeSNP3072, a high-throughput compatible SNP array, for DNA fingerprinting identification of Chinese maize varieties. Mol Breed 35:136. https://doi.org/10.1007/s11032-015-0335-0
Tobias W, Simon M, Longin C, Korzun V, Akhunov E, Ebmeyer E, Schachschneider R, Schacht J, Kazman E, Jochen CR (2013) Population structure, genetic diversity and linkage disequilibrium in elite winter wheat assessed with SNP and SSR markers. Theor Appl Genet 126:1477–1486. https://doi.org/10.1007/s00122-013-2065-1
Varshney RK, Thiel T, Sretenovic-Rajicic T, Baum M, Valkoun J, Guo P, Grando S, Ceccarelli S, Graner A (2008) Identification and validation of a core set of informative genic SSR and SNP markers for assaying functional diversity in barley. Mol Breed 22:1–13. https://doi.org/10.1007/s11032-007-9151-5
Wang XW, Wang HZ, Wang J, Sun RF, Wu J, Liu SY, Bai YQ, Mun JH, Bancroft I et al (2011) The genome of the mesopolyploid crop species Brassica rapa. Nat Genet 43:1035–1039. https://doi.org/10.1038/ng.919
Acknowledgements
This work was supported by grants from the Technology Innovation Program, BAAFS (KJCX20161503), the Key Program of the Beijing Municipal Science and Technology Committee (D171100002517001), the National Key Research and Development Program of China (2017YFD0102004), and the Earmarked Fund for China Agriculture Research System (CARS-23-A-05). We thank Robert McKenzie, PhD, from Liwen Bianji, Edanz Group China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors confirm that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig.
1 Cluster analysis of the 1167 SNP dataset for 166 Chinese cabbage inbred lines. The inbred lines of spring, summer, and autumn ecotype are shown using green, red, and yellow lines, respectively. (JPEG 6084 kb)
Supplementary Fig.
2 Cluster analysis of the 360 SNP dataset for 166 Chinese cabbage inbred lines. The inbred lines of spring, summer, and autumn ecotype are shown using green, red, and yellow lines, respectively. (JPEG 6072 kb)
Supplementary Fig.
3 DNA fingerprinting of 178 Chinese cabbage hybrid cultivars using the core SNP markers. The order of the SNP markers is the same as the order in Table 1. The order of the cultivars is the same as the order in Supplementary Table 2. (JPEG 7984 kb)
Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Li, P., Su, T., Yu, S. et al. Identification and development of a core set of informative genic SNP markers for assaying genetic diversity in Chinese cabbage. Hortic. Environ. Biotechnol. 60, 411–425 (2019). https://doi.org/10.1007/s13580-019-00138-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13580-019-00138-4