
Abstract
The objective of the present work was to optimize ritonavir (RTV)-loaded nanostructured lipid carriers (NLCs) to improve bioavailability using a quality by design (QbD)-based technique. Risk assessment was studied using “cause and effect” diagram followed by failure mode effect analysis (FMEA) to identify the effective high-risk variables for the formulation development. Quality target product profile (QTPP) and critical quality attributes (CQAs) were initially assigned for the proposed product. Central composite rotatable design (CCRD) was used to identify the individual and combined interactions of formulation variables. RTV-loaded NLC (RTV-NLC) was prepared using emulsification-ultrasonication method. The effect of formulation variables like ultrasound amplitude, lipid concentration, surfactant concentration on their responses like particle size, polydispersity index (PDI), and entrapment efficiency (EE) were studied by CCRD. The optimized formulation was subjected to lyophilization to obtain dry NLCs for solid-state analysis. DSC and PXRD investigations showed that RTV was molecularly dispersed in lipid matrix indicating amorphous form present in the formulation. FESEM and AFM depicted the spherical and uniform particles. The increase in solubility and dissolution rate is expected to be related to the molecular dispersion, amorphous state, of the drug in the nanoparticle. The optimized NLCs showed good physical stability during storage for 6 months. RTV-NLC was further subjected to in vitro studies and found a successful sustained release rate of 92.37 ± 1.03%. The parallel artificial membrane permeability assay (PAMPA) and everted gut sac model have demonstrated the permeation enhancement of RTV. In vivo study observed the enhanced bioavailability with 2.86-fold suggesting optimized NLC successfully overcome the issue of solubility.
Graphical abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Ritonavir (RTV), azapeptide protease inhibitor, is used as a boosting agent in a highly active antiretroviral therapy (HAART) regimen recommended by the world health organization (WHO) for the treatment of human immunodeficiency virus (HIV) infection [1, 2]. It is a highly lipophilic agent with log p value 3.9 and categorized in class II drug of the biopharmaceutical classification system (BCS). Its elimination half-life (t1/2) is 3–5 h and has variable bioavailability of 40–50% having poor aqueous solubility [3, 4]. Despite the promising drug molecule for the treatment of HIV, they are not effective to eradicate the infection completely from the host, and hence research work is in continuous mode all over the world. The frequent administration of RTV in the treatment of HIV is required to achieve the desired plasma level. In addition, several issues come up with the conventional dosage form including poor accessibility to lymphatics, CNS, and genitals [5].
Few works have been attempted to resolve the solubility and bioavailability issues by nanoformulations, for example, solid lipid nanoparticles [6, 7] nanosuspension [8], solid dispersion [9, 10], liposome [3, 4, 11], and polymer-based nanostructures [12], and cyclodextrin-based nanoformulation [13]. However, various authors suggested a common drawback of SLNs including drug expulsion during storage and limited drug loading [14,15,16,17,18,19] whereas polymeric nanocarrier possess undesirable cytotoxicity and unwanted accumulation of polymeric residue leads to toxicity [20]. On the other hand, liposomes require high production costs and show instability during storage is a major limitation [21].
Nevertheless, lipids are biodegradable and biocompatible which is used to completely protect the entrapped drug and release controlled manner at a prolonged period. SLNs were modified by loading oil into the lipid matrix of SLNs to form an imperfect structure so that the maximum amount of drug could encapsulate to enable a sustained rate of drug release. This modified form has been known as nanostructured lipid carriers (NLCs). They are designated as the second generation of lipid carrier next to the SLN and composed of GRAS (generally regarded as safe) status solid lipid and liquid lipids (oil) which are stabilized by a surfactant or blend of surfactant [15, 22]. On this ground, manufacturing of NLCs was preferred for encapsulating the lipophilic RTV. Here, we employed an appropriate proportion of solid lipid and liquid lipid for the manufacturing of NLCs which modulates the controlled release, drug targeting, and stability [23].
The use of high shear homogenization followed by the assistance of ultrasound waves generated by probe-sonicator using cavitation technology is the best option for the production of uniformly sized nanocarriers. The plus point of the energy-efficient technique is to control the operating parameters like power, processing time and does not require skilled personnel for the operation [24]. Besides, probe-sonicator is common equipment available in every research laboratory and can be used for the production of nanocarriers using ultrasound cavitation [25].
From our knowledge, this is the first report of production of RTV loaded NLC with several systematic steps of QbD approach to improve its bioavailability. In this framework, QTPP was assigned before the start of a process and CQA was defined based on the “cause and effect” diagram and FMEA. The CCRD was used for several experimental runs and employed for the optimization process. The optimized NLC formulation was lyophilized to convert free-flowing solid particles to study the solid-state characterizations [26]. The release from the NLC was monitored and studied by parallel artificial membrane permeability assay (PAMPA) [27]. In supporting this study, intestinal everted sac method (ex vivo model) was used for the permeability study. Thereafter, in vivo evaluation using Wistar rats was assessed for the enhancement of bioavailability.
Materials and methods
Materials
RTV (> 98% pure) was obtained from Mylan Pharmaceutical India Ltd. (Nashik, India) Compritol HD 5 ATO (solid lipid) and Capryol 90 (liquid lipid) were received from Gattefosse (Mumbai, India). Pluronic F127 was purchased from Sigma-Aldrich (Bangaluru, India). Tween® 80 and dialysis bag (M.W. cutoff 12,000–14,000 Da) was purchased from Hi-Media distributor (Mumbai, India). Other chemicals and solvents used in fabrication and evaluation were of analytical reagent (AR) grades.
Approval of animal experiments protocol
The protocol for the animal experiments was prepared according to guidelines of the Committee for Control and Supervision of Experiments on Animals (CPCSEA) and was approved by Institutional Animal Ethics Committee (IAEC) (Approval ref. No. IAEC/RCPIPER/2018–19/29) of R. C. Institute of Pharmaceutical Education and Research (RCPIPER), Shipur, India. All animal experiments were carried out according to the NIH publication No. 8023, revised 1923 guidelines for Care and Use of Laboratory animals. All the animals (Wistar male rats weighing 180–200 g approx.) were issued from the well-established animal house of RCPIPER, Shirpur, India. They were housed for study under appropriate controlled conditions of a temperature of 22 ± 5 °C and relative humidity (RH) 50 ± 10%. The animals were fed rat chow and continuous water until the day of study.
HPLC analysis of RTV
An estimation of RTV was performed using previously reported RP-HPLC method with slight modification [28]. The analysis was carried out by a reverse-phase C18 PrincetonSPHER™ column using mobile phase consisting of phosphate buffer (pH 4) and acetonitrile (50:50) at a flow rate of 1.2 mL/min. The detection was performed by UV–visible detector at 244 nm. Calibration curve was constructed over the range of 50–150 µg/mL with a regression coefficient (r2) of 0.998. The method was further validated as per International Conference on Harmonization (ICH) guidelines, Q2 (R1) [29].
Product development using QbD tool
Step 1: quality target product profile (QTPP) and critical quality attributes (CQAs)
The first step of QbD-based tool is to define QTPP for the consideration of final product. It signifies highlighting the patient-relevant product performance and characteristics that have improved the treatment. Selection of QTPP based on ICH Q8 guideline that includes dosage form, dosage strength, route of administration, pharmacokinetics, stability, and product quality attributes (particle size, zeta potential, and PDI, EE, etc.) [30].
Critical quality attributes (CQAs) are the product characteristics that depend on experimental factors, for example, drug substances, excipient, and process parameters. These factors can be critically examined and controlled to achieve the desired target of a quality product. Thus, the identification of CQA through the screening process is crucial, and based on sound knowledge and available literature. Table 1 depicts the justification of QTPP and CQAs.
Step 2: risk assessment using ranking method
In order to find the actual components utilized in the development of the product, the factors having intuition on the performance of quality products were isolated. The experimental factors were finalized using the process of risk identification analysis using cause and effect diagram and risk assessment ranking method [30]. In this method, failure mode effect analysis (FMEA) was employed to assign rank depending on risk, and further high-risk holding factors were isolated affecting CQAs of the final product.
Here, we enlisted all failDure modes for the preparation of NLCs, and their assessment was done based on the priority ranking numbers which were implemented in our previous paper [23]. Each failure mode was given a priority ranking number (PRN) and potential variables were selected based on their PRN. The PRN is a scale from 1 to 36 numbers depicted in (Table S1, Supplementary data) and calculated by the following formula,
where severity (S) is denoted as seriousness of failure, occurrence (O) is portrayed how often failures can occur, and detectability (D) is defined as the level of difficulty in detecting failures. Thus, the identified variables were employed in the production of NLCs using different levels of variables in CCRD.
Step 3: production of RTV-loaded NLCs
RTV-loaded NLC was prepared by a modified pre-emulsification probe sonication method [31]. Briefly, a weighed amount (50 mg) of RTV, Compritol HD5 ATO, and Capryol 90 was added in 2 mL chloroform to form oily phase. On the other hand, the measured amount of Pluronic F127 and Tween 80 was dissolved in double distilled water to form an aqueous phase. Both the phases were heated up to 55–60 °C to produce a clear solution. Further, the hot surfactant solution was slowly added into the oily phase during continuous stirring on a magnetic stirrer (Remi) to produce pre-emulsion. The prepared pre-emulsion was further homogenized adequately under high-speed homogenization (Ultra-Turrax, T25 Basic, Ika Werke, Stanfer, Germany) with 11,000 rpm for 6 min. The hot condition was maintained during this process and then subjected to probe sonication (PCI Analytics Pvt. Ltd., Mumbai, India). The sonicator tip was immersed in 9 mm in the NLC solution having a diameter of 17 mm. The optimized conditions of sonication device which operates with ultrasound burst of 20:15 on/off cycles and irradiated with 55 amplitude of ultrasonic intensity. The period of sonication was continued for 15 min followed by being placed in a cool place to produce the NLCs. The physical mixture of pure drug and solid lipid was prepared in order to aid in the study of solid-state characteristics and comparison purposes.
Step 4: applying experimental design in production of NLCs
The effect of ultrasound amplitude (A), lipid concentration (B), and surfactant concentration on the responses (CQA) like particle size (Y1), PDI (Y2), and EE (Y3) were evaluated by using central composite rotatable design (CCRD) coupled with response surface methodology (RSM) (Table 2). CCRD includes extra edge value (− alfa, + alfa) and is most appropriated among all the designs to explore response surfaces, second-order polynomial model, and allowing few experiments to run [32, 33].
As shown in Table 2, the independent variables such as ultrasound amplitude (A), lipid concentration (B), and surfactant concentration (C) were selected based on risk assessment and FMEA technique. The ultrasound amplitude was identified as critical process variable (CPP) and solid lipid and oil were identified as CMAs. The independent variables with low (-1), middle (0), and high (+ 1) levels associated with 10 non-center points and 5 center points were included in statistical evaluation. The alfa values (− alfa, + alfa) were auto-selected by the software. The output was analyzed using software Design Expert 12 (DX 12) [34].
Step 5: statistical data analysis
Several batches of NLCs were run according to experimental design. The analysis of responses like particle size, PDI, and EE were performed in triplicate (mean ± S.D., n = 3). The generated models were evaluated statistically and data calculations using ANOVA (analysis of variance) were suggested the levels of significance of responses were highly significant (P < 0.05). Contour plot (2D) and response surface plot (3D) were generated using Design Expert 12 (Version 12.0) software to carry out the response surface analysis [35].
Step 6: QbD-based optimization and validation of formulation
In order to optimize the formulation, the constraints were set based on minimum particle size, minimum PDI, and maximum EE for the selected response (CQA) to obtain desirability function. In the numerical method, desirability closest to 1 is considered for the optimization of the formulation. To verify the optimized condition of the formulation, the suggested optimized formulation (Table 3) was developed from the experimental variables. The observed responses were compared with predicted responses values [26].
Characteristics of prepared NLCs
Particle size and PDI measurement
The mean particle size and PDI of the NLCs were determined using a dynamic light scattering (DLS) technique using zeta sizer (Malvern Zetasizer Nano ZS90, Malvern, Worcestershire, UK). To avoid multiple scattering effects, each sample was diluted in the proportion of 1:100 using ultrapure (Milli Q) water before the measurement. The refractive indices of disperse and dispersant (water) phases were set to 1.59 and 1.330, respectively. The diluted dispersion was taken in the polystyrene cuvette and measured with pre-set equilibration time 120 s. The detection temperature was maintained at 25 °C. The results were reported as mean diameter (size, μm) of triplicate measurements for particle size and PDI.
ζ-potential measurement
The ζ-potential has a pronounced effect on the chemical and physical properties of nanocarriers. The ζ-potential of NLC formulation was measured by electrophoretic mobility of particles using a Malvern Zetasizer, Nano ZS90 (Malvern Instruments Co., Ltd., Worcestershire, UK). Each sample was diluted 100 times using ultrapure water before the measurement. A clear disposable zeta cell (DTS1060C) was used for the measurement of all samples at 25 °C. All the measurements were performed in triplicate and reported as surface potential (mV).
Drug entrapment (%) and drug loading (%)
A premier tabletop ultracentrifuge (Optima Max-XP, Beckman Coulter Inc., Brea, USA) was employed to determine percent drug entrapment from the NLCs. Briefly, the accurately weighed dispersion of NLCs was centrifuged at 20,000 RCF for 20 min, 4 °C to separate the supernatant and nanocarrier pellets. The supernatant (1 mL) was suitably diluted in methanol and scanned spectrophotometrically (UV-1900, Shimadzu, Tokyo, Japan) at 244 nm. This process was performed in triplicates and results were reported in the percentage of drug entrapment.
Drug entrapment and drug loading were calculated by the following Eqs. (1) and (2) and expressed in mean values (mean ± SD),
where Wd, Wfd, and Wnlc were the amount of total drug, amount of free drug, and amount of NLCs, respectively.
Lyophilization of RTV-NLC
For the long-term stability purpose, the RTV-NLCs were subjected to freeze-dried so that dry solid of RTV-NLCs was obtained. Mannitol 3.5% was added into formulation as cryoprotectant before loaded in freezer at − 80 °C for 24 h followed by lyophilization for 40 h using freeze dryer (Virtis Bench Top Lyophilizer, Spinco Biotech Pvt. Ltd) at 35-mT vacuum pressure. After freeze-drying process, the secondary drying was conducted at 20 °C for 12 h [36].
Percent drug content
For the determination of drug content, an accurate weighing amount of 10 mg lyophilized NLC was dissolved in methanol and subsequently sonicated and filtered through 0.22 µm. The percent drug content was analyzed using HPLC at 244 nm and calculated by following formula.
Physical characterization of NLCs
FT-IR spectra
IR spectra of pure RTV, Compritol HD5 ATO (solid lipid), PM of drug and lipids, optimized formulation, and RTV-NLC were used to analyze the intermolecular interaction of functional group between RTV and excipients in the formulation. FT-IR spectroscopy was scanned on Fourier transform spectrophotometer (FT-IR-430 Shimadzu 8400S, Tokyo, Japan) equipped with a scanning range of 4000–500 cm−1. A total of 2 mg sample was mixed with dried KBr in the ratio of 1:100, ground gently in a mortar, and squeezed into a pellet for analysis.
Crystallinity studies
The crystallinity studies were performed by differential scanning calorimetry (DSC) and powder X-ray diffraction pattern studies (PXRD).
-
a.
DSC (METTLER TOLEDO, USA) analysis was performed for the samples of pure RTV, Compritol HD5 ATO, Capryol 90, and optimized NLCs to evaluate the nature and thermal stability. Indium (In) standards were used to calibrate the instrument and nitrogen gas was supplied as purging. A sample of fixed amount (2 mg) was taken and sealed in the standard aluminum crucible. The sealed empty crucible was taken as a reference.
All the samples were analyzed over a rate of 10 °C/min from 25 to 290 °C under a nitrogen environment. Onset, peak, and end set temperature and enthalpy change were analyzed by Mettler STARe V8.10 software [37, 38].
Crystallinity (%) was calculated using Eq. (4),
-
b.
PXRD study was performed to record the diffraction spectra of a sample using PXRD (Brucker AXS D8 advance®, Karlsruhe, Germany) for and optimized batch NLCs. For the analysis, a small amount of samples were placed in sample holder and scanned with angular speed (2 h) of 2°/min at 30 mA operating voltage and 35 kV current.
The solid sample was exposed on Cu at 1.5406 A° wavelength and on a 2θ scale with a range of 3–60 °C for analysis of sample. PXRD spectra were obtained by diffractometry software coupled with a diffractometer. The crystallinity was computed by amorphous subtraction method that includes dividing the crystalline area produced by RTV-NLCs to the total area of diffractogram of RTV-NLCs [39].
The % crystallinity index (CI) was calculated using Eq. (5):
Morphology determination
AFM
To confirm the particle size and to study the morphological feature of NLC, the atomic force microscopy (AFM) was employed [34]. The optimized NLC sample was dispersed in Milli-Q water and subsequently sonicated. The thin film was prepared on AFM mica shit by spreading the dispersion volume 10 μL and incubated for air-dry overnight. The film was fixed on to AFM metal disk using double-sided tube and placed under the scanner tube. AFM analysis was performed using scanning probe microscope (SPM) with Raman spectroscopy: TriA 100 (A. P. E. Research Nanotechnology, Italy) in contact mode with silicon tip. The surface topography of RTV-NLC was recorded and analyzed in triplicate [40].
FESEM
(FESEM-S 4800, Hitachi, Japan) analysis Field emission scanning electron microscopy (FESEM) (FESEM-S 4800, Hitachi, Japan) analysis was performed in order to find out the size, shape, and surface characteristics of nanoparticles [41]. The lyophilized NLC sample was dispersed in an aqueous suspension and subsequently coated on a glass slide of approximately 30 µL. The dried glass slide was sputter (Hitachi E1010) with gold at 20 mA sputter to fabricate the particles electrically conductively for 1–2 nm thickness before imaging under accelerating voltage 30 kV [26].
1H-NMR
The 1H-NMR scan was recorded by (Bruker Avance III, 400 MHz) spectrometer at 400 MHz and at 298°K. A liquid sample of nanodispersion was introduced in NMR tube. Weighed amount of CDCl3 (D-chloroform) was added in the tube for field login and TMS, as an internal standard, was added for 0 PPM for the recording of chemical shift values of the compound [43].
Stability studies
In order to find the changes of NLC characteristics at different conditions of temperature and humidity during the storage according to the recommendation of ICH guideline Q1A (R2). For this purpose, the accelerated storage condition 4 °C, 25 °C ± 2 °C/60% RH ± 5% RH was selected and lyophilized NLCs sealed in a vial and properly and placed in stability chamber (CHM-10S, REMI Instruments Ltd., Mumbai, India) for the period of 6 months. The samples were evaluated for particle size, PDI, and EE after 2nd, 4th, and 6th months by dispersing in double-distilled water [42].
In vitro release study and pattern of kinetics
In vitro release study for prepared RTV-NLCs, PM and, RTV-suspension was performed using a previously reported method [15, 26]. Briefly, 200 ml simulated gastric solution pH 1.2 at 37 C for first 2 h followed by phosphate buffer pH at 6.8 at 37 ± 0.5 °C as dissolution medium with stirring speed of 100 RPM of the medium. At pre-set time interval, samples were withdrawn and an equal volume of freshly prepared buffer was replaced to maintain sink condition throughout the study. The retrieved samples were filtered and diluted with mobile phase accordingly and analyzed by HPLC with UV detector. The results were reported as mean ± SD after three measurements.
PAMPA
Parallel artificial membrane permeability assay (PAMPA) model was used to find out the passive permeability concentrations of drug. The experiment was performed according to the procedure adopted by Choi et al. [44]. All the requirements such as BD Gentest® (Corning Inc., MA) containing pre-coated lipid membrane, 96-well filter plate, phosphate buffer pH 6.8, and RTV-loaded NLC and RTV-suspension 2 mg/mL were collected. Initially, BD Gentest membrane (PAMPA membrane) was fitted in between donor and acceptor plate of 96-well filter plate. The upper portion of the 96-filter plate was used as donor and the lower plate was used as an acceptor. The phosphate buffer of pH 6.8 was used throughout the study. A donor sample solution of concentration 200 µg/mL was prepared by diluting RTV-NLCs and pure RTV in saline phosphate buffer (pH 6.8) solution.
A measured amount of sample solution (200 µL) was poured into the donor compartment of each well, and 300 µL phosphate buffer was poured into the receptor compartment of each well separately. The donor well was placed on the receptor well ensuring permeability membrane was in contact with buffer solution.
After 6 h of incubation at room temperature, the plate assembly was detached and samples from donor and receptor were withdrawn followed by suitably diluted. RTV permeated the membrane was analyzed by HPLC. The experiment was repeated in triplicate and the results were quoted in mean ± S.D.
A permeability difference (< 2%) signifies the integrity of membrane. The permeability coefficient (Pe) is represented by the following equation [27].
where Pe is permeability in the unit of cm/s, A is effective filter area, and A = f × 0.3 cm2 (where f = apparent porosity of the filter), VD is a donor well volume, VD = 0.3 ml, VA is a receptor well volume, VA = 0.2 ml, t is incubation time (s), CD (t) is compound concentration in donor well at time t, CA(t) is a compound concentration in receptor well at time t, and Cequilibrium is represented by the following equation.
Isolation and preparation of everted gut sac model
In order to investigate the permeability of the drug across the intestinal epithelium the most useful everted gut sac model is used. In this method, rats were kept on fasting for the period of 24 h with the supply of only water ad libitum and not supplied feed before the experiment. The whole procedure was adapted according to method described by Shekhawat and Pokharkar et al. [31]. The test animal was sacrificed by the survival dislocation method. The small intestine was excised through the abdominal incision of 4–5 cm and the underlying mesenterium was removed by manual stripping.
An appropriate segment of the intestine was identified and separated into 10 ± 0.2 cm sacs (2 cm below the pylorus, duodenum, and 20 cm below the pylorus, jejunum), flushed with water, and then aerated ice-cold KRB buffer solution. The glass rod was inserted into the intestinal segment and everted the entire intestine.
The sac was closed at one end and another end was also closed by filling 1 ml of NLC suspension and drug suspension individually. The intestinal sac was suspended individually in 50 ml oxygenated KBr buffer solution maintaining the temperature at 37 °C. At a pre-set time, the sample (0.5 mL) was collected and replaced by an equal volume of the freshly prepared buffer solution. After the experiment, the sac area was measured for the apparent permeability calculation. The withdrawal sample was measured by a UV spectrophotometer at 244 nm after diluting suitably. The experiment was performed in triplicate and the data was used in data analysis statistically.
Permeability was obtained by plotting the amount of drug transport across the sac area under study against time (min) [31]. Considering the equation of regression analysis, the slope was taken as permeation flux (J, mg/min).
The apparent permeability (Papp) was calculated mathematically by the following equation:
where Ci is the initial drug concentration (µg/mL) in the mucosal compartment, A is the surface area of the intestinal sac.
In vivo pharmacokinetic studies
Male Wistar rats approximately weighing 180–200 g were issued from the animal house of RCPIPER, Shirpur, and housed in cage separately. They were categorized into three groups and each group have six animals supplied feed and filtered water 24 h. They were kept in a place to acclimatize before the experiment. Animals were kept on fasting overnight. They were administered orally to three groups of rats at a dose of 54 mg/kg [4]. The dose receiving three groups was categorized into RTV-suspension, RTV-PM, and RTV-NLCs. The heparinized tube was kept ready for the collection of the blood sample. Approximately 0.3 mL blood sample was withdrawn from the orbital plexus at predetermined at the pre-set time of the point of 0.5,1,2,4, 8, 12, and 24 h for oral administration. The collected blood sample was ultracentrifuged (Optima Max- XP, Beckman Coulter Inc., Brea, USA) for 6000 rpm for 20 min at 4 °C to separate plasma and kept at − 20 °C until analysis.
Plasma sample extraction and bioanalysis
The plasma sample was spiked with a mixture of pure RTV and internal standard zidovudine (AZT) solution each (0.1 mL). To this solution, ethyl acetate was added for the purpose of deproteinization and vortexed properly. Afterward, this solution was centrifuged at 4000 rpm at 10 min in order to separate the ethyl acetate as a supernatant. The obtained residue was collected and analyzed by above described RP-HPLC method. The method was further validated for specificity, linearity, sensitivity, accuracy, precision, recovery, and robustness.
Pharmacokinetic parameters and statistical analysis
The calculations of pharmacokinetic parameters were established using a Microsoft excel based PK solver 2.0 add-in program. Non-compartmental analysis model was studied to determine the pharmacokinetic parameters. The relative bioavailability was determined by reported method [45, 46] and AUC0–t (t = 24) was established by the trapezoidal method. The various pharmacokinetic parameters including Cmax (max. plasma concentration), tmax (time of occurrence), AUC0-t (area under the curve, t = 24), and MRT (mean residence time) were calculated.
For statistical data analysis GraphPad Prism 6.0 (GraphPad Software Inc., CA, USA) was employed. All the results were reported in mean ± S.D. (n = 3). The data were considered to be statistically significant (P < 0.05) applying Student’s t-test and one-way ANOVA (analysis of variance) followed by Dunnett’s multiple comparisons.
Results and discussion
NLCs are an effective delivery system for the hydrophobic drug and targeting the drug across the membrane. The composition was selected based on solubility studies and formulation was developed using ultrasound waves based using QbD tool. The NLCs were prepared by microemulsification-probe sonication method in which addition of drug into the mixture of Compritol HD5 ATO (solid lipid) and Capryol 90 (liquid lipid) in the ratio of (70:30) which produces maximum room for the incorporation of drug and was stabilized by the combination of surfactants. Microparticles of lipid droplets of pre-emulsion broke down on ultrasonication leading to the formation of NLCs by acoustic cavitation [47].
QbD-based experimental framework
The significant role of the QbD-based experimental framework is the identification of specific features that define the quality of the product considering the patient-centric approach. Critical attributes of the final products that makes patient-centric, efficacious, and safe product [16].
QTPP and CQAs
The objective of QbD-based framework is to produce a quality product with understanding and consistently prove their adequate performance. QbD-based experimental framework was utilized to accomplished the production of NLCs. It is a systematic and knowledge-based framework for the development of a novel formulation to enhance the quality of the product from the initial point of the experiment. In this framework, the initial step, i.e., QTPP, was kept in mind as a goal throughout the experiments.
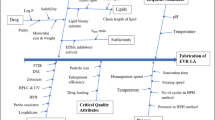
Risk assessment
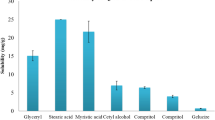
Risk assessment was performed by constructing the cause and effect diagram (Fig. 1) in accordance with ICH Q9 guidelines. The high-risk possessing factors were isolated which have an impact on the performance of the CQA. The risk factors might be the critical process parameters and critical material attributes which displayed on the cause and effect diagram. According to the risk assessment studies (Table S1), factors were categorized into high-, low-, and medium-risk levels. The factors possessing high risks were the concentration of lipids, the concentration of surfactant blend, and the process parameter, i.e., ultrasound amplitude. Lipid concentration held the highest risk in this formulation because it is the main excipient in the formulation and it was selected based on solubility. Solubility implies a higher percentage of EE and hence it was isolated. Second, the surfactant concentration used in NLC has attracted attention for the function of reduction in interfacial tension and stabilize the system [48]. The reduced particle size is the function of the optimum level of ultrasound amplitude and hence held the highest risk. However, factors falls under the low and negligible risk, they were neglected.
Influence of experimental variables

“Cause and effect” diagram portraying all the variables
A detailed description experimental variables on particle size, PDI, and EE (studies are given in supplementary data). Graphical representation of the significant effect of interrelation and interactions of independent variables on respective responses are presented as 3-dimensional (3D) response surface plot (Fig. 2).

Response surface plot depicting influence of experimental variables on particle size A, polydispersity index (PDI) B, and entrapment efficiency (EE) C
Drug loading
The NLC formulation was shown an elevated drug loading capacity, and it was found to be 2.35% w/w. This is due to the component used in the preparation of NLC, i.e., mixtures of solid (Compritol HD5 ATO) and liquid lipid (Capryol 90) contribute to achieving many imperfections that offer space to accommodate the high amount of drugs [49].
ζ-potential
The ζ-potential of all the NLC formulations was found to be in the range of − 9.8 ± 0.25 to − 13.6 ± 0.12. The NLC formulation was stabilized by the combination of nonionic surfactant such as Tween 80 and Pluronic F127 which lead to reduction in zeta potential values. The decrease in zeta potential values may lead to aggregation of particles. However, in our case, decrease in ζ-potential values was due to lowering of free energy of the system due to an interaction between nonionic surfactant (Tween 80) and drug molecule [31]. It was observed that the particles were unable to aggregate due to complete covering of particles and greater stability was maintained.
QbD-based optimization and validation study
The desirability functions data and generated the overlay plot showed the prediction values on the flag (Fig. S1, Supplementary data). The identified optimized batch showed the desirability of 0.87 which was closest to 1. The yellow region, on the overlay plot, suggested that the area of design space with feasible value and the gray region expressed the area where the response value did not fit the quality product [50].
The observed values were compared with predicted values and % bias was calculated. As per the results displayed in Table 3, the minimum % bias for the particle size and EE were observed. The formulation showed a maximum % bias for the PDI (i.e., 8.18%). It has been said that the polydispersibility index must be below 0.3, indicating uniformity of the particle distribution [23]. This might be due to the effect of human error or probably instrument error.
Lyophilization of RTV-NLCs
For the solid-state characterization and long-term stability purpose, the prepared optimized NLCs were converted into the free-flowing solid powder. Moreover, it possesses several advantages such as ease of handling and ease of administration. NLCs were lyophilized without cryoprotectant leads to agglomeration, aggregation phenomenon as the irreversible step after drying. This may be due to the chain entanglement in steric stabilizer (Tween 80) and drug particle fusion [31]. Therefore, several concentrations of mannitol (1.5%, 2.5%, and 3.5%) as a cryoprotectant were tried. The concentration of mannitol 3.5% was optimized based on the evaluation of particle size and PDI and EE.
Drug content
The assay of NLC for the percent drug content denotes the quantity of drug present in the prepared formulation. The percent drug content of lyophilized RTV-NLC was found to be 79.96 ± 0.54% w/w.
Physical characterization of NLCs
FT-IR spectroscopy
The FT-IR spectra of pure RTV, solid lipid (Compritol HD5 ATO), PM, and lyophilized NLCs are depicted in Fig. 3. The peaks (Fig. 3A) are depicted at 3356.26 cm−1 (N–H stretching), 2964.69 (C-H stretching), 1710 (C = O stretching), and 1631 (-C = C-, stretching aromatic carbon) confirming the characteristic structure of RTV [9, 10]. IR spectra of lipid mixture (Fig. 3B) shows the characteristic broadband peaks at 3650 and 3150 cm−1 which are attributed to –OH stretching. The absorption peaks observed at 1737.92 cm−1 and 1462.09 cm−1 were observed at the characteristic range of (C-O) stretching and methyl –CH deformation vibration. The spectra of PM (Fig. 3C) showed 2912.61 cm−1 (C-H) stretching, 1737.92 cm−1 (C = O) stretching, and 1467.88 corresponds to methyl (–CH) deformation vibration [51]. An optimized formulation (Fig. 3D) showed the reduced absorption peak at 3356.15 cm−1, 1710 cm−1, and 1631 cm−1, and the number of smaller absorption peaks in a range of 1813.15–2998 cm−1 get reduced which were corresponding to the crystalline fraction of drug converted into the amorphous form. This could be further confirmed in the study of DSC and PXRD.

Fourier transform infrared (FT-IR) spectra of RTV A, Compritol HD 5 B, (physical mixture) PM of RTV-Compritol HD 5 C, and optimized RTV-NLC D
Crystallinity studies
DSC study
DSC (Fig. 4) was run in order to find the drug-excipient interaction and crystallinity index of developed NLCs in a nitrogen environment. In this process, thermogram of pure RTV showed a sharp peak at 124.74 °C corresponding to their melting point represented in Fig. 4A. DSC thermogram of Compritol HD5 ATO corresponds to the melting point at 63.51 °C (Fig. 4B). Thermogram of PM showed the endothermic peak at 119.78 °C and 66 °C, respectively, which corresponds to the shifted peak of Compritol HD5 ATO and pure RTV (Fig. 4C). It indicates the crystalline RTV converted into the amorphous by complete solubilization process. Figure 4D representing the thermogram of RTV-loaded NLCs indicated the shorten and shifted peaks of the drug completely solubilized and present in an amorphous state. This is further supported by PXRD studies. Additionally, it is crucial to note that enthalpy of lipid matrix (Compritol HD 5 ATO) at 121.52 J/g as 100%, the crystallinity index (CI) of RTV-NLC was calculated using Eq. (4) and found to be 33.80% demonstrating partial crystallization of lipid [22].
PXRD study

Differential scanning calorimetry (DSC) thermogram of RTV A, Compritol HD 5 B, (physical mixture) PM of RTV-Compritol HD 5 C, and optimized RTV-NLC D
X-ray diffraction (XRD) is studied for the investigation of polymorphism and crystalline properties NCLs [52]. The diffractogram of RTV (Pure) and optimized RTV-NLCs were studied (Fig. S2). The plotted diffractogram 2 theta scale in X-axis and arbitrary count on Y-axis to identify the nature of the material. The diffractogram of pure RTV showed several peaks ranging from 5 to 30 °C and the scale indicated totally crystalline material. In brief, a diffraction angle of 2θ values at 6.838, 14.789, 17. 134, 18.128, and 21.582 corresponds to the intensity counts 67.09, 198.53, 372.38, 503.07, and 579.33, respectively, which confirms the RTV. It indicates the pure RTV is highly crystalline in nature. However, the reduced characteristic peak of ATV in the mixture of drug-lipid shows partial reduction crystalline converted into amorphous due to solubilization nature [53]. The XRD spectra of lyophilized RTV-NLC showed a large reduction in peak, and its 2θ values are 9.77, 19.90, 22.14, and 27.96 correspond to intensity count of 223.28, 303.79, 454.38, and 248.41, respectively. The other peaks of ATV were almost disappeared and reduced peaks in comparison to the pure drug during the process of NLC [4]. It clearly indicated that the role of encapsulating material (Compritol HD 5 ATO) and the intensity of the sonication process.
PXRD is used to find the polymorphism characteristics of lipid nanoparticles and changes in the crystalline order during production [54]. The crystallinity index was calculated and found to be 39.07%.
Morphological determination
AFM study
AFM study images of RTV-NLC were recorded with AFM instrument associated with RAMAN microscope and are depicted in Fig. 5a–c. Optimized formulation shows the spherical particle in 3D topographic image with an average particle size in the range of 85–200 nm. The average particle size obtained by AFM study was in good agreement with DLS measurement [34]. Histogram looks like the particle size distribution graph showing most of the particles lazing between 100 and 200 nm. The height of the particles appears lower in comparison to the original height of the particle, and this was due to the pressure of the AFM tip during the AFM measurement [55].

Image depicted atomic force microscope (AFM) analysis a, b, c, and field emission scanning electron microscope (FESEM) analysis d of the optimized NLC
FESEM
This is the modern technique to evaluate the morphology of the particles of the developed nanoparticle. Figure 5d shows spherical particles which correspond to the DLS measurement of the particle size of NLC [56]. Homogeneous size distribution was confirmed by the FESEM analysis.
1HNMR
NMR spectra (Fig. 6) of pure RTV and RTV-NLC were studied for interactions and molecular hydrogen bonding between drug molecules and lipid. As shown in Fig. 6A, the sharp proton signal at 0.8294 ppm, 1.3654 ppm, 2.9437 ppm, 5.1983 ppm, 6.9810–7.0971 ppm, and 8.2613 ppm can be attributed to doublet, 3H, -CH3; multiplate, 6H, -CH2; allylic hybridized C-H; singlet, sp2 hybridized C-H; multiplate aromatic proton, and singlet, 1H, -NH, confirming RTV. The intense signal at 7.264 ppm could be attributed to the residual impurity of solvent CDCl3.

1H Nuclear magnetic resonance (1HNMR) spectra of pure RTV and optimized RTV-NLCs
On the other hand, as can be seen in Fig. 6B presenting the spectra of NLCs, signal of drug proton disappeared, indicating the complete molecular solubilization of drug fraction in the matrix of lipids. In particular signal of aromatic ring H, sp2 hybridized C-H and allylic sp3 hybridized C-H was disappeared. This could be due to the function of the fraction of drug molecule solubilized completely in a lipid matrix. Also, this could be due to the transformation of crystalline form into amorphous molecules. These results were further supported by DSC and PXRD.
Stability study
The freeze-dried nanocarriers were evaluated after each 2nd, 4th, and 6th months for variation in particle size, PDI, and EE. An insignificant difference was observed for the particle size and PDI. In the case of EE, a slight variation was obtained after the 4th and 6th months (Table 4). The reduction in entrapment was observed due to the surface modifying property coupled with porosity inducer ability provided by Pluronic F127. The combination of Tween 80 and Pluronic F127, non-ionic emulsifiers renders the steric stabilization and electrostatic repulsion maintains the physical stability of the NLCs [57, 58].
Release simulation at physiological condition and kinetic study
In vitro RTV release studies were performed using dialysis bag from optimized NLCs, PM and, RTV-suspension. The release profile and their kinetics were compared in order to study the access in the systemic circulation. The drug release was found 92.37% for the optimized RTV-NLC and that of the RTV suspension was found 42.65% and PM for 55.21% (Fig. 7). The initial rapid release 25% for 2 h from the optimized NLC and thereafter sustained release for the next 24 h was considered as a maintenance dose. The slow release from the drug suspension is due to various reasons such as poor solubility of the drug, lack of controlled release modulator (lipid matrix), and even lack of surfactant.

In vitro drug release profile from RTV-suspension, RTV-PM, and optimized RTV-NLC
As can be seen in Fig. 7, the release rate of PM was greater than ATV suspension, this was due to the complete molecular solubilization of drug molecule in lipids, and hence, release can be controlled by lipid mixture. But release rate could not meet with the release rate of NLC formulation because the release of the drug from the formulation was regulated by combined lipid matrix and surfactant [7].
The potential ingredients in optimized NLCs have a significant role in nanosizing and solubilizing of the particle which facilitates the reducing interfacial tension. Thus, use of solubility enhancers like Tween 80 and Pluronic F127 contributed to the formulation so that dissolution and controlled release can be obtained. Thus, a combination of these surfactants in formulation could facilitate to obtain the reduced particle size modulation of release rate.
The drug release from the optimized formulation could be controlled by the union of solid lipid and liquid lipid [59]. The initial rapid release might be because of the unentrapped RTV particles stuck on the surface of the NLCs. Another decisive factor is the state of the drug substance which has an impact on the dissolution rates. During the nanosizing process, the amorphization of RTV in the formulation was ensured via ultrasound treatment. The study of DSC and PXRD characterization corroborated the amorphous state of RTV in the formulation [60].
The optimized NLC formulation was examined by various release kinetic models and best-suited model was found to be Higuchi-matrix. The correlation coefficient (R2) was found to be 0.990. The release exponent (n) and rate constant (k) were found to be 0.259 and 17.42, respectively. Thus, the release mechanism demonstrated the Fickian diffusion model since the release exponent, n is less than 0.5.
Intestinal everted sac studies
Everted sac technique can be used to assess the permeability of drugs from the intestine. The results obtained from this experiment can be compared to results of in vivo experiments [61]. The rat intestinal everted sac study was performed for the comparison of optimized NLCs, PM, and pure drug suspension at 37 ± 0.5 °C. The intestinal transport of drug particles was increased in case of optimized formulation than the drug suspension and PM. The nanonization of the particle and amorphization of the drug can be enhanced the solubility and ultimately permeability by modulating in NLC.
Drug transport through rat everted gut sac was supported by PAMPA study in order to meet the paracellular or active transport or otherwise PAMPA model fails to predict the permeability. After the everted sac permeability study, rate and extent of drug were determined as per the performance of each formulation. It was found that permeation of drug increased 2.7-fold as compared to the RTV suspension and 1.8-fold compared to RTV-lipid mixture (PM). An increase in permeability was due to the nanometric particle size, and also Pluronic F127 is a surface modifier that enhances the porosity of encapsulated the drug particle so that the release of the drug was easily influenced [58]. Additionally, the amphiphilic nature of Tween 80 used in formulation and acts as solubility enhancer contributed majority of formulation for reduction of particle size. Thus, the combined effect of surfactant, i.e., Tween 80 and Pluronic F127 could be enhanced the permeability of the RTV.
PAMPA
A non-cell PAMPA model was used in order to evaluate the permeability efficiency of RTV transcellular mechanism of passive diffusion. For transcellular diffusion, the desired characteristic, i.e., adequate lipophilicity is required to transport. Thus, the majority of the drug can access systemic circulation using passive diffusion. This is a reproducible, fast, and robust study. It is quick, easy, high throughput, and having a low cost compared to cell-based or in vivo techniques, which is a crucial advantage of this method [62]. To our best knowledge, PAMPA model was not used before this to study the permeability efficiency of RTV-NLC.
Table 5 shows an adequate permeability of RTV-NLCs in comparison to the RTV-suspension and PM. This was due to the improved log p value associated with lipid encapsulated the drug. As a result, the permeability was increased due to the higher lipid concentration in NLCs. Table 5 shows the lowest permeability value for pure RTV and highest permeability value for NLC formulation. The RTV-NLCs have Papp value is 2.34-fold than that of PM and 2.93-fold than pure RTV-suspension.
Further, it was observed that absorption occurs through the GIT and it was the function of solubility of the drug in GI milieu. Thus, the absorption and permeability of drug were due to the adequate solubility and reduced particle size of NLC formulation. Most importantly, the solubility of drug observed because of amorphous nature of drug present in NLC matrix and could be contributed to enhancing permeability.
In vivo pharmacokinetic study
The pharmacokinetic performance of the optimized RTV-NLC batch was studied using Wistar rats. PK solver 2.0 add-in program installed in Windows 8-based Microsoft Excel software was used to establish a pharmacokinetic profile. The linearity equation was utilized to assess the drug concentration in plasma using the estimated and validated HPLC method. Figure 8 depicts the plasma drug concentration against time profile showing comparison between optimized NLCs, PM, and drug suspension. A significant difference was observed in in vivo performance. The Cmax for optimized NLCs (0.472 ± 0.21) was 2.86-fold more than pure drug suspension (Cmax = 0.165 ± 0.12) and 1.85-fold more than PM (Cmax = 0.255 ± 0.03) (Table 6).

Plasma concentration profile of RTV in rats post-oral administration of RTV-suspension, RTV-PM and optimized ATV-NLCs
This could be observed because the pure drug was solubilized completely in a matrix with their molecular level. The enhanced plasma drug concentration of optimized NLCs could be due to the reduced particle size and higher absorption rate resulting in improved bioavailability. From the results, the relative bioavailability was found to be 286% as calculated from reported literature [63]. Additionally, improved pharmacokinetic performance in Wistar rats was due to the fraction of oil (liquid lipid) which augmented the higher drug encapsulation and facilitates the controlled drug release. For this reason, NLC presents as the best formulation comparison to others for RTV delivery.
Thus, higher absorption and improved bioavailability could be possible due to the higher encapsulation of drug in the lipid matrix. The drug fraction was completely solubilized and present in amorphous form makes easy to enhance dissolution rate. Moreover, reduced particle size and higher surface area were due to the ultrasound amplitude and smaller particle size could enhance the absorption in the gastrointestinal wall. However, the use of non-ionic surfactants, Tween 80, and Pluronic F127 facilitate easy absorption and improved drug transport through the lymphatic system. The higher MRT values (Table 6) of optimized RTV-NLCs depicted a longer residence time as compared to drug suspension and PM. Moreover, high protein binding capacity of the drug leads to lower metabolism [55].
Conclusion
The present study deals with the development of NLCs using QbD-based approach successfully to improve permeability and bioavailability of RTV. The DSC and PXRD studies confirmed the phase transition behavior of RTV which was observed during NLCs production. In vitro studies demonstrated that RTV-NLCs were exhibited sustained release pattern and in vivo parameters showed that the bioavailability of drug increased by 2.86 fold.
Therefore, preparation of NLCs is the best option and significantly brilliant carriers for encapsulation of lipophilic drugs. The cost-effective and potential NLC formulation can be manufactured on commercial scale using novel QbD-based approach in future. Thus, it was concluded that development of NLC has a wide scope and can be dominated as a promising delivery system for improvement oral bioavailability of RTV.
Data availability
Data available on request from the authors.
References
Kaspera R, Kirby BJ, Sahele T, Collier AC, Kharasch ED, Unadkat JD, et al. Investigating the contribution of CYP2J2 to ritonavir metabolism in vitro and in vivo. Biochem Pharmacol. 2014;91:109–18.
Patel D, Patel B, Patel C. Spectrophotometric method for simultaneous estimation of atazanavir sulfate and ritonavir in tablet dosage form. Drug Dev Ther. 2015;6:1.
Sudhakar B, Krishna MC, Murthy KVR. Factorial design studies of antiretroviral drug-loaded stealth liposomal injectable: PEGylation, lyophilization and pharmacokinetic studies. Appl Nanosci [Internet]. Springer Berlin Heidelberg; 2016;6:43–60. Available from: https://doi.org/10.1007/s13204-015-0408-8
Ahammed V, Narayan R, Paul J, Nayak Y, Roy B, Shavi G V, et al. Development and in vivo evaluation of functionalized ritonavir proliposomes for lymphatic targeting. Life Sci [Internet]. Elsevier Inc.; 2017;183:11–20. Available from: https://doi.org/10.1016/j.lfs.2017.06.022
Makwana V, Jain R, Patel K, Nivsarkar M, Joshi A. Solid lipid nanoparticles (SLN) of Efavirenz as lymph targeting drug delivery system: Elucidation of mechanism of uptake using chylomicron flow blocking approach. Int J Pharm [Internet]. Elsevier BV; 2015;495:439–46. Available from: https://doi.org/10.1016/j.ijpharm.2015.09.014
Javan F, Vatanara A, Azadmanesh K, Nabi-Meibodi M, Shakouri M. Encapsulation of ritonavir in solid lipid nanoparticles: in-vitro anti-HIV-1 activity using lentiviral particles. J Pharm Pharmacol. 2017;69:1002–9.
Kumar S, Narayan R, Ahammed V, Nayak Y, Naha A, Nayak UY. Development of ritonavir solid lipid nanoparticles by Box Behnken design for intestinal lymphatic targeting. J Drug Deliv Sci Technol [Internet]. Elsevier BV; 2018;44:181–9. Available from: https://doi.org/10.1016/j.jddst.2017.12.014
Karakucuk A, Celebi N, Teksin ZS. Preparation of ritonavir nanosuspensions by microfluidization using polymeric stabilizers: I. A Design of Experiment approach. Eur J Pharm Sci [Internet]. Elsevier BV; 2016;95:111–21. Available from: https://doi.org/10.1016/j.ejps.2016.05.010
Sinha S, Ali M, Baboota S, Ahuja A, Kumar A, Ali J. Solid dispersion as an approach for bioavailability enhancement of poorly water-soluble drug ritonavir. AAPS PharmSciTech. 2010;11:518–27.
Dhore PW, Dave VS, Saoji SD, Bobde YS, Mack C, Raut NA. Enhancement of the aqueous solubility and permeability of a poorly water soluble drug ritonavir via lyophilized milk-based solid dispersions. Pharm Dev Technol. 2017;22:90–102.
Maniyar MG, Kokare CR. Formulation and evaluation of spray dried liposomes of lopinavir for topical application. J Pharm Investig. 2019;49:259–70. https://doi.org/10.1007/s40005-018-0403-7.
Gairetta M, Bianchin MD, Kanis LA, Contri RV, Külkamp-Guerreiro IC. Development of innovative polymer-based matricial nanostructures for ritonavir oral administration. J Nanomater. 2019.
Yogananda R, Chowdary KPR. Enhancement of solubility, dissolution rate and bioavailability of efavirenz by Cyclodextrins and Solutol HS15 - a factorial study. Int J Res Pharm Chem. 2013;3:235–9.
Shete H, Patravale V. Long chain lipid based tamoxifen NLC. Part I: Preformulation studies, formulation development and physicochemical characterization. Int J Pharm [Internet]. Elsevier BV; 2013;454:573–83. Available from: https://doi.org/10.1016/j.ijpharm.2013.03.034
Patil-Gadhe A, Pokharkar V. Montelukast-loaded nanostructured lipid carriers : Part I Oral bioavailability improvement. Eur J Pharm Biopharm [Internet]. Elsevier BV; 2014;88:160–8. Available from: https://doi.org/10.1016/j.ejpb.2014.05.019
Bhise J, Kashaw SK, Sau S, Iyer AK. Nanostructured lipid carriers employing polyphenols as promising anticancer agents: Quality by design (QbD) approach. Elsevier BV; 2017;506–15. https://doi.org/10.1016/j.ijpharm.2017.04.078.
Mendes AI, Silva AC, Catita JAM, Cerqueira F, Gabriel C, Lopes CM. Miconazole-loaded nanostructured lipid carriers (NLC) for local delivery to the oral mucosa : Improving antifungal activity. Colloids Surfaces B Biointerfaces [Internet]. Elsevier BV; 2013;111:755–63. Available from: https://doi.org/10.1016/j.colsurfb.2013.05.041
Chen CC, Tsai TH, Huang ZR, Fang JY. Effects of lipophilic emulsifiers on the oral administration of lovastatin from nanostructured lipid carriers : Physicochemical characterization and pharmacokinetics. Eur J Pharm Biopharm [Internet]. Elsevier BV; 2010;74:474–82. Available from: https://doi.org/10.1016/j.ejpb.2009.12.008
Geszke-Moritz M, Moritz M. Solid lipid nanoparticles as attractive drug vehicles: Composition, properties and therapeutic strategies. Mater Sci Eng C [Internet]. Elsevier BV.; 2016; Available from: https://doi.org/10.1016/j.msec.2016.05.119
Bolhassani A, Javanzad S, Saleh T, Hashemi M, Aghasadeghi MR, Sadat SM. Polymeric nanoparticles Potent vectors for vaccine delivery targeting cancer and infectious diseases. Hum Vaccines Immunother. 2014;10:321–32.
Lembo D, Donalisio M, Civra A, Argenziano M, Cavalli R. Nanomedicine formulations for the delivery of antiviral drugs: a promising solution for the treatment of viral infections [Internet]. Expert Opin Drug Deliv. Taylor & Francis; 2018;93–114. Available from: https://doi.org/10.1080/17425247.2017.1360863
Kumbhar DD, Pokharkar VB. Engineering of a nanostructured lipid carrier for the poorly water-soluble drug , bicalutamide : Physicochemical investigations. Colloids Surfaces A Physicochem Eng Asp [Internet]. Elsevier BV; 2013;416:32–42. Available from: https://doi.org/10.1016/j.colsurfa.2012.10.031
Gurumukhi VC, Bari SB. Fabrication of efavirenz loaded nano-formulation using quality by design (QbD) based approach: Exploring characterizations and in vivo safety. J Drug Deliv Sci Technol [Internet]. Elsevier; 2020;56:101545. Available from: https://doi.org/10.1016/j.jddst.2020.101545
Patil SS, Kumbhar DD, Manwar JV, Jadhao RG, Bakal RL, Wakode S. Ultrasound-Assisted Facile Synthesis of Nanostructured Hybrid Vesicle for the Nasal Delivery of Indomethacin: Response Surface Optimization, Microstructure, and Stability. AAPS PharmSciTech. 2019;20:3–17.
Behbahani ES, Ghaedi M, Abbaspour M, Rostamizadeh K. Optimization and characterization of ultrasound assisted preparation of curcumin-loaded solid lipid nanoparticles: Application of central composite design, thermal analysis and X-ray diffraction techniques. Ultrason Sonochem [Internet]. 2017;38:271–80. Available from: https://doi.org/10.1016/j.ultsonch.2017.03.013
Gurumukhi VC, Bari SB. Quality by design (QbD)–based fabrication of atazanavir-loaded nanostructured lipid carriers for lymph targeting: bioavailability enhancement using chylomicron flow block model and toxicity studies. Drug Deliv Transl Res [Internet]. Springer US; 2021; Available from: https://doi.org/10.1007/s13346-021-01014-4
Hiremath PS, Soppimath KS, Betageri GV. Proliposomes of exemestane for improved oral delivery: Formulation and in vitro evaluation using PAMPA, Caco-2 and rat intestine. Int J Pharm. 2009;380:96–104.
Chiranjeevi K, Channabasavaraj KP, Srinivas Reddy PTN. Development and Validation of Spectrophotometric Method for Quantitative estimation of Ritonavir in Bulk and Pharmaceutical Dosage Forms. Int J ChemTech Res. 2011;3:58–62.
ICH guideline Q2(R1). ICH harmonised tripartite guideline, validation of analytical procedures: text and methodology. Curr. Step 4 version Parent Guidel 1994. 2005;13.
Pallagi E, Ambrus R, Szabó-Révész P, Csóka I. Adaptation of the quality by design concept in early pharmaceutical development of an intranasal nanosized formulation. Int J Pharm [Internet]. Elsevier BV; 2015;491:384–92. Available from: https://doi.org/10.1016/j.ijpharm.2015.06.018
Shekhawat P, Pokharkar V. Risk assessment and QbD based optimization of an Eprosartan mesylate nanosuspension: In-vitro characterization, PAMPA and in-vivo assessment. Int J Pharm [Internet]. Elsevier; 2019;567:118415. Available from: https://doi.org/10.1016/j.ijpharm.2019.06.006
Simões LS, Araújo JF, Vicente AA, Ramos OL. Design of β-lactoglobulin micro- and nanostructures by controlling gelation through physical variables. Food Hydrocoll [Internet]. Elsevier Ltd; 2020;100:105357. Available from: https://doi.org/10.1016/j.foodhyd.2019.105357
Ngan CL, Basri M, Lye FF, Fard Masoumi HR, Tripathy M, Abedi Karjiban R, et al. Comparison of Box-Behnken and central composite designs in optimization of fullerene loaded palm-based nano-emulsions for cosmeceutical application. Ind Crops Prod [Internet]. Elsevier B.V.; 2014;59:309–17. Available from: https://doi.org/10.1016/j.indcrop.2014.05.042
Ahmed S, Gull A, Alam M, Aqil M, Sultana Y. Ultrasonically tailored, chemically engineered and “QbD” enabled fabrication of agomelatine nanoemulsion; optimization, characterization, ex-vivo permeation and stability study. Ultrason Sonochem [Internet]. 2018;41:213–26. Available from: https://doi.org/10.1016/j.ultsonch.2017.09.042
Garg NK, Sharma G, Singh B, Nirbhavane P, Tyagi RK, Shukla R, et al. Quality by Design (QbD)-enabled development of aceclofenac loaded-nano structured lipid carriers (NLCs): An improved dermatokinetic profile for inflammatory disorder(s). Int J Pharm [Internet]. Elsevier BV; 2017;517:413–31. Available from: https://doi.org/10.1016/j.ijpharm.2016.12.010
Abdelwahed W, Degobert G, Stainmesse S, Fessi H. Freeze-drying of nanoparticles: Formulation, process and storage considerations. Adv Drug Deliv Rev. 2006;58:1688–713.
Zheng J, Gao Q. Tang C he, Ge G, Zhao M, Sun W. Heteroprotein complex formation of soy protein isolate and lactoferrin: Thermodynamic formation mechanism and morphologic structure. Food Hydrocoll. 2020;100.
Das S, Ng WK, Tan RBH. Are nanostructured lipid carriers (NLCs) better than solid lipid nanoparticles (SLNs): Development, characterizations and comparative evaluations of clotrimazole-loaded SLNs and NLCs? Eur J Pharm Sci [Internet]. Elsevier BV; 2012;47:139–51. Available from: https://doi.org/10.1016/j.ejps.2012.05.010
Park S, Baker JO, Himmel ME, Parilla PA, Johnson DK. Cellulose crystallinity index: Measurement techniques and their impact on interpreting cellulase performance. Biotechnol Biofuels. 2010;3:1–10.
Aji Alex MR, Chacko AJ, Jose S, Souto EB. Lopinavir loaded solid lipid nanoparticles (SLN) for intestinal lymphatic targeting. Eur J Pharm Sci. 2011;42:11–8.
Patil P, Khairnar G, Naik J. Preparation and statistical optimization of Losartan Potassium loaded nanoparticles using Box Behnken factorial design: Microreactor precipitation. Chem Eng Res Des [Internet]. Institution of Chemical Engineers; 2015;104:98–109. Available from: https://doi.org/10.1016/j.cherd.2015.07.021
Pardeshi C V., Rajput P V., Belgamwar VS, Tekade AR, Surana SJ. Novel surface modified solid lipid nanoparticles as intranasal carriers for ropinirole hydrochloride: application of factorial design approach. Drug Deliv [Internet]. 2013;20:47–56. Available from: https://doi.org/10.3109/10717544.2012.752421
Jores K, Mehnert W, Mäder K. Physicochemical investigations on solid lipid nanoparticles and on oil-loaded solid lipid nanoparticles: A nuclear magnetic resonance and electron spin resonance study. Pharm Res. 2003;20:1274–83.
Choi JU, Maharjan R, Pangeni R, Jha SK, Lee NK, Kweon S, et al. Modulating tumor immunity by metronomic dosing of oxaliplatin incorporated in multiple oral nanoemulsion. J Control Release [Internet]. Elsevier; 2020;322:13–30. Available from: https://doi.org/10.1016/j.jconrel.2020.03.012
Tiwari R, Pathak K. Nanostructured lipid carrier versus solid lipid nanoparticles of simvastatin: Comparative analysis of characteristics, pharmacokinetics and tissue uptake. Int J Pharm [Internet]. Elsevier B.V.; 2011;415:232–43. Available from: https://doi.org/10.1016/j.ijpharm.2011.05.044
Pardeshi C V., Belgamwar VS. Improved brain pharmacokinetics following intranasal administration of N,N,N-trimethyl chitosan tailored mucoadhesive NLCs. Mater Technol [Internet]. Taylor & Francis; 2020;35:249–66. Available from: https://doi.org/10.1080/10667857.2019.1674522
Kumar R, Singh A, Garg N, Siril PF. Solid lipid nanoparticles for the controlled delivery of poorly water soluble non-steroidal anti-inflammatory drugs. Ultrason Sonochem [Internet]. 2018;40:686–96. Available from: https://doi.org/10.1016/j.ultsonch.2017.08.018
Tefas LR, Rus LM, Achim M, Vlase L, Tomuță I. Application of the quality by design concept in the development of quercetin-loaded polymeric nanoparticles. Farmacia. 2018;66:798–810.
Üstündaǧ-Okur N, Gökçe EH, Bozbiyik DI, Eǧrilmez S, Özer Ö, Ertan G. Preparation and in vitro-in vivo evaluation of ofloxacin loaded ophthalmic nano structured lipid carriers modified with chitosan oligosaccharide lactate for the treatment of bacterial keratitis. Eur J Pharm Sci. 2014;63:204–15.
Shah B, Khunt D, Bhatt H, Misra M, Padh H. Application of quality by design approach for intranasal delivery of rivastigmine loaded solid lipid nanoparticles : Effect on formulation and characterization parameters. Eur J Pharm Sci [Internet]. Elsevier B.V.; 2015;78:54–66. Available from: https://doi.org/10.1016/j.ejps.2015.07.002
Aburahma MH, Badr-Eldin SM. Compritol 888 ATO: a multifunctional lipid excipient in drug delivery systems and nanopharmaceuticals. Expert Opin Drug Deliv. 2014;11:1865–83.
Kumbhar DD, Pokharkar VB. Physicochemical investigations on an engineered lipid – polymer hybrid nanoparticle containing a model hydrophilic active, zidovudine. Colloids Surfaces A Physicochem Eng Asp [Internet]. Elsevier BV; 2013;436:714–25. Available from: https://doi.org/10.1016/j.colsurfa.2013.07.044
Tran TTD, Tran PHL, Nguyen MNU, Tran KTM, Pham MN, Tran PC, et al. Amorphous isradipine nanosuspension by the sonoprecipitation method. Int J Pharm [Internet]. Elsevier BV; 2014;474:146–50. Available from: https://doi.org/10.1016/j.ijpharm.2014.08.017
Khosa A, Reddi S, Saha RN. Nanostructured lipid carriers for site-specific drug delivery. Biomed Pharmacother [Internet]. Elsevier; 2018;103:598–613. Available from: https://doi.org/10.1016/j.biopha.2018.04.055
Singh H, Jindal S, Singh M, Sharma G, Kaur IP. Nano-formulation of rifampicin with enhanced bioavailability: Development, characterization and in-vivo safety. Int J Pharm [Internet]. Elsevier BV; 2015;485:138–51. Available from: https://doi.org/10.1016/j.ijpharm.2015.02.050
Neupane YR, Srivastava M, Ahmad N, Kumar N, Bhatnagar A, Kohli K, Kanchan A. Lipid based nanocarrier system for the potential oral delivery of decitabine: Formulation design, characterization, ex vivo, and in vivo assessment. Int J Pharm [Internet]. Elsevier BV; 2014;477:601–12. Available from: https://doi.org/10.1016/j.ijpharm.2014.11.001
Wang Y, Zheng Y, Zhang L, Wang Q, Zhang D. Stability of nanosuspensions in drug delivery [Internet]. J Control Release. Elsevier BV; 2013;1126–41. Available from: https://doi.org/10.1016/j.jconrel.2013.08.006
Tan SW, Billa N, Roberts CR, Burley JC. Surfactant effects on the physical characteristics of Amphotericin B-containing nanostructured lipid carriers. Colloids Surfaces A Physicochem Eng Asp [Internet]. Elsevier BV; 2010;372:73–9. Available from: https://doi.org/10.1016/j.colsurfa.2010.09.030
Kim JK, Park JS, Kim CK. Development of a binary lipid nanoparticles formulation of itraconazole for parenteral administration and controlled release. Int J Pharm. 2010;383:209–15.
Qiao H, Chen L, Rui T, Wang J, Chen T, Fu T, et al. Fabrication and in vitro/in vivo evaluation of amorphous andrographolide nanosuspensions stabilized by D-α-tocopheryl polyethylene glycol 1000 succinate/sodium lauryl sulfate. Int J Nanomedicine. 2017;12:1033–46.
Zeng C, Zheng R, Yang X, Du Y, Xing J, Lan W. Improved oral delivery of tilianin through lipid–polymer hybrid nanoparticles to enhance bioavailability. Biochem Biophys Res Commun [Internet]. Elsevier Ltd; 2019;519:316–22. Available from: https://doi.org/10.1016/j.bbrc.2019.09.004
Veszelka S, Bocsik A, Walter FR, Hantosi D, Deli MA. Blood-brain barrier co-culture models to study nanoparticle penetration: Focus on co-culture systems. Acta Biol Szeged. 2015;59:157–68.
Dangre P, Gilhotra R, Dhole S. Formulation and statistical optimization of self-microemulsifying drug delivery system of eprosartan mesylate for improvement of oral bioavailability. Drug Deliv Transl Res. 2016;6:610–21. https://doi.org/10.1007/s13346-016-0318-7.
Acknowledgements
The authors would like to acknowledge the support of Management, Principal, and my colleagues of R. C. Patel Institute of Pharmaceutical Education and Research (RCPIPER), Shirpur.
Author information
Authors and Affiliations
Contributions
All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All animal experiments were carried out according to the NIH publication No. 8023, revised 1923 guidelines for Care and Use of Laboratory animals.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gurumukhi, V.C., Bari, S.B. Development of ritonavir-loaded nanostructured lipid carriers employing quality by design (QbD) as a tool: characterizations, permeability, and bioavailability studies. Drug Deliv. and Transl. Res. 12, 1753–1773 (2022). https://doi.org/10.1007/s13346-021-01083-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13346-021-01083-5