
Abstract
The present investigation is aimed to design a statistically optimized self-microemulsifying drug delivery system (SMEDDS) of eprosartan mesylate (EM). Preliminary screening was carried out to find a suitable combination of various excipients for the formulation. A 32 full factorial design was employed to determine the effect of various independent variables on dependent (response) variables. The independent variables studied in the present work were concentration of oil (X 1) and the ratio of S mix (X 2), whereas the dependent variables were emulsification time (s), globule size (nm), polydispersity index (pdi), and zeta potential (mV), and the multiple linear regression analysis (MLRA) was employed to understand the influence of independent variables on dependent variables. Furthermore, a numerical optimization technique using the desirability function was used to develop a new optimized formulation with desired values of dependent variables. The optimized SMEDDS formulation of eprosartan mesylate (EMF-O) by the above method exhibited emulsification time, 118.45 ± 1.64 s; globule size, 196.81 ± 1.29 nm; zeta potential, −9.34 ± 1.2 mV, and polydispersity index, 0.354 ± 0.02. For the in vitro dissolution study, the optimized formulation (EMF-O) and pure drug were separately entrapped in the dialysis bag, and the study indicated higher release of the drug from EMF-O. In vivo pharmacokinetic studies in Wistar rats using PK solver software revealed 2.1-fold increment in oral bioavailability of EM from EMF-O, when compared with plain suspension of pure drug.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Self-microemulsifying drug delivery system (SMEDDS) is an isotropic mixture of oil, surfactant, and cosurfactant that forms a microemulsion on mild agitation followed by dilutions in aqueous media, such as GI fluids [1]. The motility of the stomach and intestine favors the formation of self-emulsification in vivo [2]. SMEDDS represents the systems of forming transparent microemulsions with oil droplets size ranging from 100 to 250 nm [3]. Formation of microemulsion improves the solubilization of drug, and small size oil droplets provide larger surface area for drug absorption [4].SMEDDS has found to improve the oral bioavailability of lipophilic drugs such as Fenofibrate [5], Valsartan [6], Candesartan cilexetil [7], and Olmesartan medoxomil [8].
Eprosartan mesylate (EM) is a non-biphenyl, non-tetrazole, non-peptide selective angiotensin II receptor antagonist used in the treatment of hypertension [9]. EM has a dual pharmacological mode of action; it acts at vascular AT1 receptors (postsynaptically) and presynaptic AT1 receptors, where it inhibits sympathetically stimulated noradrenaline release. Furthermore, EM is unaffected by cytochrome P 450 and, therefore, has a low potential for metabolic drug interaction [10].
As per Biopharmaceutical Classification System (BCS), EM is a class II drug having low aqueous solubility and high intestinal permeability with high dose number (dose number is the mass of the drug divided by an uptake volume of 250 mL and the drug solubility) [11]. Orally administered EM has an absolute bioavailability of ≈13 %, with peak plasma concentration achieved 1–2 h after administration. EM is reported to exhibit extensive protein binding (≈98 %), and the volume of distribution is ≈13 L [12]. The low oral bioavailability of EM is a critical aspect of any formulations which is because of its low solubility and dissolution. Therefore, the present work aimed to design and develop SMEDDS of EM with a view to improve its bioavailability.
Formulation scientists often face the challenges during the scale-up of a product, from the formulation development to the production scale. There appears to be a great deal of uncertainty along with poor understanding, which results in unexpected failures. Manufacturing failures and inability to address regulatory specification would result in either rejection or reprocess of the batch which leads to increase in manufacturing cost. Lack of appropriate manufacturing process leads to non-compliance of regulatory guidelines and that causes delay in product development. The Food Drug Administration (FDA) in its Current Good Manufacturing Practice (cGMP) for the twenty-first century initiated quality by design (QbD) and process analytical technology (PAT) to have more controls over the manufacturing process along with a better focus on regulatory decision-making [13]. QbD is a systematic, scientific, risk-based, holistic, and proactive approach assisting both the industry and FDA to the pharmaceutical development [14]. The application of QbD principles has received lot of interest in pharmaceutical manufacturing.
Design of experiment (DoE) is a branch of applied statistics, widely employed in complex systems to study the effects of multiple factors and their interaction on system response and optimize the conditions for the desired result with minimum experiments. The application of DOE is very effective, as the outcome would be a fixed amount of information that has been collected with considerably less effort than the traditional approach. Statistical software such as Design Expert, JMP, etc., helps in design and optimization of the experiments with suitable screening models. The screening models assist in understanding the influence of variables on desired response. The full factorial design, Placket-Burman, and Box-Behnken are widely used screening models in the pharmaceutical industry. Among them, the full factorial design is considered the most efficient in estimating the influence of individual variables as well the interactive effect of each variable on desired responses [15]. The purpose of the present research is to develop a statistically optimized self-microemulsifying drug delivery system for eprosartan mesylate (EM) using 32 factorial design. A 32 full factorial design was employed to determine the effect of various independent variables on dependent (response) variables. The independent variables studied in the present work were concentration of oil (X1) and the ratio of S mix (X2), whereas the dependent variables were emulsification time (s), globule size (nm), polydispersity index (pdi), and zeta potential (mV), and the multiple linear regression analysis (MLRA) was employed to understand the influence of independent variables on dependent variables. Furthermore, a numerical optimization technique using the desirability function was used to develop a new optimized formulation with desired values of dependent variables. The optimized formulation was subjected to in vitro studies to evaluate percent drug release and in vivo studies to assess the changes in bioavailability.
Materials and methods
Materials
Eprosartan mesylate was a kind gift from Mylan Laboratory Ltd. (Nashik, India). Gift samples of Capmul MCM, Captex 200, and Acconon E were provided by ABITEC Corporation, USA. Labrasol and Transcutol-H was gifted by Gattefosse, Saint-Priest, France, and Chremophore-EL by BASF, India. Tween-80, PEG-400, PEG-200, Triton X-100, and Castor oil were purchased from Loba Chemie, India. Dialysis membrane (Mol. weight cutoff-12,000) was purchased from Hi-Media, Mumbai, India. All the chemicals used in the study were of analytical grade. HPLC grade solvents were procured from Merck, Mumbai, India. Freshly prepared doubled distilled water was used throughout the study.
Methods
Solubility studies
Solubility of the drug in an individual excipient, i.e., oils, surfactants, and cosurfactants, was studied. An excess amount of drug was placed into an empty reaction tube (Hi-Media, Mumbai, India) containing 2 mL of excipient. The resultant mixture was heated on a water bath at 40 °C and stirred vigorously using vortex mixture (V-Mixer, Scientific, India) for 5 min to facilitate solubilization of the drug. The resultant mixture was continuously agitated on rotary shaker cum incubator (Remi, India) at 40 °C for 24 h. It was then centrifuged at 10,000 rpm for 15 min. The supernatant was suitably diluted with methanol, and dissolved drug was quantified using ultraviolet spectrophotometer (UV-1800, Shimadzu, Japan) at 231 nm placing a blank [16]. Blank samples were prepared by dissolving respective oil and surfactant in methanol with the same dilution as for the test samples [17].
Preparation of liquid SMEDDS
By using different ratio of oil (20–50 %), surfactant (64–33 %), and cosurfactant (10–27 %) as shown in Table 1, multiple SMEDDS formulations of EM were prepared. The ratio of surfactant and cosurfactant was 2:1, 3:1, and 4:1. In all the formulations, the amount of eprosartan mesylate was 5 % w/w of total formulation weight. The formulation was prepared by dissolving the drug in surfactant followed by addition of cosurfactant and oil in glass vials. The resultant mixture was stirred continuously by vortex mixing and heated at 40 °C to obtain a homogeneous isotropic mixture. The SMEDDS formulations were stored at ambient temperature until further testing.
Construction of phase diagram
The phase diagram was constructed to obtain the optimum concentrations of oil, surfactant, and cosurfactant that resulted in maximum microemulsion area in phase diagram [18]. A liquid SMEDDS of 0.2 mL was placed into 300 ml of double distilled water in a glass beaker at 37 °C, and the contents were mixed gently with a magnetic stirrer at 100 rpm. The resultant emulsions were kept for 48 h at ambient temperature and observed for clarity, phase separation, and drug precipitation, and coalescence of oil droplets was judged as the unstable emulsion. All the studies were performed in triplicate with or without the drug. Phase diagram was constructed identifying the self-emulsifying region using CHEMIX ternary plot software [19].
Thermodynamic stability studies
The motive of carrying the thermodynamic stability studies was to evaluate the effect of temperature variation on SMEDDS formulations [20, 21]. The thermodynamic stability study of SMEDDS formulations was evaluated by exposing them to different stress conditions through freeze thaw cycle (−21 and +25 °C) and heating-cooling cycle (4 and 45 °C) with the storage at each temperature of not less than 48 h. The SMEDDS formulations were also subjected to centrifugation stress by centrifugation at 3500 rpm for 30 min in a microcentrifuge (Remi, India), and the extent of phase separation or any instability problems were observed [22].The stability studies were performed in triplicate for each formulation.
Design of experiments
In the present study, a 32 full factorial design was employed to evaluate the effect of selected independent variables on dependent variables. The design was based on the number of preliminary trials, carried out to find out the optimum ratio of excipients that would form microemulsion. For full factorial design study, the percentages of selected oil (Capmul MCM EP) and ratio of surfactant (Tween 80) to cosurfactant (Transcutol-H) were considered as the independent variables (factors), whereas dependent variables were emulsification time (s), globule size (nm), polydispersity index (pdi), and zeta potential. The formulations employed for the study were prepared using different levels of independent variables, i.e., low, middle, and high (Table 2). The matrix of experiments and investigated factors and responses are shown in Table 3.
Full factorial statistical modeling was used to optimize and evaluate the main effects, interaction effects, and quadratic effects of the formulation ingredients on the selected responses. Design-Expert 8.0.0.1 software (Stat-Ease Inc., USA) was used for exploring quadratic response surfaces and construction of a mathematical model. Multiple linear regression analysis (MLRA) involving independent variables and their interactions for measured responses were analyzed using model equation generated by 32 factorial design. The MLRA quadratic model equation is as follows:
where Y is the dependent variable; β 0, the intercept, is the arithmetic average of all quantities outcome of 9 runs; β 1to β 8 are the regression coefficients; X 1 and X2 are independent variables; and X 1 X 2 and X i 2(i = 1, 2) are the interaction and polynomial terms, respectively. The statistical validation of polynomials was accomplished using Yates’s ANOVA provision in the software. Afterward, feasibility, as well as grid search, was carried to find out the composition of optimum formulation.
Self-emulsification time
Self-emulsification time is the time taken by the SMEDDS to form a clear homogeneous system upon dilution. The emulsification results with complete disappearance of SMEDDS when observed visually. The efficiency of self-emulsification was assessed by using a standard USP XXIV type II dissolution apparatus (Electrolab, Mumbai, India). One gram of each formulation was added drop-wise into a 500 mL of distilled water maintained at 37 °C with gentle agitation by a rotating paddle at 50 rpm [23]. The efficiency of self-emulsification was visually assessed.
Determination of globule size and polydispersity index
The EM-loaded SMEDDS formulations were serially diluted (100-fold) with purified water followed by agitation for 1 min. The diluted samples were employed to measure the globule size and polydispersity index of emulsion using particle size analyzer (Malvern Zetasizer Nano Series ZS 90).
Zeta potential determination
The emulsion stability is directly related to the magnitude of the surface charge [24]. The zeta potential of selected formulation was determined by laser diffraction analysis using particle size analyzer (Malvern Zetasizer Nano Series ZS 90). All the formulations were diluted in a ratio of 1:100 (v/v) with distilled water and mixed thoroughly for 1 min using a magnetic stirrer [25].
In vitro drug release study
The in vitro drug release study of the optimized SMEDDS formulations (EMF-O) was carried on USP dissolution test apparatus II by dialysis bag method [26]. EMF-O formulation was instilled in the dialysis bag (Mol. weight cutoff 12,000, Hi-media Industries Inc., USA), firmly sealed and placed in 900 ml of pH 1.2, pH 4.5, and pH 6.8 buffers as the dissolution media at 37 °C. The revolution speed of the paddle was kept at 100 rpm. The 5 ml sample volumes was withdrawn at a predetermined time interval, and it is replaced with the same volume of fresh medium. The drug release profile of EM from EMF-O and a standard drug was compared in phosphate buffer pH 6.8 as a drug having a maximum solubility at this pH of the medium. The analysis of drug concentration was carried out by using UV-spectrophotometer (Shimadzu UV-1800). This study was carried out in triplicate.
Bioavailability study
Bioavailability study of optimized EMF-O formulation was carried out and compared with standard eprosartan mesylate pure drug. All experiments and protocol described in the study were approved by Institutional Animal Ethical Committee of Kamla Nehru College of Pharmacy, Butibori, India (Reg. No. KNCOP/R&D/AN-PROT/14-15/04). Wistar rats weighing 220–250 g were housed with free access to food and water. The rats were fasted overnight before the dosing with free access to water. Fasting male Wistar rats were divided into two treatment groups (n = 3), and each group orally received either the plain suspension of eprosartan mesylate or optimized EMF-O formulation. The suspension/EMF-O was suspended in 0.5 % sodium carboxyl methyl cellulose in such an amount that the dose of EM will be 10 mg/kg body weight made available in the administered volume of the suspension. After oral administration, 0.5 mL of blood was collected from retro-orbital plexus at 0.5, 1, 2, 4, 6, 8, 12, and 24 h time point. Plasma was separated from each blood sample by centrifugation at 10,000 rpm for 10 min and stored at −20 °C until analysis. Plasma (100 μL) was processed for the analysis of EM concentrations.
Extraction procedure
A single step protein precipitation method was employed for extraction of EM from rat plasma. In brief, 100 μL of drug-spiked plasma sample was pipetted into the heparinized centrifuge tube. The plasma was then deproteinized, to free the bound EM, using acetonitrile with a ratio of 1:3 (v/v) acetonitrile:plasma. The plasma:acetonitrile mixtures were vortex mixed for 3 min and then centrifuged at 5000 rpm for 5 min. Clear supernatant samples were collected and injected into the HPLC system.
HPLC analysis and sample preparation
High-performance liquid chromatography (HPLC) system consisted of LC solution data handling system (Shimadzu LC-20AD, Kyoto, Japan) equipped with SPD-20A Shimadzu UV detector (sensitivity of 0.005 absorbance unit full scale) and Rheodyne injector with 20-μL sample loop. A 25-μL Hamilton syringe was used for injecting the samples. Data acquisition was performed by using LC 2010 solution software. Chromatographic separation was carried out with Phenomenox, Gemini C18 (250 × 4.6 mm i.d., 5 μm) column at ambient temperature (28 °C) with Security Guard (4.0 × 3.0 mm i.d., 5 μm) column (Phenomenox, Torrance, CA, USA). The mobile phase for isocratic elution consisted of water and acetonitrile (55:45 v/v), pH of which is maintained at 3.4 using orthophosphoric acid (85 %). The mobile phase was prepared freshly and filtered through a Millipore vacuum filter system equipped with a 0.42-μm filter for each run and degassed by a sonicator. The isocratic elution was performed by pumping the mobile phase at a constant flow rate of 1.0 mL/min. The run time for elution was adjusted at 8 min. A sample of 20 μL was injected onto the analytical column and detected by measuring UV absorbance at 235 nm.
The developed method was found to be linear in the concentration range between 100 and 1800 ng/ml with regression coefficient (r 2) = 0.997, and the equation was Y = 111.3x +18,974. The mean recoveries of the spiked drug at 100, 900, and 1800 ng/mL were 91.53 ± 3.1, 94.82 ± 3.9, and 96.57 ± 4.5. The retention time of EM and internal standard was 2.2 and 3.1, respectively. At these retention points, there was no interference of any substance in plasma. The proposed method was found to be rapid, specific, and reproducible for determination of EM in rat plasma.
The pharmacokinetic parameters were calculated from measured EM plasma concentrations versus time profile after oral administration by a non-compartment model with the linear trapezoidal method using PK solver 2.0 software (an add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel).
Statistical analysis
Data demonstrated with arithmetic mean and standard deviation subjected to statistical assessment using Instat Graph Pad Prism software (Graph Pad Prism Version 4.00 for windows). The significant difference of various formulations was compared with one-way analysis of variance (ANOVA). The statistical probability (p) value less than 0.05 were considered to be significant.
Results and discussion
Solubility study
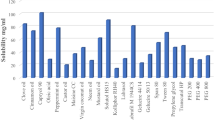
The components, i.e. oil, surfactant, and cosurfactant, used for formulating SMEDDS should have good solvent properties to achieve maximum drug solubilization. The resultant dispersion would appear as the clear and monophasic liquid at ambient temperature. The mean solubility of EM in various oils, surfactants, and cosurfactants are presented in (Fig. 1). The results suggested that among the various oils screened, Capmul MCM shows higher solubility (43.13 ± 0.71 mg/mL). Capmul MCM which is a medium chain monoglyceride (HLB 6) has partial aqueous solubility which may help in the easy dispersion of the drug in aqueous medium [27]. Capmul causes rapid release of drug in solubilized state across the entire GIT after oral administration [28]. Tween 80, a hydrophilic non-ionic surfactant (HLB 15), was found to have the maximum drug solubility (55.44 ± 0.31 mg/mL). Transcutol-H was selected as cosurfactant which shows maximum drug solubility (69.48 ± 1.15 mg/mL). Based on the solubility results, the SMEDDS formulations were developed consisting of varying concentrations of Capmul MCM (20–50 %), Tween 80 (64–33 %), and Transcutol-H (10–27 %).

Solubility data of eprosartan mesylate in various excipients
Ternary phase diagram
A ternary phase diagram was constructed with varying concentrations of oil and S/CoS (Fig. 2). Ternary phase behavior helps to predict the optimum concentration of oil and S/CoS which produce an emulsion with better stability [5]. All the SMEDDS formulations were stable at zero time and this may be attributed to the higher HLB value of Tween 80 which lowers the surface free energy required to form microemulsions and also offer mechanical protection to coalescence [29]. After storage of 48 h, all the formulations F1 to F9 showed transparency with good emulsion stability whereas formulations F10 to F12 were found to be unstable owing to phase separation and precipitation. It was observed that increasing the concentration of oil in the formulation to 50 % or above results in precipitation of emulsion with milky white appearance. This may be possible due to an insufficient concentration of surfactant to counteract the increased interfacial tension between the oil and aqueous phases.

Ternary phase diagram of SMEDDS between Capmul MCM EP, Tween 80, and Transcutol-H (the colored portion indicates the region of stable self-microemulsifying emulsion)
Thermodynamic stability studies
The thermodynamic study was done to ensure the kinetic stability of formulations and also to examine the chemical reaction occurring between the components of a formulation [30]. All the tested formulations were found to be stable to the various exposed stress conditions, i.e., freeze-thaw cycles, heating-cooling cycles, and centrifugal stress without any phase separation and precipitation in all tested conditions.
Optimization of formulation variables
The conventional practice of pharmaceutical formulation development are based on the time-consuming approach of changing one parameter at a time in which combined effects of factors and responses are not evaluated. Design of experiment (DoE) serves as an important tool to understand the complexity of the interaction between the variables and finding the optimum solution for the formulation. The present work involves a simple design with two variable studies at three experimental levels. Thus, a 32 full factorial design was applied to the formulation of SMEDDS to achieve a desired set of objectives. A total of nine formulation batches were prepared and evaluated for emulsification time, globule size, polydispersity index, and zeta potential.
Effect of factors on emulsification time
The potential ability of self-emulsification of SMEDDS formulation could be effectively estimated by determining the emulsification time. Self-emulsification occurs when the entropy change promoting dispersion is higher than the energy required for increasing the surface area of the dispersion [31]. The results of self-emulsification time are presented in Table 2. The model equation relating to self-emulsification time is as follows:
At different levels (−1, 0, 1) of S mix proportions, when oil concentration was increased, the emulsification time of SMEDDS formulation was found to increase for each level, respectively. The SMEDDS EMF1 with 20 % oil concentration shows low emulsification time (77 s); the SMEDDS EMF9 with the higher concentration of oil (40 %) shows high emulsification time (302 s). This may be possible because the higher proportion of oil requires a more time for emulsification. The polynomial equation reveals that the concentration of oil (X 1) has a strong direct influence on the emulsification time; however, the proportion of S mix (X 2) shows a little effect on the response. Each factor has a more significant effect on the response than the combined effect.
Effect on globule size
The size of the globule is crucial in self-emulsification as it determines the rate and extent of drug release as well as absorption [31]. The globule size of all nine formulations (EMF1 to EMF9) was in the range of 44.42 to 262.5 nm. With the increase in oil proportion, there was an increase in globule size. The increment in the globule size may hamper the rate of emulsification which subsequently leads to a higher emulsification time. The model equation relating to the globule size is given below;
The value of a positive coefficient of X 1 was larger, which indicate that the effect of oil concentration was the increasing influence factor on the globule size in SMEDDS formulation. The high level of S mix (4:1) in the formulations shows the negative influence on the globule size. It is indicated that the high surfactant proportion to cosurfactant in the formulations (EMF3, EMF6, and EMF9) results in a decrease in the globule size. The role of surfactant in the microemulsion is to stabilize and condense the interfacial film whereas cosurfactant causes the expansion of interfacial film. Therefore, the proportion of surfactant to cosurfactant has widely different effects on the globule size. The interaction effects of X 1 and X 2 were not significant.
Effect on polydispersity index
Polydispersity index is the ratio of standard deviation to the mean globule size. The value of pdi denotes the uniformity of globule size in the formulation [32].
The model equation relating to polydispersity index is given below.
The positive value of X 1 signifies that the higher proportion of oil in formulation results in variation in the size of oil globules which is indicated by the high polydispersity index. Whereas, the negative values of X 2 indicate that the increase in surfactant concentration leads in microemulsion having more uniform globule size. This is probably due to the presence of more surfactants molecule lowers the interfacial barrier between the oil and water resulting in more spherical and uniform oil globules. The SMEDDS formulations EMF 4, EMF 5, and EMF 6 show good uniformity in particle size with pdi (0.338, 0.324 and 0.317 respectively).
Effect on zeta potential
Zeta potential is a crucial factor in terms of stability of microemulsion. The formulation with a higher zeta potential value confers better stability, i.e., prevent aggregation of molecules. The negative value of SMEDDS formulations was probably due to the charge present on oil and surfactant molecules. All nine formulations show negative values indicating the good overall stability of the formulations. The model equation relating to the zeta potential is,
The negative value of the equation indicates that both the factors have the negative influence on the response. Zeta potential was found to be higher in the formulations having a high level of X 1; this is probably due to free fatty acids in the oil contributing significantly to charge on SMEDDS formulations. The higher level of X 2 shows the negative influence on the response; this might be due to higher proportion of surfactants leading to emulsification of free fatty acid to a greater extent and thus lowering of the zeta potential.
The results of ANOVA, presented in Table 4, indicate that all models were significant (p < 0.05) for all response parameters investigated. In addition, Design-Expert 8.0.0.1 software generates counter and 3D response surface plots for emulsification time (s), globule size (nm), polydispersity index (pdi), and zeta potential (mV) as presented in (Fig. 3).

Response surface plot and counter plot showing the combined effect of concentration of oil and Smix on a emulsification time, b globule size, c polydispersity index, and d zeta potential
Validation of optimization model
To optimize all the responses with different objectives, a numerical optimization technique by the desirability function was used to develop new optimized formulation with a desired response. The targeted ranges of the responses were limited to 100 ≤ Y 1 ≤ 250 s, 150 ≤ Y 2 ≤ 262.81 nm, 0.3 ≤ Y 3 ≤ 0.45pdi, −14.8 ≤ Y 4 ≤ −7.5 mV. The optimal values of responses were generated by numerical analysis using the Design-Expert software based on the criterion of desirability; Fig. 4 presents the 3D-response surface and counter plot of desirability region. The accuracy of the generated optimization model was estimated by preparing the SMEDDS formulation with the optimum combination of the factors as suggested by the generated model. The optimized SMEEDS formulation (EMF-O) was evaluated for various measured responses, i.e., emulsification time (s), globule size (nm), polydispersity index (pdi), and zeta potential (mV). Table 5 lists the result of experiments with predicted responses by the mathematical model and those actually observed. The optimized SMEDDS formulation of EM (EMF-O) showed emulsification time (118.45 ± 1.64 s), globule size (196.81 ± 1.29 nm), zeta potential (−9.34 ± 1.2 mV), and polydispersity index (0.354 ± 0.02). The results indicate that the generated optimized model for 32 factorial designs was well fitted.

3D response surface plot and counter plot showing the desirability region
In vitro drug release study
The in vitro drug release study of the optimized EMF-O formulation and a pure drug was determined in buffers of pH 1.2, pH 4.5, and pH 6.8. The results indicated that release of EM from EMF-O was higher in phosphate buffer of pH 6.8 than in the pH 4.2 and pH 1.2 as shown in (Fig. 5). This is possible because EM has pH-dependant solubility; an increase in the pH results in higher solubility of the drug. Optimized SMEDDS formulation release more than 92 % drug in pH 6.8 within 1 h, whereas standard drug shows only 27 % drug release in 1 h. The higher release of drug in SMEDDS may be due to the reduced particle size provides a large surface area to release drug from the solvent. Furthermore, the oil in SMEDDS may act as a carrier which allows diffusion of the drug from the dialysis membrane.

In vitro release profile of standard eprosartan mesylate and from optimized EMF-O formulations (mean ± SD; n = 3)
Bioavailability study
The in vivo pharmacokinetic behaviors of EM pure drug with SMEDDS were studied in male Wister rat. The mean plasma concentration versus time profile in male Wister rats after a single oral dose of EMF-O was compared to the drug in suspension (Fig. 6). The pharmacokinetic parameters obtained from non-compartment analysis using linear trapezoidal method are summarized in Table 6.

The mean plasma concentration versus time profile of standard drug suspension and optimized SMEDDS formulation (EMF-O) in rats after oral administration (mean ± SD, n = 3)
Relative bioavailability was calculated by using fallowing formulae
The EMF-O shows significantly increased C max and AUC0-t as compared to the pure drug suspension. However, T max of the EMF-O was relatively delayed as compared to a pure drug suspension, indicating that self-emulsification process could improve drug release and absorption in GIT. The relative bioavailability of EMF-O compared with the pure drug was significantly higher with 2.1-fold increment. The increased in the bioavailability of SMEDDS formulation might be due to the high concentration of surfactant in a formulation. A higher proportion of surfactant in formulation could improve the permeability by disturbing the cell membrane. Surfactant molecules interact with the polar head groups in the lipid bilayer of the cell membrane, affecting the overall binding forces between the groups. It also gets fitted in a lipophilic core of the bilayer and causes disruption of tight junction in the cell membrane [33]. Furthermore, oil component in formulation promotes transport of the lipophilic drug, thus bypasses the hepatic first-pass metabolism which subsequently leads to improvement in absorption and bioavailability [34]. In a nutshell, the pharmacokinetic and dissolution study reveals that optimized EMF-O formulation can lead to increase in the bioavailability of EM.
Conclusion
Self-microemulsifying drug delivery system for EM was developed and optimized successfully using quality by design approach. The full factorial design helps to predict the best possible combinations of oil and S mix. The optimized formulation (EMF-O) consists of Capmul MCM EP as oil phase with Tween 80 (surfactant) and Transcutol-H (cosurfactant). The numerical response analysis assists in understanding the influence of factors on different responses. The optimized SMEDDS (EMF-O) was evaluated for emulsification time (s), globule size (nm), polydispersity index (pdi), zeta potential (mV), and drug release. The in vitro dissolution study reveals the better release of drug in SMEDD formulation as compared with a pure drug. The developed RP-HPLC method for estimation of EM in rat plasma was reliable and reproducible. The pharmacokinetic analysis carried with PK Solver 2.0 was best to fit into the non-compartment model using the linear trapezoidal method. The in vivo study indicates the higher AUC and C max as compared to pure drug. The optimized SMEDDS formulation (EMF-O) showed 2.1-fold increments in bioavailability as compared to pure EM. Therefore, self-microemulsifying drug delivery system can be sorted as a promising drug delivery system for improvement of drug dissolution and oral bioavailability of EM.
References
Ghosh PK, Murthy RS. Microemulsion: a potential drug delivery system. Curr Drug Deliv. 2006;3:167–80.
Kommuru TR, Gurley B, Khan MK, Reddy JK. Self-emulsifying drug delivery system (SMEDDS) of coenzyme Q10: formulation, development and bioavailability assessment. Int J Pharm. 2001;212:233–46.
Kohli K, Chopra S, Dhar D, Arora S, Khar RK. Self-emulsifying drug delivery system: an approach to enhance oral bioavailability. Drug Discov Today. 2010;15:958–65.
Charman WN. Lipid, lipophilic drugs and oral drug delivery—some emerging concepts. J Pharm Sci. 2000;89(8):967–78.
Patel AR, Vavia PR. Preparation and in vivo evaluation of SMEEDS (self- microemulsifying drug delivery system) containing fenofibrate. AAPS Pharm Sci Tech. 2007;9(3):E344–52.
Dixit AR, Rajput SJ, Patel SG. Preparation and bioavailability assessment of SMEEDS containing valsartan. AAPS Pharm Sci Tech. 2010;11(1):314–21.
Nekkanti V, Karatgi P, Prabhu R, Pillai R. Solid self-microemulsifyig formulation for candesartan cilexetil. AAPS Pharm Sci Tech. 2010;11(1):9–17.
Prajapati ST, Joshi HA, Patel CN. Preparation and characterization of self-microemulsifying drug delivery system of olmesartanmedoxamil for bioavailability improvement. J Pharm. 2013;1:1–9.
Ploskar GL. Eprosartan: a review of its use in hypertension. Drugs. 2009;69(17):2477–99.
Ruilope L, Jager B. Eprosartan for the treatment of hypertension. Expert Opin Pharmacother. 2003;4(1):107–14.
Borekar S, Pawar A. Eprosartan mesylate nanosuspension with HPMC E3 for improving solubility and bioavailability. Adv Sci Eng Med. 2013;5(12):1297–304.
Robins WG, Scott JL. Eprosartan: a review of its use in the management of hypertension. Drugs. 2006;66(16):2355–77.
Suresh S, Roy S, Ahuja BK. Quality by design: an overview. Indian Drugs. 2015;52(02):5–11.
Rathore AS, Winkle H. Quality by design for biopharmaceuticals. Nat Biotechnol. 2009;27(1):26–34.
Joshi SA, Chavan SS, Sawant KK. Rivastigmine-loaded PLGA and PBCA nanoparticles: preparation, optimization, characterization, in vitro and pharmacodynamic studies. Eur J Pharm Biopharm. 2010;76:189–99.
Narkhede RS, Gujar KN, Gambhire MV. Design and evaluation of self-nanoemulsifying drug delivery systems for nebivolol. Asian J Pharm. 2014;8:200–9.
Gupta S, Chavan S, Sawant K. Self-nanoemulsifying drug delivery system for adefovirdi pivoxil: design, characterization, in-vitro and ex-vivo evaluation. Colloids Surf A Physicochem Eng Asp. 2011;392(1):145–55.
Zang L, Zang I, Zang M, Pang Y, Zhaoming L, Aili Z, Jing F. Self-emulsifying drug delivery system and the applications in herbal drugs. Drug Deliv. 2015;22(4):475–86.
Kallakunta VR, Bandari S, Jukanti R, Veerareddy PR. Oral self emulsifying powder of lercanidipine hydrochloride: formulation and evaluation. Powder Technol. 2012;221:375–82.
Negi LM, Tariq M, Talegaonkar S. Nano scale self-emulsifying oil based carrier system for improved oral bioavailability of camptothecin derivative by P-glycoprotein modulation. Colloids Surf B: Biointerfaces. 2013;111(1):346–53.
Patel PV, Patel HK, Panchal SS, Mehta TA. Self micro-emulsifying drug delivery system of tacrolimus: formulation, in vitro evaluation and stability studies. Int J Pharm Investig. 2013;3(2):95–104.
Ramasahayam B, Eedara BB, Kandadi P, Jukanti R, Bandari S. Development of isradipine loaded self-nano emulsifying powders for improved oral delivery: in vitro and in vivo evaluation. Drug Dev Ind Pharm. 2015;41(5):753–63.
Bachynsky MO, Shah NH, Patel CI, Malick AW. Factor affecting the efficiency of self-emulsifying oral delivery system. Drug Dev Ind Pharm. 1997;23:809–16.
Pongcharoenkiat N, Narsimhan G, Lyons RT, Hem SL. The effect of surface charge and partition coefficient on the chemical stability of solutes in O/W emulsion. J Pharm Sci. 2002;91:559–70.
Balakumar K, Raghavan CV, Selvan NT, Prasad RH, Abdu S. Self nanoemulsifying drug delivery system (SNEDDS) of rosuvastatin calcium: design, formulation, bioavailability and pharmacokinetic evaluation. Colloids Surf B: Biointerfaces. 2013;112:337–43.
Kang BK, Lee JS, Chon SK, Jeong SY, YuK SH, Kahng G, Lee HB, Cho SH. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beggal dogs. Int J Pharm. 2004;274:65–73.
Chambin O, Jannin V, Champion D, Chevalier C, Rochat-Gonthier MH, Pourcelot Y. Influence of cryogenic grinding on properties of self emulsifying formulation. Int J Pharm. 2004;278:79–89.
Rahman MA, Hussain A, Hussain MS. Role of excipients in successful development of self-emulsifying/microemulsifying drug delivery system (SMEDDS/SMEDDS). Drug Dev Ind Pharm. 2013;39:1–19.
Craig DQ, Barker SA, Banning D, Booth SW. An investigation into mechanism of self-emulsification using particle size analysis and low frequency dielectric spectroscopy. Int J Pharm. 1995;114:103–10.
Singh B, Bandyopadhyay S, Katare OP. Optimized self nano-emulsifying systems of ezetimibe with enhanced bioavailability potential using long chain and medium chain triglycerides. Colloids Surf B: Biointerfaces. 2012;100:50–61.
Reiss H. Entropy-induced dispersion of bulk liquids. J Colloid Interface Sci. 1975;53:61–70.
Baboota S, Shakeel F, Ahuja A, Ali J, Shafiq S. Design, development and evaluation of novel nanoemulsion formulations for transdermal potential of celecoxib. Acta Pharma. 2007;57:315–32.
Wei W, Yang W, Li Q. Enhanced bioavailability of silymarin by self-microemulsfying drug delvery system. Eur J Pharm Biopharm. 2006;63(3):288–94.
Hauss DJ, Fogal SE, Ficorilli JV, Price CA, Roy T, Jayaraj AA, Keirns JJ. Lipid-based delivery systems for improving the bioavailability and lymphatic transport of a poorly water soluble LTB4 inhibitor. J Pharm Sci. 1998;87(2):164–9.
Acknowledgment
The authors are sincerely thankful to the secretary of Kamla Nehru College of Pharmacy, Butibori, Nagpur, India, for providing the instrumental facility for carrying out the research work. The authors are also thankful to Mylan Laboratory Ltd. (Nashik, India) for providing the gift sample of eprosartan mesylate.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All institutional and national guidelines for the care and use of laboratory animals were followed.
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Dangre, P., Gilhotra, R. & Dhole, S. Formulation and statistical optimization of self-microemulsifying drug delivery system of eprosartan mesylate for improvement of oral bioavailability. Drug Deliv. and Transl. Res. 6, 610–621 (2016). https://doi.org/10.1007/s13346-016-0318-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13346-016-0318-7