
Abstract
Molecular dynamics (MD) simulations have been utilized to study peptide ion conformer establishment during the electrospray process. An explicit water model is used for nanodroplets containing a model peptide and hydronium ions. Simulations are conducted at 300 K for two different peptide ion charge configurations and for droplets containing varying numbers of hydronium ions. For all conditions, modeling has been performed until production of the gas-phase ions and the resultant conformers have been compared to proposed gas-phase structures. The latter species were obtained from previous studies in which in silico candidate structures were filtered according to ion mobility and hydrogen-deuterium exchange (HDX) reactivity matches. Results from the present study present three key findings namely (1) the evidence from ion production modeling supports previous structure refinement studies based on mobility and HDX reactivity matching, (2) the modeling of the electrospray process is significantly improved by utilizing initial droplets existing below but close to the calculated Rayleigh limit, and (3) peptide ions in the nanodroplets sample significantly different conformers than those in the bulk solution due to altered physicochemical properties of the solvent.

ᅟ
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The advent of soft ionization techniques such as matrix-assisted laser desorption ionization (MALDI) and electrospray ionization (ESI) not only brought a Nobel Prize to the field of mass spectrometry (MS) but also introduced possibilities for protein structure characterization [1,2,3]. The relatively early combination of ESI with collision-induced dissociation (CID) facilitated the determination of the primary structures of peptides and proteins [4,5,6]. The fundamental relationship between the higher-order structures of proteins and their biological functions provided a strong impetus for development of structural characterization techniques using MS [7]. With this came the consideration for the analysis of protein behavior in transitioning from the solution phase to the gas phase and ultimately vacuum environment of the mass spectrometer.
Early work using protein solutions at biologically relevant pH and employing mild mass spectrometer interface conditions illustrated that protein complexes remained as intact ions suggesting the preservation of quaternary structure during their transformation to the gas phase [8,9,10,11]. Later, ion mobility spectrometry-mass spectrometry (IMS-MS) provided insight into the degree of higher-order structure of biomolecular ions [12,13,14,15]. For some species, the ions did not exhibit “equilibrated” gas-phase structures; rather, they were suggested to resemble higher-energy, solution-phase-like conformations [13, 16, 17]. Such observations helped to introduce the concept of native MS [18,19,20,21,22,23] and supported the idea that a significant portion of non-covalent, intramolecular interactions governing solution-phase structures could be preserved during the electrospray process [13, 24].
The process of ion formation by ESI has been studied extensively over the years. Such work included the determination of ion movement in solution [25, 26], subsequent formation of the Taylor cone [27], and the emission of a fine jet of parent droplets [25, 26]. Further, as the droplets move toward the mass spectrometer inlet, the droplets shrink due to solvent evaporation and the excess like charge on the surface overcomes the surface tension resulting in droplet fission [26, 28]. The maximum number of net charges (Zr) that can be tolerated by the droplet before a fission event is determined by the Rayleigh limit (Eq. (1)) which is proportional to the temperature-dependent surface tension value of the solvent [29]. Ultimately, the repeated “shrinkage/fission” cycles guide the late-stage progeny droplets to the final steps of the ESI process resulting in the release of the dry analyte ion [26, 28].
Despite the extensive application of ESI for protein structure studies, the exact mechanism involved in the formation of anhydrous ions at the final stage of the ESI process is somewhat controversial. Two early proposed mechanisms—Dole’s charge residue model (CRM) [30] and the ion evaporation model (IEM) [31]—are normally used to explain the underlying process. CRM presents the scenario where evaporation and shrinkage of the droplets result in those having a similar size to the carried protein, and then, evaporation continues to complete dryness of the analyte ion [30, 32, 33]. In contrast, the IEM describes the ejection of a small analyte ion directly into the gas-phase environment from the surface of a droplet. Indeed, it is proposed that the IEM governs the release of charge carrier ions such as Na+ and H3O+ present on the surface of the droplet during the shrinkage/fission stages; this reduces electrostatic repulsion by direct removal of an ion [31, 34,35,36,37]. These small charge carriers can leave the droplet in the form of small ion-solvent clusters [34, 35, 37,38,39]. Konermann et al. proposed a third mechanism—the chain ejection model (CEM)—which describes the release of an unfolded protein or polymer [37]. According to the CEM, the exposure of hydrophobic surfaces—which would be hidden inside the protein core in a folded configuration—to the non-polar, gas-phase environment upon the ESI process, results in the ejection of the extended analyte from the surface of the droplet [37].
Molecular dynamics (MD) simulations have been widely used to obtain insight into the behavior of the solvent, charge carrier species, as well as the protein inside an ESI droplet [35, 37, 40]. In the work described here, extensive MD simulations are employed to examine gas-phase structure establishment by monitoring the fate of the model peptide (acetyl-PAAAAKAAAAKAAAAKAAAAK) during the late stages of the ESI process and extending to complete dryness and even further sampling of the ions’ dynamics in the gas phase. Furthermore, these ESI-generated peptide ion conformations are compared (quantitatively and qualitatively) with in silico structures that have been selected based on matches to ion collision cross sections (CCS) and deuterium uptake behavior (part 1 and part 2 installments of this work) [41, 42]. A goal is to cross validate the structure determination obtained using gas- and solution-phase modelings.
Experimental
Solution-Phase Representative Structures
The non-polarizable all-atom Amber ff14SB [43] force field was employed to generate the initial structure of the model peptide (acetyl-PAAAAKAAAAKAAAAKAAAAK) in a fully helical configuration. All the lysine residues as well as the C-terminal carboxylic acid group were assigned formal charge and neutral values representing those expected in water at pH 7. The peptide was solvated in a truncated octahedral water box using the rigid three-point TIP3P water model [44]. The system was neutralized by addition of three Cl− ions and energy minimized. Using the periodic boundary conditions, the structure was subjected to equilibration for 5 ns at isothermal-isobaric (NPT) ensemble with an average target pressure of 1 atm. The temperature was regulated by a Langevin thermostat [45, 46] with a collision frequency of 1 ps−1. Bond lengths containing the hydrogen atoms were constrained using the SHAKE algorithm [47]. The particle mesh Ewald (PME) method [48] and a cutoff value of 12 Å were employed to handle the long-range electrostatic interactions. A subsequent 0.5-μs production MD resulted in a trajectory which was sampled for further analyses. The Amber 16 [49] molecular dynamics package and the GPU-accelerated pmemd implementation were employed to perform the MD simulations.
After removal of counterions and water molecules, the cpptraj module [50] was employed to perform dihedral cluster analysis with a mask on the dihedral angles passing through 10 central amino acid residues (K(6) to K(16)). The closest structures to the mathematically generated centroids were subjected to a secondary structure analysis using the STRIDE [51] algorithm. This provided four major structure types which were selected as the solution-representative conformations.
ESI Process Simulation: Droplet Generation
The fully helical conformer from the selected closest-to-the-centroid structures was modified in order to generate gas-phase [M + 3H]3+ peptide ions with charge state arrangements of K(6)-K(11)-K(21) and K(6)-K(16)-K(21). The secondary structure elements and dihedral angle values were preserved. The empirical force field parameters for the C-terminal lysine residue terminating the peptide with a carboxylic acid group were calculated using the R.E.D. server [52,53,54,55,56,57]. Briefly, two different ACE-capped initial structures of this residue were subjected to geometry optimization at HF/6-31G(d) theory level followed by molecular electrostatic potential (MEP) computation and charge fitting steps. The peptide ion was solvated in an octahedral TIP3P water box [44]. The system was carved (in-house script) into a spherical droplet with a 2.5-nm distance from the center of mass of the peptide as the droplet radius.
The force field parameters for hydronium ion (H3O+) were generated by modifying the Baaden [58] parameters for this ion to be compatible with Amber 16 [49]. Using the surface tension value of 52.3 mJ m−2 [59] for TIP3P water model at 300 K representing the experimental effective temperature, the Rayleigh limit was determined as a maximum of 13 charges which leads to an upper limit value of 10 H3O+ ions for the droplet. Therefore, droplets with 0, 1, 4, and 10 H30+ ions located at random initial positions inside the droplets were generated. The number of water molecules ranged from 1814 to 1840 molecules.
ESI Process Simulation: Droplet Shrinkage
After extensive minimization steps, the relaxed droplet was equilibrated at 300 K for 50 ps and subsequently subjected to a desolvation method similar to the algorithm introduced by Konermann et al. [40] described as “the trajectory stitching.” To regulate the temperature during the process and prevent evaporative cooling, every 150 ps, the simulation was stopped and new, initial random velocities from a Maxwellian distribution at 300 K were assigned. Evaporated molecules present at distances larger than 6.5 nm from the center of mass of the droplet were removed to decrease the computational cost. The simulation then restarted, and this cycle continued until all H3O+ ions were ejected from the system and all water molecules evaporated. This leads to an overall simulation time ranging from 600 to 950 ns.
The simulations were performed in vacuum using the GPU-accelerated pmemd implementation in Amber 16 [49]. No long-range cutoff values for handling the electrostatics and van der Waals interactions were considered. The temperature during each 150-ps run was maintained at 300 K using Langevin dynamics [45, 46] with a collision frequency of 1 ps−1. Four replicas were generated for each system leading to an overall 32 runs (two different charge arrangements for peptides in droplets with overall charges of + 3, + 4, + 7, and + 13).
The evaporation of the last, single water molecules at a close distance to, and interacting with, charged lysine residues in the final stages of the evaporation process was computationally extensive. Additionally, these last remaining water molecules act as adducts and do not alter the conformation of the peptide ions [35, 60, 61]. Therefore, in the final stages of the simulation, the last nine remaining water molecules were eliminated manually in three out of four repetitions for the charge arrangements of K(6)-K(11)-K(21) and K(6)-K(16)-K(21). Calculation of backbone-only RMSD values on protein structure during the shrinkage simulations—obtained for the other replica (complete evaporation to dryness)—validates this methodology by showing a steady-state dynamics for the RMSD plot in the final stages of evaporation as shown in Fig. 1 .

Backbone-only RMSD values of peptide ions during the ESI process relative to the initial fully helical pre-equilibrated structures. The cpptraj module [50] was used to obtain these values. (a) Data for the K(6)-K(11)-K(21) charge arrangement. (b) Data obtained from the K(6)-K(16)-K(21) charge arrangement. Each color represents a droplet with a distinguishing total charge value at t = 0 of the simulation. The simulations were performed to complete dryness
Post-ESI Gas-Phase Structures
After complete desolvation, the remaining peptide ions were subjected to production MD simulations at 300 K for 5 ns with no cutoffs for the long-range interactions. The pairwise backbone-only RMSD calculations were performed between all structures sampled from these 32 “post-ESI” trajectories and the 300 trajectories containing the gas-phase candidate structures (with matching charge state arrangements) obtained for the first implementation of this work [41] using an in-house script and the cpptraj [50] module implemented in Amber 16 [49]. Briefly, in order to obtain these 300 trajectories, 4000 cycles of simulated annealing (SA) were performed on peptides with K(6)-K(11)-K(21) and K(6)-K(16)-K(21) charge arrangements. The pool of thermodynamically stable as well as higher-energy “annealed” structures were heated to 300 K, equilibrated, and subjected to production MD simulation at the same temperature. k-means cluster analysis using the kclust module in the MMTSB [62] toolset was performed on the resulting trajectories. CCS calculations on the closest structure to the obtained centroids provided a series of trajectories with matching CCS values to experimental values. These 300 trajectories will be referred to as “the reference trajectories.” For more detail, see the first implementation of this method [41].
Peptide Ions in Infinite Radius Droplet
To achieve a comparison between the behavior of the peptide ions in a nano-scale droplet and bulk solution of the same solvent, the two different charge arrangements of the [M + 3H]3+ fully helical peptide ions used in the ESI simulations were hydrated in a truncated octahedral water box of TIP3P [44] without the presence of any counterions. These systems were energy minimized. Using the periodic boundary conditions, the relaxed systems were subjected to a NVT ensemble heating simulation to obtain a final temperature of 300 K using a Langevin thermostat with varying collision frequencies ranging from 1.0 to 0.1 ns−1 over the course of the simulation. This was followed by an equilibration step for 1 ns with a cutoff value of 15 Å for long-range interactions at NPT conditions. The production MD simulations were further performed by continuing the equilibration conditions for 100 ns. The generated trajectories will be referred to as “infinite radius” trajectories here in order to be distinguished from the neutralized “bulk” solution simulations. After removal of all the water molecules, secondary structure analysis using the STRIDE [51] algorithm was performed on the dry peptide ions.
Results and Discussion
The Fate of the Water Droplet
The droplet geometry is a factor that must be considered in order to validate the application of the sphere-specific Rayleigh equation (Eq. ( 1 )) over the course of the evaporation process. To evaluate the droplet geometry, three I1/I3, I1/I2, and I2/I3 ratios were introduced through calculating the principal moments of inertia (MOI) [I1, I2, and I3 (I1 > I2 > I3)] for the droplet during the ESI simulation. The MOI ratios of 1 correspond to a perfect spherical geometry. Only water molecules were considered as the shape-defining species.
Figure 2 shows the I1/I3 ratios of the droplets in a series of evaporation-to-dryness simulations. The results show a steady trend at a value of 1 with slight variation from 1 for the first several hundred nanoseconds of the simulations. In addition to slight distortion in the droplet geometry, these initial variations also correspond to the frames sampled from the trajectory before applying the 6.5-nm removal cutoff at the end of each 150-ps cycle. That is, water molecules that diffuse ≤ 6.5 nm from the center of mass of the droplet during the 150 ps were considered in the MOI calculations. As shown in Figs. 2 and S1 (Supporting Information section), during the last stages of evaporation, the I1/I3 ratios exhibit a significant variation from a value of 1.

The I1/I3 ratios of the droplets during the evaporation to dryness. Each color represents a particular charge state value for the initial droplet. (a, b) The MOI ratio for droplets containing K(6)-K(11)-K(21) and K(6)-K(16)-K(21) charge arrangements, respectively
Visualizing the droplets at such late stages as well as monitoring the number of remaining water molecules (Fig. S1 in the Supporting Information section) reveals the presence of a very limited number of water molecules on the surface of the droplets. In such cases, the remaining system was not considered as a droplet and the Rayleigh equation was not applied. However, because the ejection of all H3O+ ions occurs at much earlier stages, the system asymmetry did not introduce any error in Rayleigh limit monitoring.
Therefore, due to the values obtained for MOI ratios, the droplet is considered spherical in the beginning of the simulations as required for further analysis. It is worth mentioning that, as the number of H3O+ ions increases, the droplet evaporation occurs at considerably higher rates (Fig. S1 in the Supporting Information section).
The Fate of the Hydronium Ion(s)
The majority of the ESI-representative MD simulations start from initial droplets having the maximum number of charges allowed as determined by the Rayleigh limit (+ 13 in this work); however, studies on water droplets carrying a variety of cations and anions (Na+, NH4+, Cl−, etc.) suggest that the utilization of the calculated Rayleigh limit in obtaining this upper value for droplets with diameters on the nanometer scale leads to an overestimation in the number of tolerable charges [38, 63]. This effect is profound for droplets carrying predominantly cations [38]. Previous studies also showed that the surface tension value of the simulated water models is dependent on the size of the droplet [64]. Therefore, in this work, the initial droplets were generated to exhibit a variety of total charge state values equal to (+ 13) and below the Rayleigh limit (+ 3, + 4, + 7).
Droplet geometry is related to the Rayleigh limit via the droplet radius. To calculate the radius of the droplet, the differences in maximum and minimum Cartesian coordinates of the water molecules in the spherical droplet were determined as ΔX, ΔY, and ΔZ values for each sampled frame during the ESI simulations. Due to small asymmetries observed for the droplet in the initial few hundred nanoseconds of the simulation (see Figs. 2 and S1 in the Supporting Information section), the minimum ΔX, ΔY, or ΔZ value for each frame was considered as the effective diameter yielding the effective radius (Reff) of the droplet. The Rayleigh equation was solved to obtain the minimum allowed radius (Rmin) value for a particular droplet charge state (+ 13 to + 3). This is the threshold for a droplet as any radius value below such will require a decrease in the maximum allowed charge state value by direct ejection of the charge carrier species.
The Reff values at which the ejection of the H3O+ ions occurred were extracted from the ESI-representative trajectories. Comparison between the Reff and Rmin value of the same charge state (+ 13 to + 3) revealed that the ejection of the charge carrier cations happens before the Reff shrinks to the corresponding Rmin values (i.e., before the calculated Rayleigh limit) in every case. These results are illustrated in Figs. 3 and S2 in the Supporting Information section for both peptide charge arrangements and droplets of + 3 to + 13 charge states. For droplets generated at the calculated Rayleigh limit (10 H3O+ and total charge of + 13), the initial ejection of the charge carriers occurs at the very beginning of the simulations quite possibly due to the exceeding of the apparent Rayleigh limit corresponding to the water model, droplet size, and the size-dependent surface tension values. Small fluctuations in the general decreasing trend of the droplet radius values arise from the time points during the simulations where the cutoff value of 6.5 nm has not yet been applied.

The effective radius of the droplets for ESI-representative trajectories obtained for the K(6)-K(11)-K(21) charge arrangement and the four separate droplet charge states of + 3, + 4, + 7, and + 13 (0, 1, 4, and 10 hydronium ions). The blue vertical lines represent the minimum Rayleigh radius (Rmin), while the brown vertical lines show the effective radius at which ejection of hydronium ions takes place. The Rmin value and the related effective radius at the actual ejection points are shown with the same point style
Overall, the generation of an ESI-representative simulation may require an initial droplet with a radius much larger than Rmin for a particular charge state. This strategy will increase the computational time but may be overcome by starting the simulation for the nanodroplet with a total charge state value below the calculated Rayleigh limit. Droplet evaporation then takes place, and ion ejection occurs at the apparent Rayleigh limit (or below) corresponding to the simulation parameters. This behavior is observed for the droplets of + 7 and + 4 charge states shown in Figs. 3 and S2 (Supporting Information section); the ejection of the first hydronium ion takes place after 200 and > 500 ns, respectively.
A radial distribution analysis of hydronium ions with respect to the center of mass of the droplet throughout the trajectories (Supplementary Fig. S3) shows that they prefer to locate near the surface of the droplets where the electrostatic repulsion between such same-polarity ions—that is competing with the hydration free energy—is at a minimum. The presence of these ions would disrupt the hydrogen bond network of the solvent at the surface. This effect can be dramatic for ions such as Na+ which are not capable of forming hydrogen bonds with water molecules. Therefore, one major benefit of initiating ESI-representative simulations at lower charge state values is that there will be less interruption in such hydrogen bond networks, and consequently, the surface tension values would be higher (closer to values computed for the water models). The presence of a counterion such as Cl− inside the droplet can also affect the location of the hydronium ions through a “shielding” effect, again bringing the surface tension values close to those of water models [35].
Figure 4 shows snapshots of an ESI-representative trajectory obtained from the droplet containing four H3O+ ions and peptide ions with charge arrangement of K(6)-K(16)-K(21). As the simulation proceeds, the water molecules evaporate from the surface of the droplet leading to a decrease in overall droplet size (see Figs. 3 and S2 in the Supporting Information section). At a specific radius, the repulsion between the charge carriers overcomes the surface tension value of the molecules at the surface of the droplet. The droplet then deforms and generates a cone-shaped tail directing the H3O+ ion toward the vacuum environment. The H3O+ eventually leaves the droplet bound to a cluster of water molecules as observed in a variety of other simulations that investigate the ESI process [35, 37, 40].

Diagram illustrating snapshots of an ESI-representative trajectory with four H3O+ ions at the beginning of the simulation. The hydronium ions are shown by red balls. The peptide secondary structure is represented in gray. The yellow arrows show the direction of trajectory sampling. Blue dots represent the water molecules
The Fate of the Peptide Ion
Radial distribution analysis of the peptide relative to the center of mass of all the solvent molecules in the droplet reveals that it moves toward the surface of the droplet as the simulation proceeds. As shown in Fig. 4 , the peptide exposes its hydrophobic alanine residues to the non-polar vacuum environment while solvating the highly polar, charged lysine residues in the droplet core. This is also in agreement with previous studies monitoring the behavior of small unfolded peptides in a nanodroplet [37]. As the numbers of H3O+ species in the initial droplet increase, the translocation of the peptide to the surface of the droplet occurs in the early simulation stages. Thus, the repulsion between the triply charged peptide ions and the hydronium ions appears to have an influence in locating the peptide at the surface of the droplet and, as discussed below, could lead to conformational homogeneity in the gas phase.
MD simulations of the model peptide of appropriate charge at pH = 7 showed that it sampled a series peptide structure ensembles revealing rapid dynamics between fully helical, partially helical, and random structures. Although the nanodroplet lacks the exact same properties as the bulk solution (e.g., surface tension values), it is large enough to allow structural fluctuations for the peptide ions. Such dynamics are the origin of the deviations observed in the RMSD plots in Fig. 1 . Therefore, the initial peptide structures used in the ESI-representative simulations were fully helical as the most abundant structure observed for the peptide in bulk solution (see Supplementary Fig. S4). Because of the rapid fluctuations observed in the bulk solution simulations, it is necessary to perform the ESI-representative simulations at higher numbers of repetition in order to capture as much of the variation as possible for the final structures. Limited somewhat by computation power here, 32 replicates were generated to sample these structures.
The division of replicates into four different charge carrier numbers (0, 1, 4, and 10) was utilized to investigate the effect of such numbers of these ions on the behavior of the droplet as well as the peptide ion structure. The high electrostatic repulsion among these ions for droplets exhibiting the calculated Rayleigh limit at the beginning of the simulation (10 H3O+ and a total of + 13 charge state for the droplet) can direct the peptide ion toward a point in conformational space where the structure is trapped. The generation of nanodroplets with lower initial charge state values (+ 7, + 4, + 3 with 4, 1, and 0 H3O+ ions, respectively) reduces the early Coulomb repulsion between charge species causing less disturbance in droplet properties; this permits greater peptide structural fluctuation.
The behavior of the peptide in the nanodroplet was further investigated by comparison of the peptide structure in the droplet with the results obtained from a trajectory representing an infinite radius droplet. To achieve this, the [M + 3H+]3+ peptide ions with two separate charge arrangements similar to those in the nanodroplets were solvated and subjected to dynamics. This infinite radius trajectory can provide a better understanding of the effect of the vacuum environment surrounding the nanodroplet in the ESI-representative simulations.
Secondary structure analysis using the STRIDE [51] algorithm was performed to achieve more insight into structural details of the peptide ions. These calculations were performed separately on four sections of the peptide ions including amino acid residues P(1) to K(6), A(7) to K(11), A(12) to K(16), and A(17) to K(21). The acetyl cap is ignored for such calculations. These analyses were applied on seven trajectories including infinite radius trajectories, post-ESI gas-phase production trajectories, and five radius-scanning trajectories containing sampled frames extending to the point at which the droplet radius evolves from 25 to 20, 20 to 15, 15 to 10, 10 to 5, and, finally, 5 Å to dryness.
The helicity (HR) for a structure was calculated for each peptide section. HR values were employed to generate a population-averaged helicity (HR(PA)) value according to Eq. ( 2 ):
where N represents the total number of frames in the trajectory and n is the number of frames exhibiting a particular HR value. The HR(PA) results are shown in Fig. 5 as well as Figs. S5 to S11 (Supporting Information section) for two charge arrangements of K(6)-K(11)-K(21) and K(6)-K(16)-K(21) and four different droplets of + 3, + 4, + 7, and + 13 charge state. The RMSD relative to fully helical structures for corresponding infinite radius trajectory and ESI-representative simulations (2.5 nm to dryness) are also illustrated.

The top panel presents the HR(PA) results. The plots related to droplet radius (Reff) values of 25 to < 5 Å are obtained from the ESI-representative simulations carrying zero H30+ ions. The bottom panels represent the RMSD values for the ESI-representative trajectory with a + 3 total charge state (zero H30+ ions) (black, top) and infinite radius trajectory (red, bottom). All results are calculated for the K(6)-K(11)-K(21) charge arrangement simulations
The ESI-representative trajectories in Figs. 5 and S5 to S11 as well as the RMSD plots in Fig. 1 reveal that the peptide undergoes structural fluctuations inside the nanodroplet. This behavior is in agreement with infinite radius trajectories where the peptide is surrounded by a large network of water molecules. Therefore, the selection of the initial 2.5-nm radius for the droplet was large enough to allow random walk along the peptide conformational space. However, as the number of water molecules decreases near the end of the simulations, the structure becomes kinetically trapped with significantly restricted fluctuation (steady-state regions in the RMSD plots). System entropic effects as well as solution-phase interactions between the water and the peptide that assist the random walks along the potential energy surface (PES) are absent in the vacuum environment. If the energy barriers between the multiple solution-phase structures of such a peptide ion become sufficiently high to overcome in the gas phase, it is possible that kinetically trapped conformers exhibit structural characteristics at the dryness stages that are not significantly altered in the post-ESI gas-phase production simulations.
Figure S8 in the Supporting Information section is an example of structure preservation behavior where the same secondary structures for all four sections of the peptide are observed in the droplet with radius of < 5 Å as well as the dry peptide in post-ESI production simulations. Again, this kinetic trapping behavior necessitates the replicate simulation for the ESI process in the study of biomolecular structure establishment. This conclusion does not undermine the requirement of further post-ESI simulation as cases may occur where the energy barriers are sufficiently low such that the peptide can exhibit changes in elements of secondary structure that are captured by further dynamics in vacuo. Such behavior is observed in Figs. S5, S9, and S10 (Supporting Information section) where the helicity values in post-ESI production MD simulations are different than those calculated for the last stages (< 5 Å droplet radius) of the ESI-representative simulations.
The stability of the helical structure for the model peptide is dependent on two effects: the extent of intramolecular hydrogen bonding and charge-induced helix disruption and stabilization. In the solution-phase environment, the constant competition between the formation of hydrogen bonds between the water molecules and the peptide can disrupt the hydrogen bonds within the helical structure of the peptide. This competition does not exist in the solvent-free vacuum environment. Therefore, as the number of surrounding water molecules decreases to produce a dry peptide, due to this lack of competing forces, the extent and duration of the hydrogen bonds within the helix may increase. In contrast, as the number of water molecules surrounding the peptide decreases, the shielding effect of the media decreases. Thus, in each segment of the helix, the repulsive interaction between the helix dipole and the positively charged lysine residues located N-terminal to some segments increases [65]. Therefore, as evaporation in the ESI-representative simulations occurs, HR(PA) values can initially increase—from an increase in the intramolecular hydrogen bond extent—followed by a decrease in this value due to disruption induced by charged residues (Fig. S10—residues 7 to 11—in the Supporting Information section).
In addition to the phenomena discussed above, the Coulomb repulsion arising from the presence of two charged residues before and after the peptide segments will disrupt the helical structure [65]. In the case of the model peptide here, the propensity of the proline residue located at the N-terminus to initiate a helical structure can act as another factor governing the secondary structure of the peptide [66]. Toward the C-terminus of the peptide, the proline’s helix initiation effect decreases. Figure S11 in the Supporting Information section is an example of such behavior. The HR(PA) factor constantly decreases from the first section (residues 1–6) toward the last segment at the C-terminal end (residues 17 to 21). Figures S5 and S9 in the Supporting Information section show an example of the effect of such opposing forces on the resultant secondary structure using a comparison of the HR(PA) parameter for two different charge arrangements in trajectories of the same total droplet charge state (+ 4 with one H3O+ ion at the beginning of the simulations). Although the two peptides exhibit similar trends in their HR(PA) values for the first, second, and last segments of the primary sequence, the third segments (residues 12 to 16) exhibit a different behavior. The decrease in HR(PA) is more dramatic in Fig. S5 for residues 12 to 16 in comparison with the same plot in Fig. S9. The presence of a charged lysine residue (K(11)) located N-terminal to the third segment disrupts helix formation in the third segment while this effect is not observed until the last segment for the peptide with the K(6)-K(16)-K(21) charge arrangement (Fig. S9).
Reference vs Post-ESI Trajectory Comparison
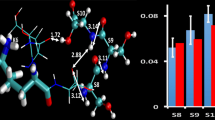
By applying the experimental CCS values as the first filtering dimension, 300 structures—originating from two different simulated annealing algorithms—were selected as “candidate” structures for triply charged peptide ions [42]. Deuterium uptake values were employed as a second criterion to provide 13 “conformer types” as the best gas-phase representative species among these 300 candidate structures of the model peptide. As noted above, one of the aims of this study is to make a comparison between the ESI-generated structures and these 13 conformer types in order to validate the methodology applied in filtering CCS-HDX results. To achieve this, a backbone-only pairwise RMSD calculation between all the frames containing the 300 reference trajectories and the post-ESI production simulations of the same peptide charge arrangements was performed (32 × 5000 ESI-generated frames with 300 × 5000 candidate-containing frames). The most similar structures (lowest RMSD value) from the 32 ESI-representative replicate trajectories to the candidate configurations were obtained. These data were compared with the 13 CCS/HDX-filtered conformer types. None of the peptide ions from the droplets of + 3 charge state (eight replicates) matched these 13 conformers. The droplets with + 13 charge states (eight replicates) had four matching structures. The ESI-representative simulations exhibiting systems with + 4 or + 7 charge states (16 replicates) each provided three structural matches. This result is consistent with the idea that the ESI-representative simulations require an initial droplet with equal or near the Rayleigh limit charge states using the presence of additional charge carrier species such as H3O+. Overall, 10 out of 32 replicates representing the ESI process were able to provide 7 out of the 13 conformer types obtained through CCS/HDX filtering. Figure 6 illustrates two examples of these matching structures with their relative RMSD values.

Gas-phase conformer types obtained from CCS/HDX method (right) and ESI-representative simulations (left) with lowest relative RMSD values. Both configurations on the left are originated from two different nanodroplets with total charge of + 13
This significant agreement between the results obtained from two different methods—the CCS/HDX filtering algorithm and the ESI-representative simulations—helps validate the methodology applied in the former analysis and provides an opportunity to replace the extensive, computationally expensive ESI simulations with the CCS/HDX filtering algorithm. The possibility of achieving high sampling rates through conformational space sampling techniques such as simulated annealing (4000 annealed structures here) [41, 42] empowers this method in providing insight into the gas-phase structures of the model peptide observed with drift tube IMS-MS measurements.
Conclusions
MD simulations can be employed to provide insight into the ion conformer establishment behavior resulting from the ESI process. In this work, several replicates of in silico nanodroplets that simulate the progeny droplets at the last stages of the ESI process were generated and subjected to extensive MD simulations to provide anhydrous peptide ions. Certain elements of the droplet behavior including droplet radius, ejection of charge carries such as H3O+ via the ion evaporation model, limitations of the Rayleigh limit in nanoscale droplet simulations, as well as the effects of the number of charge carriers were investigated. It is proposed here that the ESI-representative simulations are required to start the analysis with a number of charge carriers that is below but close to the calculated Rayleigh limit for the water mode for a specific droplet radius and simulation temperature. Additionally, it can be argued that initiating simulations with a very limited number of charge carriers is not representative of the ESI process due to the higher concentration of these species in the final stages of ESI. That said, the studies were performed here to test whether or not such conformational flexibility would be required of the modeling to provide a greater number of candidate structures for matching with the gas-phase structures which was unknown initially. In the end, these results provide added support for the accuracy of the ESI modeling.
Extensive secondary structure analyses of the model peptide in systems with varying numbers of surrounding solvent molecules were performed. Two effects including the extent and duration of intramolecular hydrogen bonding and charge-induced helix disruption and stabilization were largely able to explain the observed differences in elements of secondary structure. Notably, deviation from the trend expected for such systems can result from resulting energetic barriers leading to kinetically trapped conformers. In addition to these phenomena, because of its propensity to initiate helical structure, the effect of the proline residue on the overall peptide structure was observed to diminish in C-terminal segments.
Finally, comparison between the ESI-generated structures and the conformations obtained through CCS calculations and deuterium uptake levels provided a series of structures with a high degree of similarity. These results support the methodology applied in part 1 and part 2 installments of this work and serve to validate the developed algorithms in CCS determinations and HDX scoring. That is, for some systems, the extensive, computationally expensive ESI-representative simulations may be replaced with CCS and HDX measurements for filtering the candidate structures from extensive conformational space sampling to obtain the best gas-phase representative structures [67,68,69]. The latter method lacks some uncertainties associated with ESI simulations such as obtaining an accurate surface tension value for the water model at a specific droplet radius to determine the appropriate number of charge carrier species. That said, many biomolecular ions are not accessible to IMS-HDX-MS/MS analysis, and it should be noted that there is a growing need for improving the ESI simulations as they provide valuable insight into the mechanism of the ESI process and the establishment of gas-phase ion structure.
References
Fenn, J.B.: Electrospray wings for molecular elephants (Nobel lecture). Angewandte Chemie-International Edition. 42, 3871–3894 (2003)
Fenn, J.B., Mann, M., Meng, C.K., Wong, S.F., Whitehouse, C.M.: Electrospray ionization for mass-spectrometry of large biomolecules. Science. 246, 64–71 (1989)
Tanaka, K., Waki, H., Ido, Y., Akita, S., Yoshida, Y., Yoshida, T., Matsuo, T.: Protein and polymer analyses up to m/z 100 000 by laser ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2, 151–153 (1988)
Shevchenko, A., Wilm, M., Vorm, O., Mann, M.: Mass spectrometric sequencing of proteins from silver stained polyacrylamide gels. Anal. Chem. 68, 850–858 (1996)
Biemann, K., Scoble, H.A.: Characterization by tandem mass-spectrometry of structural modifications in proteins. Science. 237, 992–998 (1987)
Hunt, D.F., Shabanowitz, J., Yates, J.R., Zhu, N.Z., Russell, D.H., Castro, M.E.: Tandem quadrupole fourier-transform mass-spectrometry of oligopeptides and small proteins. Proc. Natl. Acad. Sci. U. S. A. 84, 620–623 (1987)
Hoaglund-Hyzer, C.S., Counterman, A.E., Clemmer, D.E.: Anhydrous protein ions. Chem. Rev. 99, 3037–3079 (1999)
Lightwahl, K.J., Schwartz, B.L., Smith, R.D.: Observation of the noncovalent quaternary associations of proteins by electrospray-ionization mass-spectrometry. J. Am. Chem. Soc. 116, 5271–5278 (1994)
Loo, J.A.: Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrom. Rev. 16, 1–23 (1997)
Smith, R.D., Lightwahl, K.J., Winger, B.E., Loo, J.A.: Preservation of noncovalent associations in electrospray ionization mass-spectrometry—multiply charged polypeptide and protein dimers. Org. Mass Spectrom. 27, 811–821 (1992)
Ganem, B., Li, Y.T., Henion, J.D.: Observation of noncovalent enzyme substrate and enzyme product complexes by ion-spray mass-spectrometry. J. Am. Chem. Soc. 113, 7818–7819 (1991)
Hudgins, R.R., Woenckhaus, J., Jarrold, M.F.: High resolution ion mobility measurements for gas phase proteins: correlation between solution phase and gas phase conformations. Int. J. Mass Spectrom. 165, 497–507 (1997)
Wyttenbach, T., Bowers, M.T.: Structural stability from solution to the gas phase: native solution structure of ubiquitin survives analysis in a solvent-free ion mobility-mass spectrometry environment. J. Phys. Chem. B. 115, 12266–12275 (2011)
Valentine, S.J., Anderson, J.G., Ellington, A.D., Clemmer, D.E.: Disulfide-intact and -reduced lysozyme in the gas phase: conformations and pathways of folding and unfolding. J. Phys. Chem. B. 101, 3891–3900 (1997)
Shelimov, K.B., Clemmer, D.E., Hudgins, R.R., Jarrold, M.F.: Protein structure in vacuo: gas-phase conformations of BPTI and cytochrome c. J. Am. Chem. Soc. 119, 2240–2248 (1997)
Wyttenbach, T., Grabenauer, M., Thalassinos, K., Scrivens, J.H., Bowers, M.T.: The effect of calcium ions and peptide ligands on the relative stabilities of the calmodulin dumbbell and compact structures. J. Phys. Chem. B. 114, 437–447 (2010)
Baumketner, A., Bernstein, S.L., Wyttenbach, T., Bitan, G., Teplow, D.B., Bowers, M.T., Shea, J.E.: Amyloid beta-protein monomer structure: a computational and experimental study. Protein Sci. 15, 420–428 (2006)
Sharon, M.: How far can we go with structural mass spectrometry of protein complexes? J. Am. Soc. Mass Spectrom. 21, 487–500 (2010)
Sharon, M., Robinson, C.V.: The role of mass spectrometry in structure elucidation of dynamic protein complexes. (2007)
Kitova, E.N., El-Hawiet, A., Schnier, P.D., Klassen, J.S.: Reliable determinations of protein–ligand interactions by direct ESI-MS measurements. Are we there yet? J. Am. Soc. Mass Spectrom. 23, 431–441 (2012)
van den Heuvel, R.H., Heck, A.J.: Native protein mass spectrometry: from intact oligomers to functional machineries. Curr. Opin. Chem. Biol. 8, 519–526 (2004)
Heck, A.J.R.: Native mass spectrometry: a bridge between interactomics and structural biology. Nat. Methods. 5, 927–933 (2008)
Rajabi, K., Ashcroft, A.E., Radford, S.E.: Mass spectrometric methods to analyze the structural organization of macromolecular complexes. Methods. 89, 13–21 (2015)
Ruotolo, B.T., Robinson, C.V.: Aspects of native proteins are retained in vacuum. Curr. Opin. Chem. Biol. 10, 402–408 (2006)
Cech, N.B., Enke, C.G.: Practical implications of some recent studies in electrospray ionization fundamentals. Mass Spectrom. Rev. 20, 362–387 (2001)
Kebarle, P., Verkerk, U.H.: Electrospray: from ions in solution to ions in the gas phase, what we know now. Mass Spectrom. Rev. 28, 898–917 (2009)
Taylor, G.I., McEwan, A.D.: The stability of a horizontal fluid interface in a vertical electric field. J. Fluid Mech. 22, 1–15 (2006)
Grimm, R.L., Beauchamp, J.L.: Evaporation and discharge dynamics of highly charged multicomponent droplets generated by electrospray ionization. J. Phys. Chem. A. 114, 1411–1419 (2010)
Rayleigh, L.: XX. On the equilibrium of liquid conducting masses charged with electricity. Philos Mag Series. 5(14), 184–186 (1882)
Dole, M., Mack, L.L., Hines, R.L., Mobley, R.C., Ferguson, L.D., Alice, M.B.: Molecular beams of macroions. J. Chem. Phys. 49, 2240–2249 (1968)
Thomson, B.A., Iribarne, J.V.: Field induced ion evaporation from liquid surfaces at atmospheric pressure. J. Chem. Phys. 71, 4451–4463 (1979)
Schmelzeisen-Redeker, G., Bütfering, L., Röllgen, F.W.: Desolvation of ions and molecules in thermospray mass spectrometry. Int. J. Mass Spectrom. Ion Process. 90, 139–150 (1989)
Fernandez de la Mora, J.: Electrospray ionization of large multiply charged species proceeds via Dole’s charged residue mechanism. Anal. Chim. Acta. 406, 93–104 (2000)
Metwally, H., McAllister, R.G., Popa, V., Konermann, L.: Mechanism of protein supercharging by sulfolane and m-nitrobenzyl alcohol: molecular dynamics simulations of the electrospray process. Anal. Chem. 88, 5345–5354 (2016)
Kim, D., Wagner, N., Wooding, K., Clemmer, D.E., Russell, D.H.: Ions from solution to the gas phase: a molecular dynamics simulation of the structural evolution of substance P during desolvation of charged nanodroplets generated by electrospray ionization. J. Am. Chem. Soc. 139, 2981–2988 (2017)
Hogan, C.J., Carroll, J.A., Rohrs, H.W., Biswas, P., Gross, M.L.: Combined charged residue-field emission model of macromolecular electrospray ionization. Anal. Chem. 81, 369–377 (2009)
Konermann, L., Ahadi, E., Rodriguez, A.D., Vahidi, S.: Unraveling the mechanism of electrospray ionization. Anal. Chem. 85, 2–9 (2013)
Caleman, C., van der Spoel, D.: Evaporation from water clusters containing singly charged ions. Phys. Chem. Chem. Phys. 9, 5105–5111 (2007)
Znamenskiy, V., Marginean, I., Vertes, A.: Solvated ion evaporation from charged water nanodroplets. J. Phys. Chem. A. 107, 7406–7412 (2003)
Konermann, L., McAllister, R.G., Metwally, H.: Molecular dynamics simulations of the electrospray process: formation of NaCI clusters via the charged residue mechanism. J. Phys. Chem. B. 118, 12025–12033 (2014)
Ghassabi Kondalaji, S., Khakinejad, M., Tafreshian, A., Valentine S., J.: Comprehensive peptide ion structure studies using ion mobility techniques: part 1. An advanced protocol for molecular dynamics simulations and collision cross-section calculation. J. Am. Soc. Mass Spectrom. 28, 947–959 (2017)
Khakinejad, M., Ghassabi Kondalaji, S., Tafreshian, A., Valentine, S.J.: Comprehensive gas-phase peptide ion structure studies using ion mobility techniques: part 2. Gas-phase hydrogen/deuterium exchange for ion population estimation. J. Am. Soc. Mass Spectrom. 28, 960–970 (2017)
Maier, J.A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K.E., Simmerling, C.: ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 (2015)
Jorgensen, W.L., Chandrasekhar, J., Madura, J.D., Impey, R.W., Klein, M.L.: Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983)
Grest, G.S., Kremer, K.: Molecular-dynamics simulation for polymers in the presence of a heat bath. Phys. Rev. A. 33, 3628–3631 (1986)
Wu, X., Brooks, B.R.: Self-guided Langevin dynamics simulation method. Chem. Phys. Lett. 381, 512–518 (2003)
Ryckaert, J.-P., Ciccotti, G., Berendsen, H.J.C.: Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 (1977)
Darden, T., York, D., Pedersen, L.: Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993)
D.A. Case, R. M.B., D.S. Cerutti, T.E. Cheatham, III T.A. Darden, R.E. Duke, T.J. Giese, H. Gohlke, A.W. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T.S. Lee, S. LeGrand, P. Li, C., Lin, T. L., R. Luo, B. Madej, D. Mermelstein, K.M. Merz, G. Monard, H. Nguyen, H.T. Nguyen, I. Omelyan, A. Onufriev, D.R. Roe, A. Roitberg, C. Sagui, C.L. Simmerling, W.M. Botello-Smith, J. Swails, R.C. Walker, J. Wang, R.M. Wolf, X. Wu, L. Xiao, P.A. Kollman: AMBER 2016. (2016, University of California, San Francisco)
Roe, D.R., Cheatham, T.E.: PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095 (2013)
Frishman, D., Argos, P.: Knowledge-based protein secondary structure assignment. Proteins. 23, 566–579 (1995)
Schmidt, M.W., Baldridge, K.K., Boatz, J.A., Elbert, S.T., Gordon, M.S., Jensen, J.H., Koseki, S., Matsunaga, N., Nguyen, K.A., Su, S.J., Windus, T.L., Dupuis, M., Montgomery, J.A.: General atomic and molecular electronic-structure system. J. Comput. Chem. 14, 1347–1363 (1993)
Gordon, M.S., Schmidt, M.W.: Advances in electronic structure theory: GAMESS a decade later. Theory and Applications of Computational Chemistry: The First Forty Years. 1167–1189 (2005)
Vanquelef, E., Simon, S., Marquant, G., Garcia, E., Klimerak, G., Delepine, J.C., Cieplak, P., Dupradeau, F.Y.: RED server: a web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids Res. 39, W511–W517 (2011)
Wang, F., Becker, J.P., Cieplak, P., Dupradeau, F.Y.: RED Python: object oriented programming for Amber force fields. Abstr. Pap. Am. Chem. Soc. 247, 1 (2014)
Dupradeau, F.-Y., Pigache, A., Zaffran, T., Savineau, C., Lelong, R., Grivel, N., Lelong, D., Rosanski, W., Cieplak, P.: The R.ED. tools: advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 12, 7821–7839 (2010)
Bayly, C.I., Cieplak, P., Cornell, W.D., Kollman, P.A.: A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges—the RESP model. J. Phys. Chem. 97, 10269–10280 (1993)
Baaden, M., Burgard, M., Wipff, G.: TBP at the water−oil interface: the effect of TBP concentration and water acidity investigated by molecular dynamics simulations. J. Phys. Chem. B. 105, 11131–11141 (2001)
Vega, C., de Miguel, E.: Surface tension of the most popular models of water by using the test-area simulation method. J. Chem. Phys. 126, 154707 (2007)
Fort, K.L., Silveira, J.A., Pierson, N.A., Servage, K.A., Clemmer, D.E., Russell, D.H.: From solution to the gas phase: factors that influence kinetic trapping of substance P in the gas phase. J. Phys. Chem. B. 118, 14336–14344 (2014)
Konermann, L., Rodriguez, A.D., Liu, J.: On the formation of highly charged gaseous ions from unfolded proteins by electrospray ionization. Anal. Chem. 84, 6798–6804 (2012)
Feig, M., Karanicolas, J., Brooks, C.L.: MMTSB tool set: enhanced sampling and multiscale modeling methods for applications in structural biology. J. Mol. Graph. 22, 377–395 (2004)
Marginean, I., Nemes, P., Vertes, A.: Order-chaos-order transitions in electrosprays: the electrified dripping faucet. Phys. Rev. Lett. 97, 4 (2006)
Zakharov, V.V., Brodskaya, E.N., Laaksonen, A.: Surface properties of water clusters: a molecular dynamics study. Mol. Phys. 95, 203–209 (1998)
Kohtani, M., Jones, T.C., Sudha, R., Jarrold, M.F.: Proton transfer-induced conformational changes and melting in designed peptides in the gas phase. J. Am. Chem. Soc. 128, 7193–7197 (2006)
Yun, R.H., Anderson, A., Hermans, J.: Proline in alpha-helix—stability and conformation studied by dynamics simulation. Proteins. 10, 219–228 (1991)
Khakinejad, M., Kondalaji, S.G., Maleki, H., Arndt, J.R., Donohoe, G.C., Valentine, S.J.: Combining ion mobility spectrometry with hydrogen-deuterium exchange and top-down MS for peptide ion structure analysis. J. Am. Soc. Mass Spectrom. 25, 2103–2115 (2014)
Khakinejad, M., Kondalaji, S., Tafreshian, A., Valentine, S.: Gas-phase hydrogen-deuterium exchange labeling of select peptide ion conformer types: a per-residue kinetics analysis. J. Am. Soc. Mass Spectrom. 26, 1115–1127 (2015)
Khakinejad, M., Kondalaji, S.G., Donohoe, G.C., Valentine, S.J.: Ion mobility spectrometry-hydrogen deuterium exchange mass spectrometry of anions: part 2. Assessing charge site location and isotope scrambling. J. Am. Soc. Mass Spectrom. 27, 451–461 (2016)
Acknowledgments
We are grateful for the financial support from the National Institutes of Health (R01GM114494). We are also grateful for conversations with Mr. Chitrak Gupta regarding coding to set up the initial water droplet from a solvent box.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
ESM 1
(PDF 1106 kb)
Rights and permissions
About this article

Cite this article
Kondalaji, S.G., Khakinejad, M. & Valentine, S.J. Comprehensive Peptide Ion Structure Studies Using Ion Mobility Techniques: Part 3. Relating Solution-Phase to Gas-Phase Structures. J. Am. Soc. Mass Spectrom. 29, 1665–1677 (2018). https://doi.org/10.1007/s13361-018-1996-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-018-1996-9