
Abstract
Pigeonpea production is severely constrained by wilt disease caused by Fusarium udum. In the current study, we discover the putative genomic regions that control resistance response to variant 2 of fusarium wilt using association mapping approach. The association panel comprised of 89 diverse pigeonpea genotypes including seven varieties, three landraces and 79 germplasm lines. The panel was screened rigorously for 3 consecutive years (2013–14, 2014–15 and 2015–2016) against variant 2 in a wilt-sick field. A total of 65 pigeonpea specific hypervariable SSR markers (HASSRs) were screened representing seven linkage groups and 29 scaffolds of the pigeonpea genome. A total of 181 alleles were detected, with average values of gene diversity and polymorphism information content (PIC) of 0.55 and 0.47, respectively. Further analysis using model based (STRUCTURE) and distance based (clustering) approaches separated the entire pigeonpea collection into two distinct subgroups (K = 2). The marker trait associations (MTAs) were established based on three-year wilt incidence data and SSR dataset using a unified mixed linear model. Consequently, six SSR markers were identified, which were significantly associated with wilt resistance and explained up to 6% phenotypic variance (PV) across the years. Among these SSRs, HASSR18 was found to be the most stable and significant, accounting for 5–6% PV across the years. To the best of our knowledge, this is the first report of identification of favourable alleles for resistance to variant 2 of Fusarium udum in pigeonpea using association mapping. The SSR markers identified here will greatly facilitate marker assisted resistance breeding against fusarium wilt in pigeonpea.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pigeonpea [Cajanus cajan (L.) Millsp.] is a major grain legume crop of India, which accounts for 70% of the global pigeonpea production. It is the second most important pulse crop next to chickpea in India, which is grown in an area of 3.86 million hectare with average annual production of 3.29 million tonnes (FAOSTAT 2014). Despite concerted breeding efforts for several decades, expected progress has not been witnessed in pigeonpea genetic improvement. The potential reasons that explain this slow progress include narrow genetic base of the cultivated pool, poor crop husbandry and exposure to a number of biotic and abiotic stresses. In recent years, genomic resources have been established as a powerful tool to increase the efficacy of crop breeding techniques. Significant progress has been achieved in developing large scale genomic and genetic resources for genetic mapping and reverse genetic analysis in pigeonpea (Varshney et al. 2010). For instance, in terms of transcriptomics resources, 10,000 expressed sequence tags (ESTs) from Sanger sequencing and 2 million short ESTs by 454/FLX sequencing have been generated followed by a comprehensive transcriptome assembly of pigeonpea (Dubey et al. 2011; Dutta et al. 2011, Kudapa et al. 2012). A total of 3072 SSR markers were synthesized from BAC-end sequences (BESs) to facilitate hybrid purity testing and genetic mapping (Bohra et al. 2011). The genome sequence has facilitated discovery of hypervariable Arhar simple sequence repeat markers (designated as HASSRs), 437 of which were experimentally validated among pigeonpea varieties (Singh et al. 2011). More recently, the utility of new 401 hypervarible SSR markers (CcGMs) was sucessfully demonstrated in pigeonpea (Bohra et al. 2017).
The pigeonpea productivity is severely limited by a range of biotic stresses such as fusarium wilt (FW), sterility mosaic disease (SMD) and pod borer (Singh et al. 2013). Among biotic stresses, fusarium wilt (Fusarium udum Butler) is the major soil borne fungal disease in pigeonpea (Butler 1906) that along with SMD is reported to account for annual losses to the tune of US$113 million (Saxena et al. 2010). The yield loss due to wilt depends on the stage at which wilting occurs; for instance 30% and 100% losses are reported when wilting occurs at the pre-harvest and pre-pod stages, respectively (Kannaiyan and Nene 1981). The existence of different variants/races of Fusarium udum poses a major challenge for breeding programs that aim to improve wilt resistance of pigeonpea (Singh et al. 2011; Tiwari and Dhar 2011). Importantly, the F. udum isolates from diverse geographical origins have been reported to show high variability in their virulence (Mishra and Dhar 2003). Prevalence of five variants of the pathogen (1, 2, 3, 4 and 5) has been reported in India through multi-locational screening of pigeonpea genotypes (Dhar 1995). Based on the distribution map of F. udum variants, variant 2 could be considered as the most virulent and widely distributed in India (Patil et al. 2013).
The management of fusarium wilt through chemical interventions is not a cost-effective and eco-friendly approach. Because of its soil borne nature and long survival of the pathogen, growing resistant varieties remains the most sustainable approach to control the disease (Mishra and Dhar 2010). Therefore, improved varieties showing resistance to major diseases are urgently needed. Based on the previous analyses that aimed to understand the genetics of FW resistance in pigeonpea, researchers have proposed the presence of a single gene of dominant (Singh et al. 1998) or recessive (Jain and Reddy 1995) nature or more than one gene (Joshi 1957) such as two complementary genes (Okiror 2002), duplicate recessive (Patil et al. 2013) in different crosses. Considering the pathogenic variability in F. udum, consistent efforts were made in the past to identify stable and broad based resistant sources through large-scale screening of pigeonpea genotypes (Okiror 1999; Sharma et al. 2012, 2016; Singh et al. 2011). Since, germplasm represents the important genetic resource that harbours resistance genes for various diseases. The effective utilization of crop diversity held in gene banks depends on our knowledge of useful traits and available markers associated with the target traits (Kumar et al. 2014).
The availability of wide-ranging molecular markers has enhanced the scope for trait mapping through linkage and linkage disequilibrium (LD) mapping strategies. The LD mapping or association analysis has been receiving unprecedented attention because of its inherent advantages over the conventional QTL mapping, which includes enhanced genetic resolution, cost-efficiency and non-requirement of the mapping populations (Myles et al. 2009; Bohra 2013). Association mapping has been demonstrated to be an extremely efficient method to genetically dissect the simple qualitative and complex quantitative traits in many other crops (Atwell et al. 2010; Sakiroglu et al. 2012; Perseguini et al. 2016). Limited efforts have been made so far to discover the DNA markers linked with the fusarium wilt resistance in pigeonpea; for instance randomly amplified polymorphic DNA (RAPD: Kotresh et al. 2006), SSR (Singh et al. 2013), single nucleotide polymorphism (SNP: Singh et al. 2016a, b). Concerning LD mapping, scanty information is available in pigeonpea that concerns mapping of determinacy trait (Mir et al. 2014). Therefore, the present study aims to identify the SSR markers tightly associated with resistance to FW variant 2 in a large set of pigeonpea genotypes, and the SSRs identified here could be used effectively in wilt resistance breeding programme.
Materials and methods
Plant material
A total of 89 pigeonpea genotypes including seven varieties, three landraces and 79 germplasm lines were used (Table 1). Seeds of these genotypes were obtained from Division of Crop Improvement, Indian Institute of Pulses Research (IIPR), Kanpur, India. These genotypes were selected based on their relevance to wilt resistance improvement programme in India.
Screening for wilt resistance
The association mapping panel was phenotyped for three cropping seasons viz. 2013/14, 2014/15 and 2015/16 against FW variant 2 in the wilt sick field (FW variant 2) at IIPR, Kanpur. Sowing was done in the month of July second fortnight in all 3 years. The experiment was conducted in randomized block design (RBD) having three replications with ICP 8863 (resistant) and Bahar (susceptible) as checks. Each replication consisted of a single row of 5 m length of each test entry with a spacing of 60 × 15 cm. The susceptible check was planted after every two test entries and resistant check was planted after every 10 test entries to monitor sickness level in the field. Recommended agronomic practices were followed. Wilt incidence was recorded fortnightly in these genotypes for 4 months (November to February), i.e. during pre-flowering, flower and pod development stages. As described by Sharma et al. (2016), the cumulative incidence for all 4 months was calculated using the following formula:
Based on the above formula, genotypes were categorized into distinct groups viz. resistant (<10.0% incidence), moderately resistant (10.1–20.0% incidence), moderately susceptible (20.1–50.0% incidence) and susceptible (>50.0% incidence). The 3 year percent wilt incidence data (Suppl. Table 1) were curated with arcsine transformation to normalize the data (Gomez and Gomez 1984).
SSR genotyping
Genomic DNA was isolated from each genotype by following the CTAB method (Agbagwa et al. 2012). To facilitate marker-trait association (MTA) detection, 65 hypervariable pigeonpea specific (HASSR) primers were used, which were previously validated on 3% agarose gels (Singh et al. 2012). Since markers selected were unmapped, the possible physical location of the SSR markers was determined by aligning these on to pigeonpea genome using BLAT option in Genome Browser (GBrowse, Cc1.0) of Legume Information System (LIS). The default parameters like minimum sequence identity percent of 90 and number of hits to return 5 were selected for alignment. Physical genome positions between markers on each chromosome were converted into centi Morgan (cM) map units by considering physical to genetic linkage ratio of 561.1 Kb/cM for 853 Mb of pigeonpea genome size (Kumawat et al. 2012). The representative genetic linkage map was drawn using GGT 2.0 software package (Van-Berloo 2008). Polymerase chain reaction (PCR) was performed as per the protocol described by Patil et al. (2016), amplicons were separated on 3% agarose gel and visualized in gel documentation unit (Alpha Innotech, India).
Statistical analyses
SSR amplicons generated for each primer were scored as allelic data in base pairs. The marker attributes like major allele frequency, gene diversity and polymorphism information content (PIC) value for all markers were computed using PowerMarker v. 3.25 (Liu and Muse 2005). Concerning clustering, DARwin v. 6.0.13 software (Perrier and Jacquemoud-Collet 2006) was employed to generate genetic distance (GD) followed by distance based unweighted neighbour joining tree and factorial analysis.
Population structure
A set of 65 SSR markers was used to understand the genetic structure and number of sub-populations in the 89 pigeonpea genotypes by employing STRUCTURE v. 2.3.3 (Pritchard et al. 2000) (http://pritch.bsd.uchicago.Edu/structure.html). Admixture model with correlated allele frequency was applied and all runs were performed with burn-in period and Monte Carlo Markov Chain replications of 20,000 and 200,000, respectively. Five independent runs were accomplished with each K values ranging from 1 to 10. Evanno’s Δk value was calculated by using Structure Harvester program by processing the STRUCTURE results (Evanno et al. 2005). Ln P(D) is the log likelihood of the observed genotype distribution in K clusters and is found by STRUCTURE simulation. Evanno’s ΔK takes into consideration the variance of ln P(D) among repeated runs and indicates the ideal K.
Association analysis
The MTAs were detected using TASSEL v. 2.1 software (Bradbury et al. 2007). The tests of significance were performed for genotypic data and 3 year wilt incidence data by following Q + K MLM (mixed linear model) method. The Q matrix was created from population structure analysis and the relative kinship matrix was calculated by TASSEL software. Alleles with minor frequency (MAF) lower than 5% were removed and a unified mixed linear model was employed for association analysis with 1000 permutations, since it accounts simultaneously for both population structure and kinship, and can effectively control the spurious associations. The MTAs were declared significant by P ≤ 0.05 with relative magnitudes suggested by the corresponding R2 value as the portion of phenotypic variation (PV) explained by the marker. Marker effects for each associated SSRs (resulting from TASSEL) were used to identify major allele (homozygous) with higher phenotypic effects on FW resistance.
Results
Extensive phenotyping against fusarium wilt
The screening of 89 pigeonpea genotypes against FW variant 2 for 3 years under wilt sick field revealed a broad range of response to FW. Based on the 3 years mean wilt incidence data, a total of 43 genotypes were found to be resistant, nine were moderately resistant, 19 were moderately susceptible and 18 were susceptible (Table 1).
Frequency distribution graphs of FW incidence for each year are illustrated in Fig. 1. The graphs depict a continuous variation with population mean slightly deviating towards resistance in all 3 years. The disease expression as reflected from FW incidence with respect to genotypes was high for year 1 and year 3, whereas relatively low disease expression in the second year might be due to adverse environmental conditions.

The frequency distribution graph showing per cent of wilt incidence for 89 pigeonpea genotypes screened against Fusarium udum (variant 2) for 3 years (2013–16) in the wilt nursery
Genetic diversity
A total of 181 alleles were detected across 89 genotypes using 65 SSR markers. The number of alleles per locus ranged from 2 to 5 with an average of 2.8 (Table 2). The PIC values ranged from 0.25 to 0.66 with an average of 0.47. Out of 65 SSRs, 29 (44.6%) were highly informative (PIC ≥0.5) and 36 (55.4%) were reasonably informative with PIC values ranging from 0.25 to 0.5. The gene diversity varied between 0.30 and 0.71 with an average of 0.55.
Population structure and cluster analysis
Population structure analysis was performed with a predefined number of sub populations (K) ranging from 1 to 10. The maximum delta K (ad hoc quantity) was reached at K = 2 suggesting the presence of two subpopulations in the entire collection (Fig. 2). Of the total 89 genotypes, 37 were assigned to sub-population 1 (Q1, red), while remaining 52 represented sub-population 2 (Q2, green) (Fig. 2). With few exceptions, a neighbour-joining (NJ) tree could categorize the entire set of 89 genotypes into two distinct clusters (Fig. 3). Subpopulations 1 and 2 corresponded to two main clusters and the colours in NJ tree were assigned according to the population structure Q1 (red) and Q2 (green). Additionally, factorial analysis was also undertaken to verify the patterns obtained in STRUCTURE and NJ analyses. Interestingly, a close agreement was observed between the results arising from STRUCTURE and factorial analysis (Fig. 3). The genotypes contained in quadrants I and II of factorial analysis belonged to subpopulation 1 in STRUCTURE analysis, whereas the subpopulation 2 of STRUCTURE was assigned to quadrants III and IV.

Population structure of 89 pigeonpea genotypes based on 65 SSR markers (K = 2) and graph of estimated membership fraction for K = 2. The maximum of adhoc measure ΔK determined by structure harvester was found to be K = 2, which indicated that the entire population can be grouped into 2 subpopulations

Unrooted neighbour joining tree and factorial analysis showing clustering of 89 pigeonpea genotypes into 2 subpopulations
Association analysis
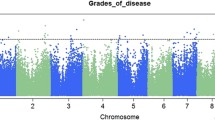
With the MLM program in the TASSEL software, MTAs were established using the FW wilt incidence data and the SSR dataset. This analysis was carried out separately for each year of wilt incidence data, and also on average incidence data of 3 years. This analysis uncovered six significant MTAs for resistance to FW variant 2 (Table 3). In the first two years (2013–2015), four SSRs namely HASSR18, HASSR128, HASSR30 and HASSR121 were found significant. In the third year (2015–16) HASSR18, HASSR174 and HASSR8 were found significant. Out of six MTAs, three markers HASSR18, HASSR128, HASSR30 were stable and significant for 3 years average wilt incidence data as depicted in Manhattan plot (Fig. 4). The phenotypic variance explained by SSRs such as HASSR18, 128, 30, 121, 174 and 8 were up to 6%, 5%, 5%, 4%, 1% and 2%, respectively across 3 years.

Manhattan plot of mixed linear model (MLM) for resistance to fusarium wilt
Allele effects were estimated for each associated SSR locus and major alleles (in homozygous state) with higher phenotypic effects were identified. Marker alleles with strong negative effect on wilt incidence reduction were shown in Fig. 5 (Suppl. Table 2). Considering six associated HASSRs, HASSR 18 with marker allele A150 bp showed stable phenotypic effect across the years. This particular allele was carried by 34 individuals, which significantly reduced the mean wilt incidence value below the threshold (<10% incidence, transformed value 0.321). However, the strong negative effects on phenotypes were noticed for HASSR128_A230 (present in 21 individuals) followed by HASSR30_A180 (54) and HASSR121_A160 (34) for the first two years and across. These SSR alleles could reduce the mean wilt incidence between 10 and 20% (<0.46). The lowest marker effects with reduced wilt incidence between 10 and 20% were noticed for HASSR174_A185 and HASSR8_A190 during the third year.

Phenotypic effects of major marker allele at six significantly associated SSR loci against FW resistance trait across the years
Marker localization and distribution map
In-order to locate the physical position of unmapped HASSR markers on pigeonpea genome the BLAT option of genome viewer from legume information system (LIS) was used. Based on alignment results, 32 HASSR markers viz. 6, 4, 4, 1, 4, 4, 1, 3 and 5 SSRs could be assigned to linkage groups 1, 2, 3, 4, 6, 7, 8, 10 and 11, respectively. No markers were found on linkage groups 5 and 9, and the remaining 33 markers corresponded to 29 different scaffolds (Table 2). The physical distances between SSR markers belonging to each chromosome were translated to genetic distances by considering the ratio of 561.1 Kb/cM (Kumawat et al. 2012). Finally, centimorgan (cM) distances between markers for each chromosome were used to draw the partial linkage map. This map enabled visualization of the probable genomic locations of FW associated SSR markers on different chromosomes (Suppl. Fig. 1). The majority of these HASSRs were assigned to distal ends of LGs such as HASSR18 on LG 1, HASSR8 on LG 11 and HASSR30 on LG 10. Similarly, HASSR121 was mapped on LG 6 in the vicinity of another HASSR, i.e. HASSR162. The remaining HASSRs like 128 and 174 could be located on unknown scaffold regions.
Discussion
In the present study, FW variant 2 was selected for association study. Based on previously published reports, variant 2 is well characterized and is the most virulent strain distributed in seven different states of India viz. Uttar Pradesh, Madhya Pradesh, Karnataka, Andhra Pradesh, Bihar, Jharkhand and Tamil Nadu (Patil et al. 2013). Management of this variant through harnessing the host resistance is key to ensure stable pigeonpea production in India. Therefore, mapping of genetic determinants of resistance against FW variant 2 was undertaken in this study. Robust disease screening of 89 pigeonpea genotypes for three cropping seasons revealed adequate variability in the wilt incidence.
The wilt incidence was recorded during pre-flowering to pod maturation stages as the reproductive phase of pigeonpea remains most susceptible to this disease (Natarajan et al. 1985). Though seedling mortality is often seen during August, adult plant wilting starts from flowering during November–December (Kotasthane and Gupta 1981). In this study, we found higher disease expression in first (2013–14) and third (2015–16) years as compared to second year (2014–15). This variable expression might be due to prevalence of congenial weather conditions favouring disease incidence in those particular years. Compared to second year, first and third years had a slightly good rainfall during November–February (see Suppl. Table 3 & Fig. 2 for weather data). This difference in rainfall might have contributed to retaining lower soil moisture required for wilt incidence. Also, spread of wilt disease from plant to plant occurs through root contacts, irrigation, rainwater and termites (Upadhyay and Rai 1992). FW is favoured by low soil temperature and increasing plant maturity (Mundkur 1935). Specifically, soil water holding capacity (30%) and soil temperatures between 20 and 30 °C favour disease development (Singh and Bhargava 1981).
In the present study, frequency distribution graph slightly skewed towards resistance in all 3 years. More recently, Sharma et al. (2016) examined environmental influences on FW incidence on 29 resistant pigeonpea genotypes across nine locations in India including IIPR, Kanpur. The authors noted higher variability in wilt incidence at Kanpur in two seasons (2007/08 and 2008/09). The current study provides evidence that variability in wilt incidence over seasons and locations might be due to GxE interactions. The authors also concluded that the genotype contributed 36.51% variation for resistance as compared to environment (29.32%). The skewed distribution observed in our study is indicative of a greater number of genetically resistant genotypes as reflected from phenotyping results (out of 89 genotypes 43 were found resistance). The varying level of pathogen’s virulence (type of race/variant prevailed over the different locations) may also yield variation in wilt incidence. Mishra and Dhar (2003) reported F. udum isolates from diverse geographical origins to exhibit greater variability in their virulence.
The SSR markers are still preferred for genetic analyses due to their abundance in genome, multi-allelic and co-dominant nature and ease of assay (Gupta and Varshney 2000). Moderate to high mean values for PIC (0.47) and gene diversity (0.55) were obtained in the current study with 65 SSR markers. As was reported recently in a panel of 94 pigeonpea genotypes (Bohra et al. 2017), we also observed a strong agreement among results obtained using both model and distance based clustering methods. The presence of moderate to low genetic diversity in cultivated pigeonpea genotypes was congruent with earlier studies using various marker systems in pigeonpea such as RAPD (Ratnaparkhe et al. 1995), restricted fragment length polymorphism (RFLP) (Nadimpalli et al. 1994), amplified fragment length polymorphism (AFLP) (Punguluri et al. 2006), diversity arrays technology (DArT) (Yang et al. 2006), intron spanning region (ISR) (Kudapa et al. 2012), SSR (Bohra et al. 2011) and SNP (Kassa et al. 2012).
The population structure acts as a strong confounding factor in association studies (Bohra 2013), especially with respect to traits that are important in diversifying selection and family relatedness associated with recent co-ancestry (Nordborg and Weigel 2008). As a result, spurious associations occur frequently in cases where the population structure is not factored in (Gupta et al. 2005). The efficiency of eliminating false positive associations that arise due to complex patterns of the population stratification/relatedness in experimental sample depends on the use of optimum statistical method (Zhang et al. 2010). Therefore, to perform genome wide MTA study, selection of appropriate statistical method is crucial. In this context, the general linear model (GLM) and the mixed linear model (MLM) are the two commonly used methods. Among these, the superiority of MLM approach over the GLM is evident in several studies (Huang et al. 2010; Yang et al. 2010). Among grain legumes, this approach has been widely and successfully used in various crops such as common bean (Shi et al. 2011), soybean (Hu et al. 2014) and chickpea (Thudi et al. 2014). Therefore, the MLM owing to its ability to simultaneously account for both population structure and kinship control false associations was employed in the present study. The MTAs for 3 year wilt incidence data revealed six SSR markers (HASSR18, HASSR128, HASSR30, HASSR121, HASSR174 and HASSR8) to be significantly associated with wilt resistance with 1–6% PV as depicted from Manhattan plot. HASSR18, which explained 5 to 6% of PV across the years was found to be the most stable and significant marker. Similarly, lower R2 values (up to 8%) were assigned to SNP markers in pigeonpea by Mir et al. (2012) while mapping determinacy trait using association mapping approach. Compared to the conventional QTL mapping using biparental crosses, the relatively low R2 values in association mapping is not unexpected since the biparental QTL analysis often leads to overestimation of variation explained by the QTLs (Varshney et al. 2012).
Allele effects on phenotype for each associated SSR locus were also calculated, and consequently, HASSR 18 _A150 showed stable and negative effect on phenotypic as reduction in the mean wilt incidence value below the threshold (<10% incidence) as compared to alleles of remaining SSR loci that reduced mean wilt incidence between <10 and 20%. By declaring the genotypes with mean wilt incidence of <10% as resistant and 10.1–20.1% as moderately resistant, HASSR18_A150 could be more efficient in detecting resistant individuals as compared to remaining marker alleles viz., HASSR128_A230, HASSR30_A180, HASSR121_A160, HASSR174_A185 and HASSR8_A190. Similarly, the favourable SSR alleles with major effects on phenotype were determined for kernel length, width and weight in wheat (Breseghello and Sorrells 2006) and for plant height in sorghum (Wang et al. 2012).
Reports on identification of stable DNA markers for resistance to FW in pigeonpea are still limited. For instance, Kotresh et al. (2006) reported RAPD marker linked to FW resistance in pigeonpea through bulked sergregant analysis. Singh et al. (2013) reported six ASSR markers significantly associated with fusarium wilt resistance based on Kruskal–Wallis ANOVA and simple regression analysis. Later, Singh et al. (2016a, b) while confirming the efficacy of these associated SSR markers concluded that the markers ASSR1, ASSR23 and ASSR148 could be more useful in marker assisted breeding (MAB) in pigeonpea. Among the three informative ASSRs, one of the markers ASSR23 was located on LG6 (Kumawat et al. 2012). By using a sequencing-based bulked segregant analysis (Seq-BSA) approach, Singh et al. (2016a, b) reported association of four candidate nsSNPs in four genes viz., C.cajan_07078 (PHD figure protein), C.cajan_07124 (rRNA processing protein), C.cajan_02962 (NADH dehydrogenase) and C.cajan_03203 (retrovirus like polyprotein) with FW resistance. Of the four nsSNPs, the SNP in the candidate genes C.cajan_03203 showed deleterious effect on the protein structure and was speculated to be involved in plant defence against FW pathogen through root expression study. These four genes are located on the linkage groups CcLG02 and CcLG11. Interestingly, in the present study two associated HASSR markers, i.e. HASSR121 (PV 4%) and HASSR 8 (PV 2%) for FW were physically located at LG06 and LG11, respectively. These results are in agreement with ASSR23 markers assigned to LG06 (Kumawat et al. 2012) and candidate genes for FW, C.cajan_02962 and C.cajan_03203 assigned to LG11 (Singh et al. 2016a, b). However, it remains difficult to pinpoint the genomic regions shared between the two studies unless we have the complete information about the kind of race/variant prevailing on those particular areas. We assume that the genomic regions identified in this study could be novel and remain more relevant to breeding against variant 2 that is prevalent at IIPR, Kanpur.
To our knowledge, this study represents the first report on the discovery of stable and significant MTAs for F. udum variant 2 based on FW incidence recorded over multiple years. Since we have employed unmapped SSR markers for the association study, we have drawn a representative genetic linkage map of these SSRs. This map helped to assign significantly associated markers on LG01, LG06, LG10 and LG11. The results strongly indicate the involvement of several genomic regions in conditioning FW resistance in pigeonpea. Similarly, SNP-based association analysis in common bean revealed occurrence of multiple significant loci across all 11 chromosomes for resistance to common bacterial blight (Shi et al. 2011). In our study, most of the associated SSR markers were placed on distal ends of linkage groups. Similarly, Perseguini et al. (2016) also reported telomeric localization of associated SSR and SNP markers for resistance to angular leaf spot in common bean.
In summary, here we identify six significantly associated SSR markers PV ranging between 1 and 6% for resistance to FW variant 2, which after validation in different genetic backgrounds might prove immensely useful in MAB. Given this, validation of these SSRs in the bi-parental mapping populations is underway in order to further confirm their utility in MAB for FW resistance in pigeonpea.
References
Agbagwa IO, Datta S, Patil PG, Singh P, Nadarajan N (2012) A protocol for high-quality genomic DNA extraction from legumes. Genet Mol Res 11:4632–4639
Atwell S, Huang YS, Vilhjalmsson BJ, Willems G, Horton M, Li Y et al (2010) Genome wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 465:627–631
Bohra A (2013) Emerging paradigms in genomics-based crop improvement. Sci World J 2013:585467
Bohra A, Dubey A, Saxena RK, Penmetsa RV, Poornima KN, Kumar N et al (2011) Analysis of BAC-end sequences (BESs) and development of BES-SSR markers for genetic mapping and hybrid purity assessment in pigeonpea. BMC Plant Biol 11:56
Bohra A, Jha R, Pandey G, Patil PG, Saxena RK, Singh IP et al (2017) Genome wide hypervariable SSR markers for molecular breeding applications in pigeonpea [Cajanus cajan (L.) Millspaugh]. Front. Plant Sci 8:377
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633
Breseghello F, Sorrells ME (2006) Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics 172:1165–1177
Butler EJ (1906) The wilt disease of pigeonpea and pepper. Agric J India 1:25–26
Dhar V (1995) Disease resistant lines with stable resistance identified as donor parents under AICPIP – Pathology programme from 1984–1994. Consolidated report of kharif pulses, plant pathology. All India co-ordinated pulses improvement project (ICAR). Kanpur, IIPR, P.IV
Dubey A, Farmer A, Schlueter J, Cannon SB, Abernathy B, Tuteja R et al (2011) Defining the transcriptome assembly and its use for genome dynamics and transcriptome profiling studies in pigeonpea (Cajanus cajan L.) DNA Res 18:153–164
Dutta S, Kumawat G, Singh BP, Gupta DK, Singh S, Dogra V et al (2011) Development of genic-SSR markers by deep transcriptome sequencing in pigeonpea [Cajanus cajan (L.) Millspaugh]. BMC Plant Biol 11:17
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
FAOSTAT (2014) Food and agriculture organization of United Nations. http://faostat.fao.org
Gomez KA, Gomez AA (1984) Statistical procedure for agricultural research, 2nd edn. Wiley, NewYork
Gupta PK, Rustgi S, Kulwal PL (2005) Linkage disequilibrium and association studies in higher plants: present status and future prospects. Plant Mol Biol 57:461–485
Gupta PK, Varshney RK (2000) The development and use of microsatellite markers for genetic analysis and plant breeding with emphasis on bread wheat. Euphytica 113:163–185
Hu Z, Zhang D, Zhang G, Kan G, Hong D, Yu D (2014) Association mapping of yield-related traits and SSR markers in wild soybean (Glycine soja Sieb. and Zucc.) Breed Sci 63:441–449
Huang X, Wei X, Sang T, Zhao Q, Feng Q, Zhao Y et al (2010) Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet 42:961–967
Jain KC, Reddy MV (1995) Inheritance of resistance to fusarium wilt in pigeonpea (Cajanus cajan L.) Indian J Genet Plant Breed 55:434–443
Joshi AR (1957) Genetics of resistance to disease and pests. Indian J Genet Plant Breed 17:305–317
Kannaiyan J, Nene YL (1981) Influence of wilt at different growth stages on yield loss in pigeonpea. Trop Pest Manage 27:141
Kassa MT, Penmetsa RV, Carrasquilla-Garcia N, Sarma BK, Datta S, Upadhyaya HD et al (2012) Genetic patterns of domestication in pigeonpea (Cajanus cajan (L.) Millsp.) and wild Cajanus relatives. PLoS One 7:e39563. doi:10.1371/journal.pone.0039563
Kotasthane SR, Gupta O (1981) Trend of wilting in pigeonpea varieties of variable maturity. Abstracts of papers of the third symposium of plant pathology, Indian Phytopathological Society, 14–18 Dec 1981. New Delhi, India, p 161–162
Kotresh H, Fakrudin B, Punnuri SM, Rajkumar BK, Thudi M, Paramesh H et al (2006) Identification of two RAPD markers genetically linked to a recessive allele of a fusarium wilt resistance gene in pigeonpea (Cajanus cajan L. Millsp.) Euphytica 149:113–120
Kudapa H, Bharti AK, Cannon SB, Farmer AD, Mulaosmanovic B, Kramer R et al (2012) A comprehensive transcriptome assembly of pigeonpea (Cajanus cajan L.) using Sanger and second-generation sequencing platforms. Mol Plant 5:1020–1028. doi:10.1093/mp/ssr111
Kumar Y, Kwon SJ, Coyne CJ, Hu J, Grusak MA, Kisha TJ et al (2014) Target region amplification polymorphism (TRAP) for assessing genetic diversity and marker trait associations in chickpea (Cicer arietinum L.) germplasm. Genet Resour Crop Evol 61:965–977
Kumawat G, Raje RS, Bhutani S, Pal JK, Mithra ASVCR, Gaikwad K et al (2012) Molecular mapping of QTLs for plant type and earliness traits in pigeonpea (Cajanus cajan L. Millsp.) BMC Genet 13:84
Liu K, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
Mir RR, Kudapa H, Srikanth S, Saxena RK, Sharma A, Azam S (2014) Candidate gene analysis for determinacy in pigeonpea (Cajanus spp.) Theor Appl Genet 127:2663–2678
Mir RR, Saxena RK, Saxena KB, Upadhyaya HD, Kilian A, Cook DR et al (2012) Whole-genome scanning for mapping determinacy in pigeonpea (Cajanus spp.) Plant Breed 132:472–478
Mishra S, Dhar V (2003) Variability in isolates of Fusarium udum Butler, the wilt pathogen of pigeonpea. In: Proceedings of ISMPP Zonal Conference (Eastzone). IISR, Lucknow
Mishra S, Dhar V (2010) Identification of broad base and stable sources of resistance to fusarium wilt in pigeonpea. Indian Phytopathol 63(2):165–167
Mundkur BB (1935) Influence of temperature and maturity on the incidence of sun hemp and pigeonpea wilt at Pusa. Indian J Agric Sci 5:606–618
Myles S, Peiffer J, Brown PJ, Ersoz ES, Zhang Z, Costich DE et al (2009) Association mapping: critical considerations shift from genotyping to experimental design. Plant Cell 21:2194–2202
Nadimpalli RG, Jarret RL, Phatak SC, Kochart G (1994) Phylogenetic relationships of the pigeonpea (Cajanus cajan) based on nuclear restriction fragment length polymorphism. Genome 36:216–223
Natarajan M, Kannaiyan J, Willey RW, Nene YL (1985) Studies on the effects of cropping system on fusarium wilt of pigeonpea. Field Crops Res 10:333–346
Nordborg M, Weigel D (2008) Next-generation genetics in plants. Nature 456:720–723
Okiror MA (1999) Evaluation of pigeonpea (Cajanus cajan) germplasm for resistance to fusarium wilt. Indian J Agric Sci 69:600–601
Okiror MA (2002) Genetics of resistance to Fusarium udum in pigeonpea (Cajanus cajan (L.) Millsp.) Indian J Genet Plant Breed 62:218–220
Patil PG, Byregowda M, Patil BR, Das A, Mary Reena GA, Sowjanya MS et al (2016) Microsatellite markers linked to sterility mosaic disease resistance in pigeonpea (Cajanus cajan L. Millsp.) Legume Genomics Genet 7(6):1–9
Patil P, Singh IP, Dhar V, Chaudhary RG, Datta S, Chaturvedi SK et al (2013) Variant specific studies on inheritance for resistance to fusarium wilt in pigeonpea [Cajanus cajan (L.) Millsp.] Legume Res 36(6):576–581
Perrier X, Jacquemoud-Collet JP (2006) DARwin software. Centre de cooperation internationale en recherche agronomique pour le developpement (CIRAD), Paris
Perseguini JMKC, Oblessuc PR, Rosa JRBF, Gomes KA, Chiorato AF, Carbonell SAM et al (2016) Genome-wide association studies of anthracnose and angular leaf spot resistance in common bean (Phaseolus vulgaris L.). PLoS ONE 11(3). doi: 10.1371/journal.pone.0150506
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945
Punguluri SK, Janaiah K, Govil JN, Kumar PA, Sharma PC (2006) AFLP fingerprinting in pigeonpea (Cajanus cajan (L.) Millsp) and its wild relatives. Genet Resour Crop Evol 53:423–531
Ratnaparkhe MB, Gupta VS, Ven-Murthy MR, Ranjekar PK (1995) Genetic fingerprinting of pigeonpea [Cajanus cajan (L.) Millsp.] and its wild relatives using RAPD markers. Theor Appl Genet 91:893–898
Sakiroglu M, Sherman-Broyles S, Story A, Moore KJ, Doyle JJ, Charles Brummer E (2012) Patterns of linkage disequilibrium and association mapping in diploid alfalfa (M. sativa L.) Theor Appl Genet 125:577–590
Saxena RK, Saxena KB, Kumar RV, Hoisington DA, Varshney RK (2010) Simple sequence repeat-based diversity in elite pigeonpea genotypes for developing mapping populations to map resistance to fusarium wilt and sterility mosaic disease. Plant Breed 129:135–241
Sharma M, Ghosh R, Telangre R, Rathore A, Saifulla M, Mahalinga DM et al (2016) Environmental influences on pigeonpea Fusarium udum interactions and stability of genotypes to fusarium wilt. Front Plant Sci 7:253. doi:10.3389/fpls.2016.00253
Sharma M, Rathore A, Mangala UN, Ghosh R, Sharma S, Upadhyay HD et al (2012) New sources of resistance to fusarium wilt and sterility mosaic disease in a mini-core collection of pigeonpea germplasm. Eur J Plant Pathol 133:707–714
Shi C, Navabi A, Yu K (2011) Association mapping of common bacterial blight resistance QTL in Ontario bean breeding populations. BMC Plant Biol 11:52
Singh AP, Bhargava SN (1981) Survival studies on three species of fusarium causing wilt of pigeonpea. Phytopathol Z 100:300–311
Singh IP, Dhar V, Chaudhary RC, Pandey DK (1998) Genetic of fusarium wilt resistance in pigeonpea. In: national symposium on management of biotic and abiotic stresses in pulse crops, 26–28th June 1998. IIPR, Kanpur, p 15
Singh NK, Gupta DK, Jayaswal PK, Mahato AK, Dutta S, Singh S et al (2012) The first draft of the pigeonpea genome sequence. J Plant Biochem Biotechnol 21:98–112
Singh VK, Khan AW, Saxena RK, Kumar V, Kale SM, Sinha P et al (2016a) Next-generation sequencing for identification of candidate genes for fusarium wilt and sterility mosaic disease in pigeonpea (Cajanus cajan). Plant Biotechnol J 14:1183–1194
Singh AK, Rai VP, Chand R, Singh RP, Singh MN (2013) Genetic diversity studies and identification of SSR markers associated with fusarium wilt (Fusarium udum) resistance in cultivated pigeonpea (Cajanus cajan). J Genet 92:273–280
Singh F, Singh IP, Mazumder MD (2011) Identification of fusarium wilt-resistant sources of long-duration pigeonpea (Cajanus cajan). Indian J Agric Sci 81:1046–1051
Singh D, Sinha B, Rai VP, Singh MN, Singh DK, Kumar R et al (2016b) Genetics of fusarium wilt resistance in pigeonpea (Cajanus cajan) and efficacy of associated SSR markers. Plant Pathol J 32:95–101
Thudi M, Upadhyaya HD, Rathore A, Gaur PM, Krishnamurthy L, Roorkiwal M et al (2014) Genetic dissection of drought and heat tolerance in chickpea through genome-wide and candidate gene-based association mapping approaches. PLoS One 9:e96758
Tiwari S, Dhar V (2011) Prevalence of new variants of Fusarium udum in India. Indian Phytopathol 64:243–246
Upadhyay RS, Rai B (1992) Wilt disease of pigeonpea. In: plant disease of international importance. In: Singh US, Mukhopadyaya Kumar A, Chaube HS (eds) . Prentice Hall, Engelwood cliffs, pp 388–404
Van-Berloo R (2008) Computer note: GGT 2.0: versatile software for visualization and analysis of genetic data. J Hered 99(2):232–236
Varshney RK, Paulo MJ, Grando S, Van Eeuwijk FA, Keizer LCP et al (2012) Genome-wide association analyses for drought tolerance related traits in barley (Hordeum vulgare L.) Field Crops Res 126:171–180
Varshney RK, Penmetsa RV, Dutta S, Kulwal PL, Saxena RK, Datta S et al (2010) Pigeonpea genomics initiative (PGI): an international effort to improve crop productivity of pigeonpea (Cajanus cajan L.) Mol Breed 26:393–408
Wang YH, Bible P, Loganantharaj R, Upadhyaya HD (2012) Identification of SSR markers associated with height using pool-based genome-wide association mapping in sorghum. Mol Breed 30:281–292
Yang S, Pang W, Ash G, Harper J, Carling J, Wenzl P et al (2006) Low level of genetic diversity in cultivated pigeonpea compared to its wild relatives is revealed by diversity arrays technology. Theor Appl Genet 113:585–595
Yang X, Yan J, Shah T, Warburton ML, Li Q, Li L et al (2010) Genetic analysis and characterization of a new maize association mapping, panel for quantitative trait loci dissection. Theor Appl Genet 121:417–431
Zhang Z, Ersoz E, Lai CQ, Todhunter RJ, Tiwari HK, Gore MA et al (2010) Mixed linear model approach adapted for genome-wide association studies. Nat Genet 42:355–360
Acknowledgements
This work was financially supported by the Indian Council of Agricultural Research-Network Project on Transgenic in Crops- Sub Project- Functional Genomics in Pigeonpea.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Barbara Naganowska
Electronic supplementary material
Suppl. Fig. 1
Representative partial map showing genome positions of markers associated with resistance to wilt (indicated with arrow). SSR markers were located on the reference genome using the BLAT options of genome browser in Legume information system. The marker Mb positions were converted and represented in cM map units and map was drawn with GGT software (Van-Berloo 2008) (GIF 5812 kb)
Suppl. Fig. 2
Weather graph depicting differences for temperature, relative humidity and rainfall for three cropping seasons (July–April) (Major differences were indicated by circle in the graph) (GIF 6094 kb)
Suppl. Table 1
Phenotypic data on percent wilt incidence for 89 pigeonpea genotypes for 3 years (DOC 106 kb)
Suppl. Table 2
Phenotypic effects of major marker allele at six significantly associated SSR locus against FW (DOC 36 kb)
Suppl. Table 3
Comparison of weather data for the 4 months (November to December for each year) during which phenotyping against FW was carried out (DOC 35 kb)
Rights and permissions
About this article

Cite this article
Patil, P.G., Dubey, J., Bohra, A. et al. Association mapping to discover significant marker-trait associations for resistance against fusarium wilt variant 2 in pigeonpea [Cajanus cajan (L.) Millspaugh] using SSR markers. J Appl Genetics 58, 307–319 (2017). https://doi.org/10.1007/s13353-017-0400-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13353-017-0400-y