
Abstract
Competitive allele-specific PCR (KASPar) assay is a user-friendly system that provides flexibility in the numbers of single nucleotide polymorphisms (SNPs) and genotypes. Based on Illumina-GA-IIx genomic data from 10 genotypes with a broad genetic background, 3183 SNPs were selected for KASPar assays development, and 568 were finally converted and selected for Brassica rapa germplasm characterization (17.8%) on the basis of reproducibility, missing data rate, and uniform genetic distribution. High levels of polymorphism of these markers across 231 B. rapa genotypes were verified, illustrating by high polymorphic information content (averaged 0.34), minor allele frequency (0.37), genetic diversity (0.45), and the low observed heterozygosity (0.10). Based on the SNP dataset, structure and principal coordinates analysis, and neighbor-joining phylogenetic methods were used to examine the population structure and gave highly consistent results. The 231 accessions were divided into the four primary subspecies, representing 99 accessions from B. rapa ssp. pekinensis, 85 from B. rapa ssp. chinensis, 30 from B. rapa ssp. rapifera, and 17 from B. rapa ssp. oleifera and were further subdivided into 12 lower-order clusters according to different morphotypes. The genetic variability and pairwise fixation index analysis revealed that the ssp. pekinensis accessions possess the most extensive genetic variation among the four subspecies. The KASPar system is highly useful for validating SNPs and will be valuable for genetics research and breeding applications in B. rapa.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Brassica crops are important sources of food, feed, and fuel in many parts of the world and provide a unique opportunity for investigating plant genome evolution. The six widely cultivated Brassica species, including three diploid species B. rapa (A genome), B. nigra (B genome), and B. oleracea (C genome) and three amphidiploid species B. juncea (A and B genomes), B. napus (A and C genomes), and B. carinata (B and C genomes), are a classic example of the importance of polyploidy in plant evolution, as illustrated by the triangle of U (Nagaharu, 1935). Furthermore, the B. rapa crops, especially the two subspecies ssp. pekinensis (Chinese cabbage) and ssp. chinensis (Pak choi) (Labana and Gupta, 1993), play critical roles in vegetable production and supply in China and other Asian countries. In China, B. rapa is the top-ranked vegetable in terms of cultivated area and total yield and is extremely important to the country’s agricultural economy.
B. rapa crops are predominantly cross-pollinated diploid (2n = 20), with an estimated genome size of 485 Mb (Wang et al. 2011). As a hybrid crop, B. rapa is a model plant for genetic studies due to its high recombination rate and rich genetic diversity. In terms of breeding, the selection of diverse genetic resources possessing different agronomic characteristics and understanding the genetic relationships between these breeding materials are crucial for cultivar improvement. However, in many cases, we know very little about the ecology and population structure of these genetic materials. Also, centuries of artificial selection for desirable traits have resulted in an overall loss of genetic diversity in many of the early self-pollinated inbred lines, which are important materials for B. rapa crops breeding. Therefore, it is imperative to understand the genetic diversity present within the available breeding lines using genome-wide molecular markers.
Over the past three decades, several different DNA marker technologies have been used to detect genetic diversity in the cultivated B. rapa gene pool, such as random amplified polymorphic DNA (RAPD), amplified fragment length polymorphism (AFLP), and simple sequence repeats (SSRs) (Song et al. 1988; Powell et al. 1996; Chen et al. 2000; Guo et al. 2002; He et al. 2003). At present, SNPs are the markers of choice for genome-wide analyses due to their higher density level across the genome and high genetic stability, and also because they can be readily adapted to automated genotyping methods. A number of high-throughput, cost-effective SNP genotyping platforms have been developed such as the GoldenGate (Fan et al. 2003) and Infinium platforms (Steemers and Gunderson 2007), TaqMan (Livak et al. 1995), and the KASPar platform (KBiosciences, www.kbioscience.co.uk). Many of these platforms have been applied to important crop species such as barley, wheat, maize, soybean, cowpea, and pea (Allen et al. 2011; Cortés, et al. 2011; Hiremath et al. 2012). KASPar is a user-friendly system that provides flexibility in the numbers of SNPs and genotypes to be used for assays. Because of the importance of KASPar assays in genotyping variable numbers of samples with variable numbers of SNPs, they have been developed for wheat, common bean, and chickpea (Allen et al. 2011; Cortés, et al. 2011; Hiremath et al. 2012).
The aim of this study was to use the newly KASPar technology to develop a flexible and cost-effective SNP genotyping platform in B. rapa. In particular, by using a broad panel of 231 materials across four subspecies, B. rapa ssp. pekinensis, B. rapa ssp. chinensis, B. rapa ssp. rapifera, and B. rapa ssp. oleifera, the newly developed SNP markers were conducted to characterize the polymorphism information of the markers and genotype the germplasm. Moreover, their use in marker-associated research, germplasm charterization, population structure, and phylogenetic relationships assessment across the four subspecies was emphatically evaluated.
Materials and methods
Germplasm and DNA isolation
A total of 231 B. rapa inbred lines, which were collected from different areas in China, as well as Japan, Korea, Thailand, Pakistan, Nepal, Vietnam, Russia, England, the USA, and Australia, were used in this study (Supplementary Table 1).
Total DNA was extracted from two to three young leaves following a standard DNA isolation protocol (Su et al. 2014). DNA quality and concentration were measured with a NanoDrop 2000 UV spectrophotometer (Thermo Scientific, MA, USA), and working solutions were prepared at a concentration of 10 ng/μL.
Single nucleotide polymorphism and KASPar genotyping
SNPs were identified by using Illumina GA IIx technology on 10 genotypes, including two autumn-, two summer- and one spring-type accessions from ssp. pekinensis, two accessions from ssp. chinensis var. communis, and one each from subspecies chinensis var. communis (naibaicai), chinensis var. communis (heiyebaicai) and rapifera, which were selected from the 231 inbred lines. A total of 10.8 GB of 36-base short single-end DNA sequencing reads was generated from these genotypes. The identification of SNPs between the 10 genotypes and the reference genome of Chiifu-401-42 (v1.5) (Wang et al. 2011) was performed using the GATK software (McKenna et al. 2010).
High-quality SNP candidates were then selected for the KASPar assays. The strict criteria used for the selection of high-quality SNPs for KASPar assays included the following: (1) the minor allele frequency (MAF) value among the 10 genotypes must ≥ 0.1, (2) read depth ≥ 5, and (3) the potential SNP candidates are evenly distributed across the whole genome. Finally, a total of 3183 high-quality SNPs were filtered and found suitable for KASPar assays.
For each SNP, two allele-specific forward primers and one common reverse primer were designed by LGC (Laboratory of the Government Chemist). Using these primers, KASPar assays were performed in final reaction volumes of 1 μL in 1536-plates (No. KBS-0751-001, KBioscience), containing 1× KASP reaction mix (KBS-1016-011, KBioscience), 12 nM each allele-specific forward primer, 30 nM reverse primer, and 4 ng of genomic DNA. The Gene Pro Thermal cycler (Hydrocycler) was used for amplification with the following cycling conditions: 15 min at 94 °C, 10 touch-down cycles of 20 s at 94 °C and 60 s at 65–57 °C (the annealing temperature for each cycle being reduced by 0.8 °C per cycle), and 26–42 cycles of 20 s at 94 °C and 60 s at 57 °C. Fluorescence detection of the reactions was performed using an Omega Fluorostar scanner (BMG PHERAstar), and the data were analyzed using KlusterCaller 1.1 software (KBioscience). Detailed instructions can be downloaded at www.kbioscience.co.uk.
All markers were comprehensively screened over the 231 genotypes, and 1167 SNPs (36.7%) with scorable allele calls were successfully developed. However, after excluding markers that (1) were monomorphic, (2) had > 20% missing values across all genotypes, (3) had ambiguous SNP calls, (4) had a MAF < 0.1, and (5) had loci for which > 10 inbred samples showed the “ab” genotype, the total number of usable SNP markers was reduced to 568 (17.8%). Polymorphic markers were classified into eight segregation patterns (ab × cd, ef × eg, hk × hk, lm × ll, nn × np, aa × bb, ab × cc and cc × ab). For the SNP marker, its normal segregation pattern was aa × bb.
Marker polymorphism and population structure analysis
The polymorphic information content (PIC) and gene diversity values for the SNP markers in this study were calculated by using PowerMarker software (http://statgen.ncsu.edu/powermarker/). To assess genetic diversity within different subspecies or variant clusters, we used Genalex 6.3 (Peakall and Smouse, 2006) to estimate MAF, observed heterozygosity (ObsHET), and the FST values.
Population structure was investigated using two different methods: principal coordinates analysis (PCoA) and STRUCTURE (Pritchard et al. 2000). PCoA was carried out based on the modified Rogers’ distances (Wright 1978). The modified Rogers’ distances (d w ) were calculated as d w = \( \frac{1}{\sqrt{2m}}\ \sqrt{\sum_{i=1}^m\sum \limits_{j=1}^m{\left({p}_{ij}-{q}_{ij}\right)}^2} \), and genetic similarities were calculated as 1 − d w to produce principal coordinate scores, which were then used to investigate different population groups within the collection of 231 germplasm accessions. Patterns revealed by the first three coordinates of each accession were plotted using the G3D procedure.
STRUCTURE was run on the full dataset of 568 SNPs using an admixture model and the default settings. The most likely value of k was determined by the delta k method (Evanno et al. 2005) implemented in STRUCTURE HARVESTER (Earl and VonHoldt 2011).
The matrix of genetic distances was used to create a neighbor-joining (N-J) tree with Mega 5 (Tamura et al. 2011). Based on these categories, population and subpopulation genetic structure were further analyzed by conducting an analysis of molecular variance (AMOVA) using Arlequin (Excoffier et al. 1992; Peakall and Smouse, 2006). To assess differentiation significance, alleles were randomly permuted 1000 times among individuals (Edgington 1995).
Results
Identification of candidate SNPs and development of SNP markers for the KASPar assays
Based on the 10 resequencing genotypes, a total of 149,100,294 usable single-end reads (each of which was 36 bp in length), with an average depth of 3.9× and coverage of 81.1% were generated (Supplementary Table 2). The Q20 ratio averaged 90.3%, and the guanine+cytosine (GC) content was 40.5%. Through comparisons with the reference genome of the inbred Chiifu-401-42 (v1.5), we detected a total of 709,037 SNPs, which translated to an overall density across the genome of 2488 SNPs/Mb.
A total of 3183 high-quality SNPs were filtered for the development of KASPar assays, and finally, 568 SNP markers which satisfied the criteria as described were selected for further analysis of the 231 genotypes. These SNP markers developed based on different ssp. pekinensis, ssp. chinensis, and rapifera genotypes, which were also polymorphic and useful for oleifera accessions (Supplementary Table 3). The physical distribution of the 568 loci on the 10 chromosomes was determined from their mapped positions on the Chiifu-401-42 genome sequence (Fig. 1). Most of the SNP loci were found to be distributed evenly throughout the genome, with an average density of 2.0 SNPs/Mb. Only two gaps (> 3 Mb intervals) were found to be present on chromosome 5.

Distribution of the 568 SNP marker loci on the 10 chromosomes of Brassica rapa
Marker polymorphism analysis in B. rapa
Data obtained from the 231 B. rapa genotypes were used to calculate the PIC value of each SNP marker. The PIC for the 568 markers across all accessions ranged from 0.10 to 0.38, with an average of 0.34 for all examined accessions. In particular, the percentage of PIC values between 0.3 and 0.4 was 87.9% (Fig. 2a; Supplementary Table 4), which suggested that these markers were strongly polymorphic. Loci (46.1%) had MAF values of 0.4–0.5, 49.8% had MAF values of 0.3–0.4, and 4.6% had values of 0–0.3 (Fig. 2b; Supplementary Table 4). The ObsHET of the 568 variation loci ranged from 0 to 0.54, with an average of 0.10. As the 231 lines included in this study have been selfed for many generations and can all be expected to be largely homozygous, very little heterozygosity should be present in these lines. Indeed, only 2.9% of the lines had ObsHET values > 0.2 (Fig. 2c; Supplementary Table 4). The genetic diversity within the germplasm collection was also assessed, and was found to range from a low of 0.11 to a high of 0.5, with an average of 0.45 (Fig. 2d; Supplementary Table 4).

PIC (a), MAF (b), observed heterozygosity (c), and genetic diversity (d) values for the 568 SNP markers based on data from 231 inbred lines
Population classification analysis
PCoA was initially performed based on the 568 high-quality SNPs to investigate population structure on the entire dataset of 231 genotypes. The proportion of genotypic variance explained by the three principal coordinates was 14.21, 5.07, and 3.92%, respectivly (Supplementary Fig. 1). Plotting with both the 3D (Supplementary Fig. 1) and 2D (Fig. 3a) plot of the PCoA confirmed the presence of the four major populations, which is in agreement with traditional classification schemes. Population I is referred to as the ssp. pekinensis, which included 99 accessions, population II included 85 accessions from ssp. chinensis, while populations III and IV included 30 and 17 inbred lines, respectively, from ssp. rapifera and ssp. oleifera.

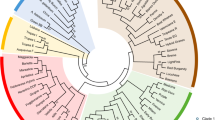
Analyses of population structure in B. rapa. a Principal coordinates analysis of population structure for the 231 B. rapa accessions. b Neighbor-joining tree of all inbred lines calculated from 568 SNP markers. The 12 divergent groups are shown in colored shapes. The scale bar indicates the simple matching distance. c Populations structure analysis. All 231 germplasm accessions were further divided into 12 subpopulations. Pop1, ssp. pekinensis; Pop2, ssp. chinensis; Pop3, ssp. rapifera; Pop4, ssp. oleifera. Subpop1, spring-type Chinese cabbage lines of ssp. pekinensis (Spr); Subpop2, autumn-type (Aut); Subpop3, summer-type (Sum); Subpop4, ssp. chinensis var. parachinensis (P); Subpop5, ssp. chinensis var. communis (naibaicai) (N); Subpop6, ssp. chinensis var. communis (heiyebaicai) (H); Subpop7, ssp. chinensis var. communis (C); Subpop8, ssp. chinensis var. japonica (J); Subpop9, ssp. chinensis var. taicai (T); Subpop10, Nar, ssp. chinensis var. narinosa (Nar); Subpop11, ssp. rapifera (R); Subpop12, ssp. oleifera (O)
To further assess relationships among these accessions, we used STRUCTURE and observed a gradual increase in log likelihood from k = 2–14. The best delta k was 5, then followed by k = 3, and the third was k = 4 (Supplementary Fig. 2). When k = 3, the 231 genotypes were divided to three groups: ssp. pekinensis, ssp. chinensis, and a mixed group of ssp. rapifera and ssp. oleifera (Fig. 3b); when k = 4, the STRUCTURE divided the set of materials to the four traditional subspecies, ssp. pekinensis, ssp. chinensis, ssp. rapifera, and ssp. oleifera, which was consistent with the phyletic classification results obtained from PCoA (Fig. 3b). When k = 5, on the basis of k = 4, the ssp. pekinensis was further divided to two groups, a spring-ecotype group and a mixed group of autumn- and summer-ecotype group (Fig. 3b).
We also showed phylogeny on an unrooted N-J tree calculated from pairwise genetic distances (Fig. 3c). All 231 genotypes were classified into four main groups with 12 lower-level clusters (Fig. 3c). Group I, comprising 99 inbred lines from ssp. pekinensis, was further grouped into three clusters based on cultivation season: spring-ecotype cluster (comprised of 29 spring-ecotype lines), autumn-ecotype cluster (comprised of 23 autumn-ecotype, one summer-ecotype and one spring-ecotype lines, respectively), and summer-ecotype cluster (comprised of 34 summer-ecotype, 11 autumn-ecotype and two spring-ecotype inbred lines, respectively) (Fig. 3c). Group II, 85 inbred lines from ssp. chinensis, was further divided into eight variety clusters; ssp. chinensis var. communis (36 lines), ssp. chinensis var. narinosa (13 lines), ssp. chinensis var. parachinensis (17 lines), ssp. chinensis var. taicai (five lines), ssp. chinensis var. japonica (two lines), ssp. chinensis var. communis (seven lines), and ssp. chinensis var. communis (five lines) (Fig. 3c). Groups III and IV included 30 and 17 accessions, respectively, of ssp. rapifera and ssp. oleifera (Fig. 3c).
Marker diversity analysis among the different populations
The polymorphism of these 568 SNP markers was further analyzed among the four subspecies. As shown in Table 1, 567, 556, 475, and 480 SNP markers were polymorphic in the subspecies pekinensis, chinensis, rapifera, and oleifera, respectively. The polymorphism rates of the SNP markers for these four groups were 99.8, 97.9, 83.6, and 84.5%, respectively. Notably, four SNP markers were found to be polymorphic exclusively in ssp. pekinensis (Table 1).
The average PIC values of ssp. chinensis, ssp. rapifera, and ssp. oleifera were 0.28, 0.28, and 0.25, respectively, while a higher PIC value, that was 0.32, was detected in ssp. pekinensis. The corresponding MAFs and gene diversities were 0.26, 0.27, 0.24, and 0.33 and 0.35, 0.36, 0.32, and 0.41, respectively, while the observed heterozygosity showed a different trend, with values of 0.09, 0.24, 0.17, and 0.05, respectively (Table 1).
Population structure in B. rapa crops
The 231 genotypes were divided into four primary populations and then 12 subpopulations. To test if populations are significantly different, we hierarchically analyzed variation with an AMOVA analysis. The result indicated that 49.82% of the genetic variation resided between germplasms within different subpopulations and then followed by 20.71, 15.78, and 13.68%, which was each caused by variations within samples, variations between populations, and variations between subpopulation within population (Supplementary Table 5). Thus, we concluded that most of the variance was observed at the subpopulation level.
To test if populations and subpopulations are significantly different, we performed a randomization test. From the output, we can see four histograms representing the distribution of the randomized strata. The observed results in the output show that there was significant population/subpopulation structure considering all levels of the population/subpopulation strata (Fig. 4). Furthermore, pairwise estimates of FST showed that the highest level of genetic differentiation was between the ssp. pekinensis and the ssp. chinensis populations (FST = 0.15) and the lowest was between the ssp. chinensis and the ssp. oleifera populations (FST = 0.07) (Supplementary Table 6).

Significance testing of population and subpopulation differentiation. The black line represents the observed data. The above graphs show significant population differentiation at all levels given that the observed line does not fall within the distribution expected from the permutation
Discussion
SNP selection and KASPar assay in B. rapa
A number of marker systems, such as RAPD, AFLP, SSR, diversity array technology markers (DArT), and single feature polymorphism (SFP), have been developed for germplasm characterization of different crops (Song et al. 1988; Powell et al. 1996; Chen et al. 2000; Guo et al. 2002; He et al. 2003). Recently, SNP markers have also been developed and converted to cost-effective genotyping platforms such as KASPar and BeadXpress assays (Allen et al. 2011; Cortés, et al. 2011; Hiremath et al. 2012; Roorkiwal et al. 2013). KASPar assays provide flexibility in terms of number of SNPs used for genotyping. This feature provides upper edge to KASPar assays as compared to other SNP genotyping assays. KASPar assays have been found suitable for diversity estimation in common bean, chickpea, and peanu (Allen et al. 2011; Cortés, et al. 2011; Hiremath et al. 2012); however, this assay has not been used for large scale germplasm characterization in B. rapa.
In this study, candidate SNPs for KASPar assays were initially selected on the basis of reproducibility, signal strength, and their utility for defining the different genotypes. The candidate loci were further screened based on PIC, MAF, missing data rate, and uniform genetic distribution. Moreover, we also considered polymorphism bias, which will be present if the genetic background of the selected materials is narrow. Here, genomic sequence data from 10 genotypes with a broad genetic base was analyzed and used for SNP selection (Supplementary Table 2). Although, we did not use oleifera genotypes for SNP selection, some markers were also polymorphic and useful for oleifera accessions (Supplementary Table 3).
Of the 3183 selected SNPs, 1167 were successfully converted into KASPar assays (36.7%) and 568 were finally selected for germplasm characterization (17.8%). The failure of the remaining SNP markers to be validated is likely due to technical issues, incorrect primer design, and/or the need to optimize PCR conditions. The average PIC, MAF, genetic diversity, ObsHET, and PredHET value were 0.35, 0.37, 0.45, 0.10, and 0.45, respectively (Fig. 2; Supplementary Table 4). Especially, the average PIC value is much higher than recently developed KASPar assay or Illumina SNP array for pigeonpea, maize, and wheat representing as 0.16, 0.09, and 0.33, respectively (Saxena et al. 2012; Tian et al. 2015; Tobias et al., 2013). All these parameters suggest a high discriminatory ability and reliable high depth resolution for these SNPs. In addition, the higher PIC value of the 231 inbred lines may indicate a higher diversity in our experimental set but must certainly also be attributed to the larger number of lines included in our study. Previously, SSR markers were detected within morphotypes represented by multiple accessions and the mean PIC values were 0.48 (Brussels sprouts), 0.54 (broccoli), 0.57 (cauliflower), and 0.65 (cabbage) (Federico et al. 2008). It must be noted that for biallelic markers such as SNPs, the PIC ranges from 0 to 0.5; however, for multiallelic markers like SSRs, the PIC values can exceed 0.5 and approach 1. SSR markers have been used for variety identification for more than 10 years because of their high discriminatory power and associated relatively easy-performed experimental techniques. Compared with SSRs, SNPs are bi-allelic and high-throughput, making them easy to read, compare, and integrate between different data sources. In addition, with the development of a variety of SNP genotyping platforms, SNPs are thus ideal for DNA fingerprinting, genetic diversity analysis, and molecular marker-assisted selection (MAS) in breeding.
Utility of KASPar assays for marker-associated research and germplasm characterization
The current availability of the 568 markers in B. rapa could provide high or significant marker density in many of the populations to be adequate to allow a thorough scan of the genome for QTL discovery, association analysis, and map-based cloning and anchoring of the genome sequencewith the genetic map. Seven of the 231 plant accessions in this study are reported to be parents of several mapping populations segregating for various economically important traits, such as heading color and downy mildew resistance (with QDX (Br053) and ZDJ (Br062) as parents), leaf color (ZYC (Br164) and ZDJ (Br062)), verticillium wilt resistance (CR-WM (Br020) and JDY (Br049)), and club-root resistance (ZL6 (Br048) and 20395SD (Br051)) (Yu et al. 2011; Zhang et al. 2012; Wang et al. 2014; Su et al. 2014). For instance, a total of 213 SNP markers showed polymorphism between inbred lines of QDX and ZDJ (data not shown). Therefore, our study provides a list of polymorphic markers that can be not only used to assess the genetic diversity of B. rapa germplasm resources but will also be helpful in enriching the recently-developed AFLP/SSR/InDel-based genetic linkage maps for intraspecific mapping population.
Assessing the relationships within germplasm collections can assist in the selection of more distantly related lines for use in breeding programs. Here, SNP genotyping data was used to quantify the genetic diversity and distances within the B. rapa germplasm collection (Fig. 3b, c). A detailed individual-by-individual genetic distance matrix was concluded, which could be of great use to plant breeders (Supplementary Table 7). Pair-wised genetic distance among the 231 accessions ranged from 0.01 to 2.46, with an averaged value of 0.68. In modern breeding systems, the selection of excellent germplasm resources and the use of hybrid vigor are effective to cultivate improved varieties of B. rapa crops. For instance, Xin No. 3 is a cross hybrid between the inbred lines JDY (Br049) and QDX (Br053), both of which show no outstanding agronomic characters. However, Xin No. 3 is one of the most popular Chinese cabbage varieties cultivated in autumn, sharing 90 and 50% of the planting areas in Beijing and North China, respectively. Here, we noticed that the value of pair-wised evolutionary distance between JDY and QDX reached 0.66, which is far above average value among autumn-ecotype (0.52). Thus, we speculated that there are many factors accounting for hybrid vigor, but a distant phylogenetic relationship of the parents should be one of the causes. These results, which coincided with our current hybrid breeding practices, verified the significance of germplasm characterization in heterosis breeding.
Population differentiation and phylogenetic characterization
By using the SNP genotyping data, the overall genetic diversity among the 231 accessions was measured as high as 0.45 (Supplementary Table 4), indicating that extensive genetic variation is present within this subspecies. Furthermore, AMOVA is a powerful tool to test hypotheses of population structure (Grünwald and Hoheisel 2006). Our AMOVA analysis revealed that most of the variance (49.82%) arise from within different subpopulations, and there is significant population/subpopulation structure considering all levels of the population/subpopulation strata (Fig. 4), which provided another important evidence that we indeed have some sort of population or subpopulation structure among B. rapa.
The major allele frequency (not minor allele frequency (MAF)) was averaged 0.63 (Supplementary Table 4), suggesting that there are large number of loci that are not fixed in the population. Besides, the FST between different groups, which indicate the level of population differentiation, was estimated between 0.07 and 0.15 (Supplementary Table 6), which is much lower than that in rice (0.55) (Huang et al. 2012), and cucumber (0.41) (Qi et al. 2013), but is comparable to maize (0.11) (Hufford et al. 2012). Wright, 1978 suggested that when FST value ranged from 0 to 0.05, populations differentiate at a low level; 0.05–0.15, at a modest level; and > 0.15, at a high level. Thus, we believed that there is a large amount of variation present in B. rapa crops and genetic differences did existed among different B. rapa populations, but the degree was relatively low. One of the possible reasons is that all subspecies of B. rapa could be free to mate with each other, like maize, and this reduced the effect of genetic isolation. On the other hand, all the germplasms used in the study were inbred lines that have been self-fertilized for at least six generations. Selfing tends both to reduce the level of genetic variability within populations and to increase the amount of genetic differentiation among populations, which keep population differentiation at a modest level.
In our study, three complementary methods, PCoA, STRUCTURE, and a N-J tree, were used to analyze the population structure and individual relationships within our germplasm collection, and the 231 accessions were divided into four groups, as traditional classification (Fig. 3b, c). Pairwise FST statistics within subspecies showed that genetic differentiation was not evenly distributed across the four populations (Supplementary Table 6). We noticed that only the FST value between ssp. pekinensis and ssp. chinensis just reached 0.15, which suggested a considerable genetic differentiation between the two populations. However, considering the big phenotypic differences between ssp. pekinensis and ssp. chinensis (heading vs. non-heading), the FST value is quite low. This fact makes us infer that the big phenotypic differences might be controlled by only a handful of genes or genes that acted in a genetic pathway.
Artificial selection of spring-ecotype of Chinese cabbage
Chinese cabbage was first recorded in China in the eighteenth century. Only two decades ago, it was originally an autumn crop, but now it has grown all year round, represented as autumn-, summer-, and spring-ecotype (Ke. 2010). About a century ago, some germplasms from Chiifu, ShanDong, China, were spreaded to Japan and Korea. Due to the difference of ecologic condition, consumer demand, and long time artificial selections, those germplasms possessing bolting-resistance genetic resources were further domesticated to spring-ecotype at the second half of the twentieth century.
To investigate genetic relationships among the three clusters and to search for evidence of selection of spring-ecotype Chinese cabbage, we conducted the STRUCTURE analysis based on Chinese cabbage accessions. At a k of 4, the spring-ecotype was seperated from the Chinese cabbage group, showing that the spring-ecotype shared quite different genetic composition with the autumn- and summer-ecotype. Meanwhile, summer- and autumn-ecotypes were further seperated from each other by using the k of 6, but a certain level of mixed genetic composition was still found within the two clusters. The above results showed that the three clusters were clearly distinguished, although different degrees of introgression were detected in these groups.
To further delineate the evolutional roadmap of spring-ecotype, information of N-J tree was explored. From the observed genetic distances (Fig. 3c), the traditional autumn- and summer-ecotypes were closer to the root of ssp. pekinensis, and the spring-ecotype positioned at the most distant point from the root. It indicated that the spring-ecotype was the most modern ecotype and was selected from the other two traditional ecotypes. In the future, more detailed genotyping data or resequencing data of the traditional landrace from ShanDong, China will be valuable for exploring the impact of genomic selection on domestication.
Molecular characterization of ssp. chinensis var. taicai and ssp. chinensis var. japonica
B. rapa ssp. chinensis var. taicai was reported to have originated from wild ssp. rapa in Europe but was now only existed in ShanDong and JiangSu, China (Ke. 2010). The highly variable characteristics of var. taicai make many researchers deem it as a separate subspecies, not a variety, of B. rapa. To further discuss the argument, pairwised FST values between var. taicai and the four B. rapa subspecies, pekinensis, chinensis, rapifera, and oleifera, were each calculated. We noticed that the FST values of var. taicai vs. ssp. oleifera and var. taicai vs. ssp. rapifera were 0.094 and 0.056, respectivley, while the values of var. taicai vs. ssp. pekinensis and var. taicai vs. ssp. chinensis were 0.021 and 0.017, respectivley. Compared with subspecies rapifera and oleifera, we believed that ssp. chinensis var. taicai did have enough genetic differentiation to make it as a separate subspecies of B. rapa (Supplementary Table 8; Fig. 3c). In addion, more detailed genotyping data or resequencing data of the ssp. chinensis var. taicai will be valuable for exploring its origination. This makes it a valueable source for introducing genetic diversity into new varieties in B. rapa breeding programs.
It is interesting that the two var. japonica accessions, JSJ (Br165) and JSC (Br166), previously considered to be a variety of ssp. chinensis, clustered with two Japanese ssp. rapifera accessions (Br205, Br206) and one ssp. chinensis var. communis accession (Br163) (Fig. 3c). A remarkable characteristic of var. japonica crop is tillering. The stem branches at the basal region without elongation, and then numerous leaves grow (Hirai and Matsumoto, 2007). We predicted that either the var. japonica has experienced complicated genetic introgression from ssp. rapifera and ssp. chinensis var. communis or it may be a product of hybridization between the two groups.
In summary, this study provides an extensive resource of cost-effective and polymorphic KASPar markers of B. rapa and their application in population stucture characterization. The 568 SNP markers, coupled with future new developed KASPar markers, will make it possible for breeders to genotype thousands of accessions rapidly and economically and to provide great help in MAS breeding.
References
Allen AM, Barker GLA, Berry ST (2011) Transcript-specific, single-nucleotide polymorphism discovery and linkage analysis in hexaploid bread wheat (Triticum aestivum L.) Plant Biotechnol J 9:1086–1099
Chen YP, Cao JS, Miao Y, Ye WZ (2000) Analysis of genetic polymorphism in vegetable crops of Brassica campestris by RAPD markers. J Zhejiang Univ (Agric Life Sci) 26:131–136
Cortés AJ, Chavarro MC, Blair MW (2011) SNP marker diversity in common bean (Phaseolus vulgaris L.) Theor Appl Genet 123:827–845
Earl DA, Vonholdt BM (2011) Structure harvester: a website and program for visualizing structure output and implementing the evanno method. Conserv Genet Resour 4(2):359–361
Edgington ES (1995) Randomization tests. CRC Press, Boca Raton, FL
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Fan JB, Oliphant A, Shen R, Kermani BG, Garcia F, Gunderson KL, Hansen M, Steemers F, Butler SL, Deloukas P, Galver L, Hunt S, McBride C, Bibikova M, Rubano T, Chen J, Wickham E, Doucet D, Chang W, Campbell D, Zhang B, Kruglyak S, Bentley D, Haas J, Rigault P, Zhou L, Stuelpnagel J, Chee MS (2003) Highly parallel SNP genotyping. Cold Spring Harb Symp Quant Biol 68:69–78
Federico L, Amy V, Thomas C (2008) Development of a set of public SSR markers derived from genomic sequence of a rapid cycling Brassica oleracea L. genotype. Theor Appl Genet 117:977–985
Grünwald NJ, Hoheisel G-A (2006) Hierarchical analysis of diversity, selfing, and genetic differentiation in populations of the oomycete aphanomyces euteiches. Phytopathology 96(10):1134–1141
Guo JX, Zhou NY, Ma RC, Cao MQ (2002) Genetic diversity in Brassica rapa revealed by AFLP molecular markers. J Agric Biotechnol 10:138–143
He YT, Chen BY, Fu TD, Li DR, Tu JX (2003) Origins and evolution of Brassica campestris L. in China. Acta Genet Sin 30:1003–1012
Hirai M, Matsumoto S (2007) Brassica rapa. In: Vegetables. Springer, Berlin, pp 185–190
Hiremath PJ, Kumar A, Penmetsa RV (2012) Large-scale development of cost-effective SNP marker assays for diversity assessment and genetic mapping in chickpea and comparative mapping in legumes. Plant Biotechnol J 10:716–732
Huang XH, Kurata N, Wei XH, Wang ZX, Wang A, Zhao Q, Zhao Y, Liu KY, Lu HY, Li WJ, Han B (2012) A map of rice genome variation reveals the origin of cultivated rice. Nature 490:497–501
Hufford MB, Xu X, van Heerwaarden J, Pyhäjärvi T, Chia J-M, Cartwright RA, Elshire RJ, Glaubitz JC, Guill KE, Kaeppler SM, Lai J, Morrell PL, Shannon LM, Song C, Springer NM, Swanson-Wagner RA, Tiffin P, Wang J, Zhang G, Doebley J, McMullen MD, Ware D, Buckler ES, Yang S, Ross-Ibarra J (2012) Comparative population genomics of maize domestication and improvement. Nat Genet 44(7):808–811
Ke GL (2010) Chinese cabbage breeding. China Agricluture Press, Beijing, pp 18–24
Labana KS, Gupta ML (1993) Importance and origin in breeding oilseed Brassicas Springer-Verlag. Berlin, Germany, pp 1–20
Livak KJ, Flood SJA, Marmaro J, Giusti W, Deetz K (1995) Oligonucleotides with fluorescent dyes at opposite ends provide a quenched probe system useful for detecting PCR product and nucleic acid hybridization. Genome Res 4:357–362
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303
Nagaharu U (1935) Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Jap J Bot 7:389–452
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in excel population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Powell W, Morgante M, Andre C, Hanafey M, Vogel J, Tingey S, Rafalaski A (1996) The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol Breed 2(3):225–238
Pritchard JK, Stephens P, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Qi JJ, Liu X, Shen D, Miao H, Xie BY, Li XX, Zeng P, Wang SH, Shang Y, Gu XF, Du YC, Huang SW (2013) A genomic variation map provides insights into the genetic basis of cucumber domestication and diversity. Nature Genet 45:1510–1515
Roorkiwal M, Sawargaonkar SL, Chitikineni A, Thudi M, Saxena RK, Upadhyaya HD, Vales MI, Riera-Lizarazu O, Varshney RK (2013) Single nucleotide polymorphism genotyping for breeding and genetics applications in chickpea and pigeonpea using the beadxpress platform. The Plant Genome 6(2):494–494
Saxena RK, Varma Penmetsa R, Upadhyaya HD, Kumar AS, Carrasquilla-Garcia NO, Schlueter JA, Farmer AN, Whaley AM, Sarma BK, May GD, Cook DR, Varshney RK (2012) Large-scale development of cost-effective single-nucleotide polymorphism marker assays for genetic mapping in pigeonpea and comparative mapping in legumes. DNA Res 19(6):449–461
Song M, Osborn TC, Williams PH (1988) Brassica taxonomy based on nuclear restriction fragment length polymorphism (RFLPs). 2. Preliminary analysis of sub-species within B. rapa (syn campestris) and B. oleracea. Theor Appl Genet 76:593–600
Steemers FJ, Gunderson KL (2007) Whole genome genotyping technologies on the BeadArray ™ platform. Biotechnol J 2:41–49
Su TB, Yu SC, Wang J, Zhang FL, Yu TJ, Zhang DS, Zhao XY, Wang WH (2014) Loss of function of the carotenoid isomerase gene BrCRTISO confers Orange color to the inner leaves of Chinese cabbage (Brassica rapa L. ssp. pekinensis). Plant Mol Biol Rep 33:648–659. https://doi.org/10.1007/s11105-014-0779-0
Tamura K, Peterson D, Peterson N, Stecher G, Nei M et al (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Tian H-L, Wang F-G, Zhao J-R, Yi H-M, Wang L, Wang R, Yang Y, Song W (2015) Development of maizeSNP3072, a high-throughput compatible SNP array, for DNA fingerprinting identification of Chinese maize varieties. Mol Breed 35(6):136
Tobias W, Simon M, Longin C, Korzun V, Akhunov E, Ebmeyer E, Schachschneider R, Schacht J, Kazman E, Jochen CR (2013) Population structure, genetic diversity and linkage disequilibrium in elite winter wheat assessed with SNP and SSR markers. Theor Appl Genet 126(6):1477–1486
Wang X, Wang H, Wang J et al (2011) The genome of the mesopolyploid crop species Brassica rapa. Nat Genet 43:1035–1039
Wang WH, Zhang DE, Yu SC, Liu J, Wang D, Zhang FL, Yu YJ, Zhao XY, Lu GX, Su TB (2014) Mapping the BrPur gene for purple leaf color on linkage group A03 of Brassica rapa. Euphytica 199:293–302
Wright S (1978) Evolution and genetics of populations, variability within and among natural populations, vol 4. The University of Chicago Press, Chicago
Yu SC, Zhang FL, Zhao XY, Yu YJ, Zhang DS (2011) Sequence-characterized amplified region and simple sequence repeat markers for identifying the major quantitative trait locus responsible for seedling resistance to downy mildew in Chinese cabbage (Brassica rapa ssp. pekinensis). Plant Breed 130:580–583
Zhang SJ, Yu SC, Zhang FL, Zhao XY, Yu YJ, Zhang DS (2012) Inheritance of downy mildew resistance at different developmental stages in Chinese cabbage via the leaf disk test. Hort Environ Biotechnol 3(5):397–403
Funding
This work was supported by grants from the National Key Research and Development Program of China (2016YFD0101701), the National Plan for Science and Technology Support (2014BAD01B08), the Technology Innovation Program, BAAFS (KJCX20161503), the earmarked fund for China Agrculture Research System (CARS-23-A-05), and the National Natural Science Foundation of China (Nos. 31772297 and 31772307).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Supplementary Fig. 1
3-D plot of principal coordinate analysis (PCoA) of the 231 accessions. (DOCX 120 kb)
Supplementary Fig. 2
Delta K calculation (DOCX 50 kb)
Supplementary Table 1
(XLSX 25 kb)
Supplementary Table 2
(XLSX 12 kb)
Supplementary Table 3
(XLSX 142 kb)
Supplementary Table 4
(XLSX 52 kb)
Supplementary Table 5
(XLSX 8 kb)
Supplementary Table 6
(XLSX 9 kb)
Supplementary Table 7
(XLSX 695 kb)
Supplementary Table 8
(XLSX 9 kb)
Rights and permissions
About this article

Cite this article
Su, T., Li, P., Yang, J. et al. Development of cost-effective single nucleotide polymorphism marker assays for genetic diversity analysis in Brassica rapa. Mol Breeding 38, 42 (2018). https://doi.org/10.1007/s11032-018-0795-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-018-0795-0