
Abstract
Growth and fatness traits are economically important in the pig industry. To dissect the genetic architecture of these traits in commercial pigs, we conducted a genome-wide association study (GWAS) for carcass weight, backfat thickness, and body weight in two commercial populations: Duroc × (Landrace × Yorkshire) (DLY) and Duroc populations. To enhance the detection power, three GWAS approaches including single-trait GWAS, multi-trait GWAS and meta-analysis were used in this study. A total of 13 suggestive loci were identified on nine chromosomes. The most significant locus was detected at 272.05 Mb on SSC1, and it was associated with backfat thickness at the first rib in the DLY population. Three genes at the identified loci (TBC1D1, BAAT and PHLPP1) were highlighted as functionally plausible candidate genes for pig growth and fatness traits. Genome-wide significant locus was not evidenced in this study, indicating that large populations are required to identify QTL with minor effects on growth and fatness traits in commercial pig populations, in which intensively artificial selections have been imposed on these traits and small genetical variances usually retain in these traits.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Growth and fat deposition are economically important traits in the global pig industry, and they have been intensively selected for several decades (Bosi and Russo 2010). Decoding the genetic architectures of these traits would accelerate their genetic gain in the pig-breeding program by marker-assisted selection and therefore benefit the global pig industry. To understand the molecular bases of phenotypic traits in pigs, many quantitative trait locus (QTL) mapping studies have been conducted in the past 3 decades. Up to now, 1363 and 2470 QTL/associations respectively for growth and fatness traits have been deposited in the pig QTL database (http://www.animalgenome.org/cgi-bin/QTLdb/index, Release 33, August 26, 2017). Notably, a list of major QTL for body weight and fat deposition on pig chromosomes (SSC for Susscrofa chromosome) 1, 2, 4, 6, and 7 have been repeatedly detected in multiple populations (Walling et al. 2000; Sato et al. 2003; Ma et al. 2009; Ai et al. 2012). However, due to a low density of microsatellite markers, traditional QTL mapping usually resulted in a poor resolution with large confidence intervals of 20 or more cM, hindering the application of the identified QTL in pig breeding programs (Riquet et al. 2011).
With the commercialization of Illumina PorcineSNP60 Beadchip in 2008 (Ramos et al. 2009), genome-wide association studies (GWAS) have been widely explored to detect associations between high-density SNPs and economically important phenotypes in pigs, including growth and fatness traits (Fontanesi et al. 2012; Okumura et al. 2013; Fowler et al. 2013; Onteru et al. 2013; Do et al. 2014; Fontanesi et al. 2014; Jiao et al. 2014; Long et al. 2015; Zhu et al. 2014; Howard et al. 2015; Qiao et al. 2015; Wang et al. 2015; Sato et al. 2016). Although most GWAS were conducted in experimental populations, some were performed in commercial populations, including Large White (Fontanesi et al. 2012; Fowler et al. 2013; Onteru et al. 2013; Fontanesi et al. 2014), Duroc (Okumura et al. 2013; Fowler et al. 2013; Do et al. 2014; Jiao et al. 2014; Long et al. 2015; Howard et al. 2015; Wang et al. 2015; Sato et al. 2016), and Pietrain (Fowler et al. 2013). The aim of this study was to detect genomic loci associated with backfat thickness, body weight, and carcass weight in two commercial pig populations via three GWAS approaches.
Materials and methods
Compliance with ethical standards
All procedures involving animals followed the guidelines for the care and use of experimental animals established by the Ministry of Agriculture of China. The ethics committee of Jiangxi Agricultural University specifically approved this study.
Animals and phenotypic recording
The first population was derived from a cross between Duroc boars and Landrace × Yorkshire hybrid sows (hereafter referred to as DLY), consisting of 304 intact females and 306 castrated males. These 610 animals were raised under a common management system on a farm in Xiushui county, Jiangxi province, and were fed ad libitum with a corn and soybean meal based diet containing 3,100 kJ DE, 16% CP, and 0.78% lysine. All animals accessed to fresh water and were slaughtered at the age of 180 ± 3 day in a commercial abattoir. Hot carcass weight (CW), backfat thicknesses at the first rib (FBF) and at the last rib (LBF) were measured postmortem according to the method described in our previous study (Ai et al. 2012). The average backfat thickness (ABF) was the mean of FBF and LBF. The second population was from a purebred Duroc population comprising 983 intact boars. These boars were also fed with a consistent diet containing 3,100 kJ DE, 16% CP, and 0.78% lysine on a farm in Xingxin city, Guangdong province, and freely accessed to fresh water and feed. LBF was recorded for each boar at body weight of 90–110 kg, and then adjusted to the backfat thicknesses at 100 (BF100) and 115 kg (BF115) using the formulae proposed by National Swine Improvement Federation of United States of America (NSIF, http://www.nsif.com/), respectively:
where BF and BW are the recorded backfat thickness (mm) and body weight (kg) respectively.
Meanwhile, body weight was adjusted to a standardized value at the age of 160 days (BW160) using the following formula also from NSIF:
where BW and Age are the recorded body weight (kg) and its weighting age of days.
Genotyping and quality control
Genomic DNA of each animal was extracted from ear or tail tissue using a routine phenol/chloroform extraction method, and was diluted to a final concentration of 50 ng/μl. All individuals from both populations were genotyped using Illumina PorcineSNP60 BeadChips (Illumina, San Diego, CA, USA) following the manufacturer’s protocol. The unmapped SNPs and those with multiple genomic locations on the pig reference genome assembly (Sscrofa10.2) were retained and assigned to chromosome 0, and their positions were operationally determined in an interval of 20 kb according to their names in ascending order as described previously (Guo et al. 2013). PLINK was used for quality control of the data based on the following criteria: (1) SNP call rate > 0.90, (2) minor allele frequency (MAF) > 0.01, (3) not significant deviation from Hardy–Weinberg equilibrium (P < 10−5), and (4) animal call rate > 0.9 (Purcell et al. 2007). The qualified animals and SNPs were retained for further statistical analysis.
Statistical analysis
The means and standard deviations of the recorded traits were calculated by the mean and SD functions of R (R3.2.0, R Foundation for Statistical Computing, Austria), respectively. The phenotypic differences between sexes were tested by the t-test function in the DLY population. The correlation coefficients among these traits were calculated by the cor function, and their differences between sexes were determined by a Fisher’s transformation (Fisher 1925). The lm function was used to determine the fixed factors and covariates in the GWAS model.
A family-based score test (FASTA) implemented in the GenABEL package of R (Aulchenko et al. 2007) was used to detect the association between SNPs and traits (Chen and Abecasis 2007). FASTA explored two-stage analyses. The first stage estimated the phenotypic expectation vector (E(y)), the polygenic additive variance \( \left({\widehat{\sigma}}_u^2\right) \) and the residual variance \( \left({\widehat{\sigma}}_e^2\right) \) using the following mixed model:
where y is the phenotype vector; μ is the population mean; b is the vector of fixed effects (sex and batch) in the DLY population (an additional fixed effect of carcass weight was used in the analysis for backfat thicknesses), and is year and month of birth in the Duroc population; u is the predictor vector of polygenic addictive random effect following N(0, G σ u 2), where G is the genomic kinship matrix calculated on the basis of the identity-by-state of the SNPs on the autosomes (Amin et al. 2007) and σ u 2 is the polygenic additive variance; 1, X, and Z are the incidence vector and matrices for μ, b, and u, respectively.
During the second stage, the association between SNPs and traits was evaluated by a FASTA test. The statistic (T 2) for each SNP was calculated using the maximum likelihood estimates (E(y), \( {\widehat{\sigma}}_u^2 \) and \( {\widehat{\sigma}}_e^2 \)) according to following formula:
where g is the genotype vector for the tested SNP, E(g) is a vector with identical elements that give the unconditional expectation of each genotype, and I is an identity matrix.
T 2 statistics followed a \( \lambda \bullet {\chi}_1^2 \) distribution, where λ is the residual inflation. λ was estimated from the genomic data using the following formula:
where the numerator is the median of the statistic across the genome, and the denominator is the median of \( {\chi}_1^2 \).
The estimate of λ can adjust the value of T 2 at the tested locus and make the corrected statistic following a chi-square distribution with 1 degree of freedom. This procedure is known as genomic control (Devlin and Roeder 1999). A permutation approach with 10,000 iterations was explored to obtain the threshold P values for suggestive (5% chromosome-wide), 5%, and 1% genome-wide significance levels of GWAS (Churchill and Doerge 1994).
Multi-trait GWAS were performed to detect pleiotropic polymorphisms using the method proposed by Bolormaa et al. (2014). Briefly, a statistic, which approximately follows a chi-square distribution with the number of traits tested as its degrees of freedom, was calculated for each SNP according to the following formula:
where T i is a vector of the signed t values of the ith SNP from the above-mentioned single-trait GWAS and \( {T}_i^{\hbox{'}} \) is the transpose of vector T i ; V −1 is the inverse of the correlation matrix of the traits, and the correlation between two traits was calculated using the estimated effects of the qualified SNPs (signed t values). The threshold P values for significant levels were the same as those using in the single-trait GWAS.
A meta-analysis can increase statistical power especially for a locus with small effect by collectively using information from multiple independent studies (Evangelou and Ioannidis 2013). We used the METAL software (Willer et al. 2010) to perform the meta-analysis of GWAS in the two populations. The pooled effect and its standard error of a tested SNP were weighted by the inverse of variance, and they were calculated according to the following formula, respectively:
where n is the population number, and β i and s i are the allele effect and its standard error in the ith population respectively.
A statistic (β divided by s) of Z-test is used to evaluate the evidence of associations between SNPs and traits in the meta-analysis. The threshold P values for suggestive, 5%, and 1% genome-wide significance levels were the means of their counterparts used in the two populations, respectively.
To determine the approximate positions of unmapped SNPs surpassing the significant threshold, a two-point linkage analysis was performed using CRI-MAP software (Green et al. 1990) in a White Duroc × Erhualian F2 resource population (Guo et al. 2013), in which all individuals were also genotyped by Illumina PorcineSNP60 BeadChips. Haplotypes of target regions were inferred by PLINK (Purcell et al. 2007), and linkage disequilibrium (LD) blocks were constructed by Haploview4.2 (Barrett et al. 2005) under the default parameters. To highlight candidate genes for each significant locus, the functions of all annotated genes within an interval of 1.5 Mb around the top SNP at each locus were searched via the GeneCards web (Rebhan et al. 1997). If a gene is associated with any traits related to growth and fatness in human, mouse, or other farm animals, it was considered a candidate gene for the detected locus.
Results
Statistics of phenotypic data
Table 1 lists descriptive statistics of the recorded traits in the two populations. In the DLY population, the castrated male’s backfat was significantly thicker (P < 0.01) than the intact female’s, while carcass weight was not different (P = 0.79) between sexes. The simple correlation coefficients between the recorded traits in two populations are shown in Table 4. In the DLY population, all traits were significantly and positively correlated with each other (P < 0.01), and all correlation coefficients were higher in males than in females (P < 0.05). All traits were also significantly correlated with each other (P < 0.01) in the Duroc population, while BW160 was negatively correlated with BF100 and BF115.
Qualified SNPs and animals in the GWAS
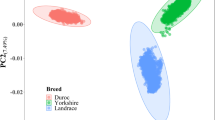
A total of 607 pigs and 39,166 SNPs including 4,830 unmapped SNPs passed the quality control in the DLY population, and 983 individuals and 38,077 SNPs including 4,833 unmapped SNPs in the Duroc population. The average physical distances between adjacent SNPs were 75.4 and 77.8 kb respectively in the two populations. A common set of 29,508 qualified SNPs between the two populations was used in the meta-analysis of GWAS, and the average physical distance between adjacent common SNPs was 101.7 kb. Population stratification existed in both populations as revealed by a principal component analysis (Fig. S2). To adjust the relationships between animals, a random polygenic effect estimated with a genomic kinship was included in the GWAS model.
Single-trait GWAS results
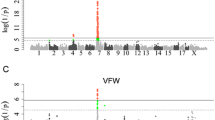
In single-trait GWAS analyses, we only identified four chromosomal regions associated with backfat thickness traits at the suggestive significant level in the two populations (Table 2 and Fig. 1). Two loci associated with FBF were identified in the DLY population. One was at 272,049,666 bp on SSC1, with the top SNP located in a 408-kb LD block (Fig. 2a), and the other was at 78,209,229 bp on SSC8. The other two loci, each at 176,159,526 bp in a 541-kb LD block (Fig. 2b) on SSC1 and at 143,372,799 bp on SSCX, were associated with both BF100 and BF115 in the Duroc population.

Manhattan plots of single-trait GWAS for fatness and growth traits in DLY and Duroc populations. The solid and dotted horizontal lines indicate the 5% genome-wide and chromosome-wise (suggestive) significant threshold values respectively

Linkage disequilibrium (LD) blocks and candidate genes within the significantly associated regions on SSC1. a DLY pigs (around 272 Mb), and b Duroc pigs (around 176 Mb). From top to below are Manhattan plots, annotated genes within the chromosomal region that were retrieved from the Ensemble genome browser (www.ensembl.org/Sus_scrofa/Info/Index) and LD blocks, respectively
Although we did not detect any mapped SNPs significantly associated with CW in the DLY population and with BW160 in the Duroc population, two unmapped SNPs including MARC0026068 and MARC0062432 appeared to be associated with the two traits in the two populations at the suggestive level, respectively (Fig. 1). MARC0026068 and MARC0062432 were tightly linked to ALGA0117611 (rec = 0.0114, LOD = 65.12) at 139,538,709 bp on SSC6 and MARC0030372 (rec = 0.0029, LOD = 188.99) at 9,651,795 bp on SSC7, respectively. Therefore, we inferred that there were two loci each associated with CW on SSC6 and with BW160 on SSC7.
GWAS meta-analysis results
As shown in Table 3 and Fig. 5, only two suggestive loci were identified in the meta-analysis of GWAS. One was associated with backfat thickness around 34,634,619 bp on SSC7, and the top SNP was M1GA0009933. The other was associated with body weight at 156,381,110 bp on SSC2, and the top SNP was ALGA0104903. In addition to the two loci, an unmapped SNP DIAS0001614 was also associated with body weight at the suggestive significant level. This SNP was tightly linked to ASGA0105629 (rec = 0.0000, LOD = 126.43) at 156,102,759 bp on SSC2.
Multi-trait GWAS results
No significant locus was identified in the DLY population (Table 3, Fig. 6). Three suggestive loci were detected in the Duroc population, each at 79,601,958 bp on SSC8, at 54,624,831 bp on SSC12 and at 5,800,383 bp on SSCX, and the top SNPs at these three loci were MARC0059309, ASGA0085214, and ASGA0080690 respectively.
Potential loci with P-value less than 0.001
Some potential chromosomal regions with P-value (corrected by genomic control) less than 0.001 are listed in Tables 5 and 6.
Discussion
Comparison of our findings with previous reports
In this study, we conducted single-trait GWAS, multi-trait GWAS, and meta-analyses of GWAS for backfat thickness, carcass weight, and body weight in two commercial pig populations. To our knowledge, this is the first meta-analysis of GWAS for backfat thickness and body weight in commercial pigs. A total of 13 loci on nine chromosomes were identified in this study, six of which have also been detected in other commercial populations. The locus for carcass weight at 139,538,709 bp on SSC6 is proximal to a locus for average dairy gain (ADG) in Duroc (Sato et al. 2016) and Yorkshire (Onteru et al. 2013) populations; and is also close to a locus for days to 100 kg of body weight in a Duroc population (Wang et al. 2015). Another locus for body weight at 160 days at 9,651,795 bp on SSC7 is adjacent to a reported locus for ADG in an Italian Large White population (Fontanesi et al. 2014). The locus for backfat thickness at 34,634,619 bp on SSC7 corresponds to a reported locus for backfat thickness in two Duroc populations (Zhu et al. 2014; Sato et al. 2016). Another locus for backfat thickness at 176,159,526 bp on SSC1 confirms the previously reported locus for this trait at 178.02 Mb on this chromosome (Zhu et al. 2014). A multi-trait locus at 54,624,831 on SSC12 is close to a locus for ADG around 53.06 Mb in Duroc pigs (Howard et al. 2015), and another multi-trait locus at 28,284,785 bp on SSC6 was evidenced to a locus for ADG in an Italian Large White population (Fontanesi et al. 2014). These consistent loci provide potential markers for selective breeding of growth and fatness traits in commercial pig populations.
Large populations and high-density markers are required to map loci for growth and fatness traits in commercial populations
We used hundreds of pigs in the DLY (n = 607) and Duroc (n = 983) populations, but only a total of 13 suggestive loci were identified in this study. This could be explained by intensive artificial selection on commercial populations. It is well known that intensive artificial selection has been imposed on growth and backfat thickness in commercial breeds like Duroc, Landrace, and Large White during the past decades (Schiavo et al. 2016), resulting in relatively small genetic variance in these traits. Therefore, major loci for these traits could have been fixed in commercial populations by strong artificial selection. Large population size is required to detect minor loci, and hundreds of animals are not enough to identify such small effects. Moreover, the average physical distance between adjacent SNPs was more than 75 kb in the two populations, and some loci with minor effects could not be identified due to insufficient marker coverage of the 60 K BeadChip (Becker et al. 2013).
Comparison of results from the three GWAS approaches
Except that one locus was identified on SSC8 by single-trait GWAS in the DLY population (78.2 Mb) and by multi-trait GWAS in the Duroc (79.6 Mb), no other common locus was detected between the two populations or across the three GWAS approaches (Tables 2 and 3, Figs. 1, 5 and 6). This may be due to different genetic backgrounds between the two populations and different theoretical assumptions of the three GWAS methods. As mentioned in the 'Materials and methods' section, the Duroc population was a purebred population, but the DLY population was derived from a cross of three breeds including Duroc, Landrace, and Large White. Therefore, the two populations had different genetic backgrounds. In this study, three GWAS methods were used to identify genomic loci for growth and fatness traits from different facets. Single-trait GWAS in single and multiple populations are powerful in detecting QTL for target traits in each population and across populations, respectively. Multi-trait GWAS is efficient in mapping QTL with pleiotropic effects on target traits in each population.
Candidate genes at the identified loci
SSC1
The top SNP (ALGA0008905) at the suggestive locus for backfat thickness on SSC1 in the DLY population located in a 408-kb LD block (Fig. 2A), and the BAAT (bile acid-CoA:amino acid N-acyltransferase) gene was about 460 kb upstream of the SNP and 49.55 kb upstream of the LD block. BAAT is involved in bile acid metabolism and indirectly facilitates lipid- and fat-soluble vitamin absorption (Hadžić et al. 2012). Therefore, we considered BAAT a candidate gene for backfat thickness. Another locus was evidenced in the Duroc population, and the lead SNP (MARC0034873) at this locus mapped at 176.16 Mb and resided in the PHLPP1 (PH domain leucine-rich repeat-containing protein phosphatase 1) gene within a 541-kb LD block (Fig. 2B). PHLPP1 encodes a phosphatase, and can terminate Akt signaling that is able to regulate insulin levels (Dong et al. 2014). It has been reported that PHLPP1 abundance was increased in adipose tissue and skeletal muscle of obese individuals, and variants in this gene have been associated with BMI and insulin resistance in humans (Andreozzi et al. 2011) and average daily weight in pigs (Howard et al. 2015). Thus, we propose PHLPP1 as a candidate gene at this locus. We noted that MC4R, a well-characterized gene for fatness, growth, and feed intake in pigs (Kim et al. 2000; Óvilo et al. 2006; Fan et al. 2009), is located 2.39 Mb downstream of PHLPP1. It is worthwhile conducting further investigations to test whether MC4R is also a candidate gene for this locus.
SSC8
We detected a suggestive locus associated with BW160 at 32.36 Mb on SSC8 in the Duroc population (Table 4). TBC1D1 (TBC1 (tre-2/USP6, BUB2, cdc16) domain family, member 1) gene is 1.31 Mb upstream of the top SNP at this locus. This gene has been implicated in human obesity (Stone et al. 2006; Meyre et al. 2008; Scherag et al. 2010), and functional mutations in this gene led to leanness and protected from diet-induced obesity in mice (Chadt et al. 2008). Furthermore, it was associated with ham weight in Italian Large White pigs (Fontanesi et al. 2011). Therefore, it is a promising candidate gene for BW160.
Conclusion
We conducted single-trait GWAS, multi-trait GWAS, and meta-analysis of GWAS for backfat thickness, carcass weight, and body weight in two commercial pig populations. We show that single-trait GWAS is powerful to detect trait-specific QTL and multi-trait GWAS is efficient for mapping pleiotropic QTL, while meta-analysis can improve the detection power for common loci across multiple small populations. In total, we identified only 13 suggestive loci with minor effects on the tested traits. This indicates that intensive selection for lean meat production during the past decades could cause the fixation of major loci for growth and fatness in commercial breeds such as Duroc, Landrace, Duroc, and Pietrain. Therefore, large populations are required to identify QTL for growth and fatness traits in these breeds. TBC1D1, BAAT, and PHLPP1 are three functionally plausible candidate genes for pig growth and fatness traits.
References
Ai H, Ren J, Zhang Z, Ma J, Guo Y, Yang B, Huang L (2012) Detection of quantitative trait loci for growth- and fatness-related traits in a large-scale white Duroc × Erhualian intercross pig population. Anim Genet 43:383–391
Amin N, van Duijn CM, Aulchenko YS (2007) A genomic background based method for association analysis in related individuals. PLoS One 2:e1274
Andreozzi F, Procopio C, Greco A, Mannino GC, Miele C, Raciti GA, Iadicicco C, Beguinot F, Pontiroli AE, Hribal ML, Folli F, Sesti G (2011) Increased levels of the Akt-specific phosphatase PH domain leucine-rich repeat protein phosphatase (PHLPP)-1 in obese participants are associated with insulin resistance. Diabetologia 54:1879–1887
Aulchenko YS, Ripke S, Isaacs A, van Duijn CM (2007) GenABEL: an R library for genome-wide association analysis. Bioinformatics 23:1294–1296
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21:263–265
Becker D, Wimmers K, Luther H, Hofer A, Leeb T (2013) A genome-wide association study to detect QTL for commercially important traits in Swiss large white boars. PLoS One 8:e55951
Bolormaa S, Pryce JE, Reverter A, Zhang Y, Barendse W, Kemper K, Tier B, Savin K, Hayes BJ, Goddard ME (2014) A multi-trait, meta-analysis for detecting pleiotropic polymorphisms for stature, fatness and reproduction in beef cattle. PLoS Genet 10:e1004198
Bosi P, Russo V (2010) The production of the heavy pig for high quality processed products. Ital J Anim Sci 3:309–321
Chadt A, Leicht K, Deshmukh A, Jiang LQ, Scherneck S, Bernhardt U, Dreja T, Vogel H, Schmolz K, Kluge R, Zierath JR, Hultschig C, Hoeben RC, Schurmann A, Joost HG, Al-Hasani H (2008) Tbc1d1 mutation in lean mouse strain confers leanness and protects from diet-induced obesity. Nat Genet 40:1354–1359
Chen WM, Abecasis GR (2007) Family-based association tests for genomewide association scans. Am J Hum Genet 81:913–926
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Devlin B, Roeder K (1999) Genomic control for association studies. Biometrics 55:997–1004
Do DN, Ostersen T, Strathe AB, Mark T, Jensen J, Kadarmideen HN (2014) Genome-wide association and systems genetic analyses of residual feed intake, daily feed consumption, backfat and weight gain in pigs. BMC Genet 15:27
Dong L, Jin L, Tseng HY, Wang CY, Wilmott JS, Yosufi B, Yan XG, Jiang CC, Scolyer RA, Zhang XD, Guo ST (2014) Oncogenic suppression of PHLPP1 in human melanoma. Oncogene 33:4756–4766
Evangelou E, Ioannidis JP (2013) Meta-analysis methods for genome-wide association studies and beyond. Nat Rev Genet 14:379–389
Fan B, Onteru SK, Plastow GS, Rothschild MF (2009) Detailed characterization of the porcine MC4R gene in relation to fatness and growth. Anim Genet 40:401–409
Fisher RA (1925) Statistical methods for research workers. Oliver and Boyd, Edinburgh
Fontanesi L, Colombo M, Tognazzi L, Scotti E, Buttazzoni L, Dall'Olio S, Davoli R, Russo V (2011) The porcine TBC1D1 gene: mapping, SNP identification, and association study with meat, carcass and production traits in Italian heavy pigs. Mol Biol Rep 38:1425–1431
Fontanesi L, Schiavo G, Galimberti G, Calo DG, Scotti E, Martelli PL, Buttazzoni L, Casadio R, Russo V (2012) A genome wide association study for backfat thickness in Italian large white pigs highlights new regions affecting fat deposition including neuronal genes. BMC Genomics 13:583
Fontanesi L, Schiavo G, Galimberti G, Calo DG, Russo V (2014) A genomewide association study for average daily gain in Italian large white pigs. J Anim Sci 92:1385–1394
Fowler KE, Pong-Wong R, Bauer J, Clemente EJ, Reitter CP, Affara NA, Waite S, Walling GA, Griffin DK (2013) Genome wide analysis reveals single nucleotide polymorphisms associated with fatness and putative novel copy number variants in three pig breeds. BMC Genomics 14:784
Green P, Falls K, Crooks S (1990) CRI-MAP Documentation. Washington University School of Medicine, St. Louis
Guo YM, Zhang XF, Ren J, Ai HS, Ma JW, Huang LS (2013) A joint genomewide association analysis of pig leg weakness and its related traits in an F2 population and a Sutai population. J Anim Sci 91:4060–4068
Hadžić N, Bull LN, Clayton PT, Knisely AS (2012) Diagnosis in bile acid-CoA: amino acid N-acyltransferase deficiency. World J Gastroenterol 18:3322–3326
Howard JT, Jiao S, Tiezzi F, Huang Y, Gray KA, Maltecca C (2015) Genome-wide association study on legendre random regression coefficients for the growth and feed intake trajectory on Duroc boars. BMC Genet 16:59
Jiao S, Maltecca C, Gray KA, Cassady JP (2014) Feed intake, average daily gain, feed efficiency, and real-time ultrasound traits in Duroc pigs: II. Genomewide association. J Anim Sci 92:2846–2860
Kim KS, Larsen N, Short T, Plastow G, Rothschild MF (2000) A missense variant of the porcine melanocortin-4 receptor (MC4R) gene is associated with fatness, growth, and feed intake traits. Mamm Genome 11:131–135
Long Y, Ruan GR, Su Y, Xiao SJ, Zhang ZY, Ren J, Ding NS, Huang LS (2015) Genome-wide association study identifies QTLs for EBV of backfat thickness and average daily gain in Duroc pigs. Genetika 51:371–378
Ma J, Ren J, Guo Y, Duan Y, Ding N, Zhou L, Li L, Yan X, Yang K, Huang L, Song Y, Xie J, Milan D, Huang L (2009) Genome-wide identification of quantitative trait loci for carcass composition and meat quality in a large-scale white Duroc x Chinese Erhualian resource population. Anim Genet 40:637–647
Meyre D, Farge M, Lecoeur C, Proenca C, Durand E, Allegaert F, Tichet J, Marre M, Balkau B, Weill J, Delplanque J, Froguel P (2008) R125W coding variant in TBC1D1 confers risk for familial obesity and contributes to linkage on chromosome 4p14 in the French population. Hum Mol Genet 17:1798–1802
Okumura N, Matsumoto T, Hayashi T, Hirose K, Fukawa K, Itou T, Uenishi H, Mikawa S, Awata T (2013) Genomic regions affecting backfat thickness and cannon bone circumference identified by genome-wide association study in a Duroc pig population. Anim Genet 44:454–457
Onteru SK, Gorbach DM, Young JM, Garrick DJ, Dekkers JC, Rothschild MF (2013) Whole genome association studies of residual feed intake and related traits in the pig. PLoS One 8:e61756
Óvilo C, Fernández A, Rodríguez MC, Nieto M, Silió L (2006) Association of MC4R gene variants with growth, fatness, carcass composition and meat and fat quality traits in heavy pigs. Meat Sci 73:42–47
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575
Qiao R, Gao J, Zhang Z, Li L, Xie X, Fan Y, Cui L, Ma J, Ai H, Ren J, Huang L (2015) Genome-wide association analyses reveal significant loci and strong candidate genes for growth and fatness traits in two pig populations. Genet Sel Evol 47:17
Ramos AM, Crooijmans RP, Affara NA, Amaral AJ, Archibald AL, Beever JE, Bendixen C, Churcher C, Clark R, Dehais P, Hansen MS, Hedegaard J, Hu ZL, Kerstens HH, Law AS, Megens HJ, Milan D, Nonneman DJ, Rohrer GA, Rothschild MF, Smith TP, Schnabel RD, Van Tassell CP, Taylor JF, Wiedmann RT, Schook LB, Groenen MA (2009) Design of a high density SNP genotyping assay in the pig using SNPs identified and characterized by next generation sequencing technology. PLoS One 4:e6524
Rebhan M, Chalifa-Caspi V, Prilusky J, Lancet D (1997) GeneCards: integrating information about genes, proteins and diseases. Trends Genet 13:163
Riquet J, Gilbert H, Servin B, Sanchez MP, Iannuccelli N, Billon Y, Bidanel JP, Milan D (2011) A locally congenic backcross design in pig: a new regional fine QTL mapping approach miming congenic strains used in mouse. BMC Genet 12:6
Sato S, Oyamada Y, Atsuji K, Nade T, Sato S, Kobayashi E, Mitsuhashi T, Nirasawa K, Komatsuda A, Saito Y, Terai S, Hayashi T, Sugimoto Y (2003) Quantitative trait loci analysis for growth and carcass traits in a Meishan x Duroc F2 resource population. J Anim Sci 81:2938–2949
Sato S, Uemoto Y, Kikuchi T, Egawa S, Kohira K, Saito T, Sakuma H, Miyashita S, Arata S, Kojima T, Suzuki K (2016) SNP- and haplotype-based genome-wide association studies for growth, carcass, and meat quality traits in a Duroc multigenerational population. BMC Genet 17:60
Scherag A, Dina C, Hinney A, Vatin V, Scherag S, Vogel CI, Muller TD, Grallert H, Wichmann HE, Balkau B, Heude B, Jarvelin MR, Hartikainen AL, Levy-Marchal C, Weill J, Delplanque J, Korner A, Kiess W, Kovacs P, Rayner NW, Prokopenko I, McCarthy MI, Schafer H, Jarick I, Boeing H, Fisher E, Reinehr T, Heinrich J, Rzehak P, Berdel D, Borte M, Biebermann H, Krude H, Rosskopf D, Rimmbach C, Rief W, Fromme T, Klingenspor M, Schurmann A, Schulz N, Nothen MM, Muhleisen TW, Erbel R, Jockel KH, Moebus S, Boes T, Illig T, Froguel P, Hebebrand J, Meyre D (2010) Two new loci for body-weight regulation identified in a joint analysis of genome-wide association studies for early-onset extreme obesity in French and German study groups. PLoS Genet 6:e1000916
Schiavo G, Galimberti G, Calo DG, Samore AB, Bertolini F, Russo V, Gallo M, Buttazzoni L, Fontanesi L (2016) Twenty years of artificial directional selection have shaped the genome of the Italian large white pig breed. Anim Genet 47:181–191
Stone S, Abkevich V, Russell DL, Riley R, Timms K, Tran T, Trem D, Frank D, Jammulapati S, Neff CD, Iliev D, Gress R, He G, Frech GC, Adams TD, Skolnick MH, Lanchbury JS, Gutin A, Hunt SC, Shattuck D (2006) TBC1D1 is a candidate for a severe obesity gene and evidence for a gene/gene interaction in obesity predisposition. Hum Mol Genet 15:2709–2720
Walling GA, Visscher PM, Andersson L, Rothschild MF, Wang L, Moser G, Groenen MA, Bidanel JP, Cepica S, Archibald AL, Geldermann H, de Koning DJ, Milan D, Haley CS (2000) Combined analyses of data from quantitative trait loci mapping studies. Chromosome 4 effects on porcine growth and fatness. Genetics 155:1369–1378
Wang K, Liu D, Hernandez-Sanchez J, Chen J, Liu C, Wu Z, Fang M, Li N (2015) Genome wide association analysis reveals new production trait genes in a male Duroc population. PLoS One 10:e0139207
Willer CJ, Li Y, Abecasis GR (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26:2190–2191
Zhu D, Liu X, Rothschild M, Zhang Z, Zhao S, Fan B (2014) Genome-wide association study of the backfat thickness trait in two pig populations. Front Agric Sci Eng 1:91–95
Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (31660303, 31525023 and 31460590).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Communicated by: Maciej Szydlowski
Electronic supplementary material
Tables 4, 5, and 6
ESM 1
(DOCX 45 kb)
ESM 2
Quantile–quantile plots for the recorded traits in the two populations. (GIF 118 kb)
ESM 3
Schematic diagram of the first two principal components based on the genomic kinships. a DLY, and b Duroc. (GIF 71 kb)
ESM 4
Manhattan plots of meta-analyses for backfat thickness (ABF from DLY and BF100 from Duroc) and body weight (carcass weight from DLY and BW160 from Duroc). The solid and dotted horizontal lines indicate the 5% genome-wide and chromosome-wise (suggestive) significant threshold values, respectively. (GIF 117 kb)
ESM 5
Manhattan plots of multi-trait GWAS in DLY and Duroc populations. (GIF 95 kb)
Rights and permissions
About this article

Cite this article
Guo, Y., Qiu, H., Xiao, S. et al. A genome-wide association study identifies genomic loci associated with backfat thickness, carcass weight, and body weight in two commercial pig populations. J Appl Genetics 58, 499–508 (2017). https://doi.org/10.1007/s13353-017-0405-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13353-017-0405-6