
Abstract
Multidrug-resistant tuberculosis (MDR-TB) is becoming a global health crisis. The World Health Organization has released new guidelines for the use of tuberculosis-active drugs for the treatment of patients with MDR-TB. Despite documented activity against tuberculosis isolates, doses and exposure targets are yet to be optimized. Our objective was therefore to review the clinical pharmacokinetic and pharmacodynamic literature pertaining to drugs recommended to treat MDR-TB and to identify target areas for future research. To date, published research is limited but studies were identified that evaluated the pharmacokinetics and pharmacodynamics of these drugs. Exposure targets were assessed and summarized for each drug. Exposure-based targets (e.g., area under the concentration curve/minimum inhibitory concentration) appear to be most commonly associated with predicting drug efficacy. Dose variation studies based on these targets were largely inconclusive. Future research should focus on determining the risks and benefits of dose optimization to meet exposure targets and improve patient outcomes. The role of therapeutic drug monitoring also remains yet to be confirmed, both from a clinical perspective as well as a resource allocation perspective in regions where MDR-TB is active.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Dose optimization may improve treatment outcomes for patients with multidrug-resistant tuberculosis. |
Exposure targets provide a basis for optimizing drug regimens.. |
1 Introduction
Tuberculosis is a serious infectious disease and is listed as one of the top ten causes of deaths worldwide [1]. It affects both adults and children and has a greater prevalence in developing regions in Africa and Asia. Despite treatment progress and a global decrease in incidence of approximately 2% per year, the emergence of resistant forms of tuberculosis is quickly becoming a global health crisis [1, 2]. Multidrug-resistant tuberculosis (MDR-TB) is defined as a form of tuberculosis infection caused by bacteria that demonstrate resistance to (at least) the first-line treatment options of isoniazid and rifampin [1, 3]. These drugs are considered to be the most potent for treating tuberculosis and therefore any resistance that precludes their use can be detrimental to treatment success and result in poor patient outcomes. In 2017, it was estimated that there was an incidence of MDR-TB of > 450,000 new cases worldwide. The burden of these cases is largely (~ 50%) found within the countries of India, China, and the Russian Federation. Optimizing efficacy of MDR-TB treatment is therefore a primary concern for government, policymakers, researchers, and clinicians, worldwide [1, 4, 5].
The World Health Organization (WHO) released updated guidelines for the treatment of resistant forms of tuberculosis in 2019 [6]. Drugs recommended to treat MDR-TB are provided in Table 1. The drugs have been stratified into three different groups, based on current knowledge about effective combinations and efficacy against MDR-TB. For a patient presenting with MDR-TB, it is recommended that all three Group A agents are used and that one agent from Group B is selected to ensure four active drugs. If only one or two Group A agents can be used, then both Group B agents are to be selected. If four active drugs are unable to be selected from Groups A and B, then Group C agents are to be added to complete the regimen. It is noted, however, that the evidence is not clear with respect to using Group C agents as part of the primary regimen [6]. The guidelines also recommend regimens of different durations. The majority of patients are recommended to complete a longer treatment duration regimen, consisting of a total treatment duration of 18–20 months. In patients who have not been treated for > 1 month with second-line tuberculosis agents or in those patients who have confirmed tuberculosis sensitivity to fluoroquinolones and second-line injectable drugs, a shorter duration of 9–12 months may be considered [6]. In either case, treatment of MDR-TB involves co-administration of multiple drugs over long periods of time, which may have implications based on the clinical pharmacokinetic and pharmacodynamic profiles of recommended drugs.
As the global spotlight moves towards treatment of resistance forms of tuberculosis, including MDR-TB, there has been an increase in published literature pertaining to the clinical pharmacokinetics and pharmacodynamics of recommended agents. Understanding how these drugs work at these levels and being aware of potential clinical implications resulting from pharmacokinetic and pharmacodynamic properties may help to design more effective and safe drug regimens in the future [7, 8]. There is currently a debate in the literature about drug resistance, specifically whether or not resistance is a problem arising from non-adherence or variations in drug exposure [9,10,11]. It is argued that tailoring doses to patients in order to optimize drug exposure may be just as important (if not more) than selecting the drugs to make up the treatment regimen [9, 11]. As programmatic tuberculosis treatment is not routinely monitored, the utility and feasibility of monitoring exposure is not yet known [8]. Given this current controversy and the increase in literature describing pharmacokinetic/pharmacodynamic outcomes, this review aims to critically evaluate the pharmacokinetic and pharmacodynamic literature pertaining to drugs used for the treatment of MDR-TB.
2 Literature Search
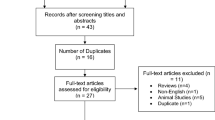
A search of Pubmed and EMBASE was conducted to identify relevant literature for this review. Databases were searched using combinations of the keywords—tuberculosis OR MDR-TB OR multidrug resistant tuberculosis, AND pharmacokinetics OR pharmacodynamics, AND each individual drug name (as identified in Table 1). The search was conducted from database inception until November 24, 2019. The search was limited to articles published in English and those that were conducted in humans. Articles were downloaded and reviewed if they reported on clinical pharmacokinetic or pharmacodynamic outcomes associated with at least one of the drugs recommended to treat MDR-TB in MDR-TB patients. Articles solely reporting on limited sampling strategies were excluded. Conference abstracts and unpublished articles were not reviewed.
3 Results of Literature Search
An overview of the drugs used to treat MDR-TB and known parameters predictive of activity are provided in Table 1 [6, 12].
3.1 Group A Medicines
Group A medicines include levofloxacin or moxifloxacin, bedaquiline, and linezolid [6]. All three (either levofloxacin or moxifloxacin) are to be included as part of the regimen for patients with MDR-TB. As such, these medicines form the backbone of MDR-TB treatment. Clinical studies have found high activity of these medicines against MDR-TB, which has translated into important outcomes, such as clinical cure.
The pharmacokinetics and pharmacodynamics of levofloxacin and moxifloxacin are well established in many populations, as these drugs are used to treat a variety of bacterial infections. For treatment of MDR-TB with levofloxacin, it has been shown that increased doses lead to higher exposures in MDR-TB patients [13]. It has also been shown that the free area under the concentration time curve over minimum inhibitory concentration (fAUC/MIC) is a useful parameter for predicting efficacy [14, 15]. Recent literature shows that these exposure targets are not commonly met in both adults and children [16, 17]. One study conducted in MDR-TB patients aimed to determine the achievement of the fAUC/MIC of levofloxacin dosed at 15 mg/kg. The study found high variability in the AUC and 4 of 20 patients did not achieve a target fAUC/MIC > 100 [18]. Another study conducted in children with MDR-TB similarly assessed exposure with levofloxacin doses of 15–20 mg/kg [19]. A simulation model was developed to estimate exposures (AUC and maximum concentrations [Cmax]) in children receiving the 20 mg/kg dose. Findings showed exposures were lower than target for all age ranges. Using the simulation model, the authors predicted that those children weighing 8–11 kg may require doses up to 40 mg/kg to achieve target exposures based on adult data [19]. Malik et al. found only 27% of children aged 2–10 years met concentration targets when given WHO recommended doses, yet 80% achieved with higher doses [20]. Another study included data from 37 pharmacokinetic/pharmacodynamic studies in adults (32 studies) and children (5 studies) with MDR-TB [21]. Similar to the other reported studies, this study found that the standard daily dosing of 1,000 mg levofloxacin did not achieve an fAUC/MIC > 100 in 80% of the patients with an MIC and minimum bactericidal concentration of 1 mg/l. However, those patients with an MIC of 0.5 mg/l all achieved target parameters, suggesting usual doses may be suitable for less resistant organisms [21]. Conversely, Deshpande et al. [22] found doses of 25 mg/kg (1500 mg/day) were favorable for achieving target responses in MDR-TB patients and Mase et al. [23] reported target attainment in children with doses of 15–20 mg/kg. Kumar et al. [24] assessed covariates on pharmacokinetic parameters in Indian children. Significant findings for levofloxacin included a higher Cmax in females (11.5 µg/ml vs 7.3, p = 0.017). From a formulation perspective, it has been shown that dispersible tablets may have up to 41% greater exposure than regular adult tablets when given to children aged < 5 years [25]. These studies show that despite good evidence to use levofloxacin in MDR-TB, there is still urgent research needed to optimize dosing and formulations that will achieve exposure targets, especially for organisms with greater resistance and high MICs.
Pharmacokinetic and pharmacodynamic variability has also been shown with moxifloxacin and MDR-TB patients. Thee et al. [16] conducted a prospective study in children with MDR-TB aimed to assess the pharmacokinetics and safety of a 10 mg/kg dose. Findings showed serum concentrations were lower than in adults treated with 400 mg per day, thus concluding higher doses may be necessary for this age group. Chang et al. [26] conducted a study in adults and found high patient variability, including the fact that patient weight influenced apparent clearance (Cl/F) for moxifloxacin, which prompted the authors to conclude that dosing should consider patient weight. Simulations based on this study data support a 200 mg daily dose of moxifloxacin for patients weighing < 50 kg and 400 mg daily for those weighing ≥ 50 kg. Ganatra et al. [27] studied dosing further and found that high dose moxifloxacin (600–800 mg/day) may be optimal to overcome increasing resistance at typical critical concentrations of 0.5 mg/l. Gumbo et al. [28] studied the effect of increasing moxifloxacin doses on target attainment rates (400, 600, or 800 mg/day). Findings showed best attainment was achieved with the 800 mg/day dose, compared to both the 600 and 400 mg/day doses (93% vs 86% and 59%, respectively). These studies and others demonstrate that higher doses may be needed to overcome resistant organisms. However, higher doses must be well studied to ensure any increase in efficacy is not offset by compromising tolerability [26,27,28,29]. Heinrichs et al. [30] showed that tissue penetration into the lung is excellent for MDR-TB patients treated with moxifloxacin.
Bedaquiline is a Group A drug used for the treatment of MDR-TB after demonstrating efficacy in clinical trials [31]. It demonstrates linear pharmacokinetics following a one-time dose up to 700 mg and multiple doses up to 400 mg daily [32]. It is well absorbed with a time to maximum concentration of 5 h. It undergoes oxidative metabolism via CYP3A4 and is known to have a very long terminal half-life (~ 173 h). Achievement of therapeutic concentrations in cerebral spinal fluid is speculated to be low [33]. Body weight and albumin level are known to influence bedaquiline and metabolite (M2) disposition [34]. Population pharmacokinetic modeling (4-compartment disposition model) found bedaquiline to be widely distributed (apparent volume > 10,000 l) with a low clearance. CL/F was approximately 50% higher for black patients, as opposed to other races, and females had a lower apparent volume (Vc/F) than males (by approximately 15%). Based on the results of this study, bedaquiline’s long terminal elimination half-life is speculated to be due to redistribution from tissue compartments [35]. An additional study found bedaquiline to be detected in plasma at 200 days post treatment, suggesting there could be a propensity for resistance development [36]. To date, the exposure response relationship of bedaquiline remains to be adequately characterized and should be a priority for future research [8]. Some work to date suggests that this may be possible [37]. Initial work has suggested that activity of bedaquiline is concentration dependent and that the AUC/MIC may therefore be the best parameter as a response marker [38]. Pharmacokinetic/pharmacodynamic modeling suggests that the addition of bedaquiline to MDR-TB treatment regimens may also allow for shorter durations of treatment (18 months) while maintaining high efficacy rates [39]. Related, an interim analysis from an ongoing phase 2 study shows that bedaquiline given for at least 24 weeks, in addition to an individualized background regimen, may be a suitable option for Japanese patients [40].
The addition of linezolid to tuberculosis regimens increases culture conversion and clinical cure [6, 41]. However, linezolid has a narrow therapeutic index with the frequency of adverse events directly proportional to the dose and duration of therapy [41, 42]. Mitochondrial toxicities including peripheral or optic neuropathy and hematological toxicity are most common and may correlate with trough concentrations [43]. Optimal pharmacokinetic/pharmacodynamic targets have not yet been established but efficacy appears to be driven by the AUC/MIC [44]. It is also noted that an AUC/MIC ratio of 100 when co-administered with a companion drug is required to prevent drug resistance [45]. Brown et al. [46] studied different dosage regimens to optimize efficacy and safety. More frequent dosing (300 mg twice daily) offered more bacterial kill but increased toxicity, compared to less frequent dosing (600 mg daily).
Alffenaar et al. [47] studied alternative doses of linezolid to investigate if smaller doses could minimize adverse effects but maintain adequate exposure. Eight patients were enrolled and received linezolid 300 mg (reduced dose) orally twice daily for 3 days, followed by 600 mg (standard dose) orally twice daily. Findings showed seven patients reached a target AUC24/MIC ratio > 100. Despite no clear evidence that this ratio will predict therapeutic efficacy, it is concluded that a reduced dose may be appropriate for future study [47]. Similar findings were also demonstrated in a systematic review with meta-analysis [48]. Results from this report suggest 300 mg orally twice daily may meet both efficacy and safety targets but the authors recognize the data are largely driven by a single center and require further study. A study that assessed linezolid concentrations in resected lung tissue found low tissue penetration of linezolid into diseased lung but the clinical implications are unclear [49]. However, the authors caution against lowering doses < 600 mg per day.
3.2 Group B Medicines
Group B medicines include clofazimine and cycloserine or terizidone [6]. It is recommended to add one or both medicines to treatment regimens. Clofazimine is a riminophenazine dye used for the treatment of leprosy but has activity against MDR TB [50]. A recent systematic review identified nine observational studies that included patients treated with clofazimine for drug resistant tuberculosis [50]. Clofazimine-inclusive regimens produced favorable outcomes in 65% of patients with MDR-TB. Despite these positive outcomes, little dose optimization data exist for the use of clofazimine in MDR-TB and this should be a priority for future research [8].
Current guidelines recommend the use of cycloserine or terizidone in MDR-TB treatment regimens [6]. Both agents are partial N-methyl-d-aspartate receptor agonists and antagonists of pyridoxal 5′-phosphate (P5P) reactions. Terizidone is converted in vivo to cycloserine via hydrolysis [8]. Chang et al. [26] conducted a population pharmacokinetic study using cycloserine doses of 250–500 mg twice daily. The authors determined a one-compartment model with first order absorption best represented the data obtained. Findings showed a slower absorption rate constant, slower Cl/F, and smaller Vd/F in patients with MDR-TB compared to health volunteers but that total daily doses of 500–750 mg cycloserine were likely to achieve a Cmax within the therapeutic range [26]. Mpagama et al. [17] found 2 h concentrations at 14 days of cycloserine to achieve expected concentrations and 12 (52%) of the patients had concentrations that exceeded the upper limit of the 2 h concentration range (20–35 µg/ml). Three (13%) patients had concentrations below expectations. Kumar et al. [24] found age (> 12 years) to be a significant predictor of higher Cmax and AUC values in Indian children. Court et al. [51] found no major influence of co-administration with terizidone.
Deshpande et al. [52] completed a systematic search to identify pharmacokinetic and pharmacodynamics studies performed with cycloserine MDR-TB but found no studies assessing exposure targets. The same authors then completed exposure effect and dose fractionated studies in a hollow fiber system of tuberculosis. MICs were also determined in 415 tuberculosis isolates and a susceptibility breakpoint was established to be 64 mg/l. Efficacy was determined to be associated with the percentage of time above MIC. Monte Carlo simulations showed a dose of 750 mg twice daily was most effective to achieve targets to treat pulmonary tuberculosis, while a dose of 500 mg twice daily was likely sufficient for meningitis; these doses are proposed to achieve target exposure in the lung cavities of 92% of patients and 85% of those patients with meningitis, respectively [52].
Little data exist regarding the pharmacokinetics and pharmacodynamics of terizidone, likely due to its conversion to cycloserine in vivo. Mulubwa et al. [53] reported the development and validation of a chromatographic method to measure concentrations of terizidone concentrations. The authors then followed up with a population pharmacokinetic study. Samples were analyzed from 78 MDR-TB patients undergoing intensive phase therapy. Sampling occurred over 24 h and terizidone was found in 272 samples from 39 patients. Mean and median concentrations were 49.3 µg/ml and 51.8 µg/ml, respectively.
3.3 Group C Medicines
Group C drugs are to be added to the regimen for completion and when Group A and B drugs cannot be used [6]. Despite known activity against MDR-TB, clinical evidence for these drugs is largely scarce. Pharmacokinetic and pharmacodynamic data are also not well established [8].
Delamanid is a relatively new Group C drug indicated for the treatment of MDR-TB in adults. Stinson et al. [54] conducted a study to determine MICs of Delamanid and critical concentrations for susceptibility. A total of 460 isolates were used, of which 316 originated from patients enrolled in a global clinical trial, 76 from trials conducted in South Africa and 68 that were obtained outside clinical trials (45 Japanese and 23 South African). MICs ranged from 0.001 to 0.05 µg/ml. This translated into an MIC50 (MIC that inhibits 50% of isolates) of 0.004 µg/ml and an MIC90 (MIC that inhibits 50% of isolates) of 0.012 µg/ml. Based on the MICs and other pharmacokinetic data, the authors report a critical concentration of 0.2 µg/ml [54]. This breakpoint is supported by pharmacokinetic data that showed 95.8% of patients receiving 100 mg twice daily delamanid had a Cmax of ≥ 0.2 µg/ml and 79.8% had a minimum concentration of ≥ 0.2 µg/ml [55]. The results from this study showed higher sputum culture conversion with twice daily delamanid dosing compared to placebo (41.9% more patients, p = 0.04) [55].
Sturkenboom et al. [56] conducted a systematic review to determine the optimal dose and frequency of amikacin in MDR-TB regimens. The authors identified five studies that assessed pharmacokinetic, pharmacodynamic, and pharmacokinetic/pharmacodynamic parameters in association with clinical outcomes. Findings from four pharmacokinetic studies showed variability in how studies described Cmax and that the route of administration (intravenous or intramuscular) has an impact on pharmacokinetic parameters [56]. The authors described one included study that used a hollow fiber system model of tuberculosis to identify amikacin pharmacokinetic/pharmacodynamic markers that associate best with efficacy [56]. It was found that Cmax/MIC was favored to kill Mycobacterium tuberculosis. A Cmax/MIC ratio of 10.1 at the site of infection was found to be the optimal dosing target for intermittent therapy that optimized cure for MDR-TB. This resulted in a serum Cmax/MIC ratio of 75 [56, 57]. Of note, an AUC0–24/MIC ratio of 103 also reached maximal effective concentrations [58]. Studies showed an increased risk of toxicity (including ototoxicity) was associated with cumulative AUC corresponding with cumulative days of treatment [56, 59]. These findings suggest that higher intermittent dosing, as opposed to daily dosing, may be more beneficial to reach target Cmax/MIC values and optimize killing. This could therefore be a target for future research.
The carbapenem antibiotics (imipenem-cilastatin and meropenem) are recommended as Group C drugs and are gaining importance as therapeutic options [5, 60]. A systematic review summarized the clinical evidence for these drugs, in addition to ertapenem [61]. Seven studies of varying design met the inclusion criteria and showed positive treatment success for regimens containing a carbapenem to treat both MDR-TB and extensively resistant tuberculosis. Culture conversion rates specifically ranged from 60 to 95%. Tolerability of carbapenems as part of anti-tuberculosis regimens was high [61]. Despite little pharmacokinetic/pharmacodynamic data published for imipenem-cilastatin or meropenem in MDR-TB patients, some data exist for ertapenem. van Rijn et al. [62] published a larger systematic review that included 35 relevant articles. In vivo studies were identified for imipenem, meropenem, and ertapenem. The authors concluded that there is insufficient evidence to recommend one carbapenem drug over another at this time. From a pharmacokinetic/pharmacodynamic perspective, Van Rijn et al. [63] also evaluated drug exposure in 12 MDR-TB patients receiving doses of 1,000 mg once daily. In relation to an MIC of 0.25 mg/l, almost all (11/12) patients exceeded 40% time-free concentration > MIC (Tfree > MIC). Nine patients met the criteria for an MIC of 0.5 mg/l and two patients with an MIC of 1 mg/l. The exposure target of 40% tfree > MIC may also be able to be extrapolated to other carbapenems [63].
Pyrazinamide is indicated for both drug susceptible and MDR-TB [6]. Chirehwa et al. [64] conducted a study to determine pharmacokinetic/driven optimal dosing regimens in MDR-TB. Simulations from MDR-TB data show that drug exposure is associated with weight bands and that doses of 1,500 mg (33–50 kg), 1,750 mg (51–70 kg), and 2,000 mg (> 70 kg) ensure at least 90% of patients reach an AUC0–24 h target of 363 mg*h/l. This means that some patients may need higher doses than those currently recommended for treatment of TB. Mugabo and Mulubwa [65] developed a population pharmacokinetic model of pyrazinamide and pyrazinoic acid based on 51 adult patients with MDR-TB. This study found that pharmacokinetic parameters were not affected by age, HIV status, or sex.
The Group C drug para-aminosalicylic acid may offer efficacy for both MDR-TB [6]. Chang et al. [26] developed a population pharmacokinetic model for this drug in patients with MDR-TB. The results showed an absorption rate constant of 0.51/h, an apparent clearance of 30.8 l/h, and apparent volume of distribution of 79.4 l. Through simulation, it was determined that doses of 4.95–6.6 g of p-aminosalicylic acid twice daily (or 3.3 g three times daily) would be most likely to achieve concentrations (Cmax) within the target range (20–60 µg/ml) [26]. It should be noted, however, that slow phenotypes of N-acetyltransferase (NAT1*14 and NAT1*3 alleles) might result in higher p-aminosalicylic acid exposures [66].
Ethionamide is recommended as a Group C drug for MDR-TB [6]. Deshpande et al. [67] conducted a multi-dose hollow fiber system model of tuberculosis to identify the AUC0-24h/MIC ratio that produced optimal outcomes. It was found that optimal exposure occurred with an AUC0–24 h/MIC ratio > 56.2. Doses of 20 mg/kg/day were shown through Monte Carlo experiments to achieve this target in 95% of patients, as long as the MIC was < 2.5 mg/l. Findings showed ethionamide may be an important drug for MDR-TB treatment up to MICs of 2.5 mg/l. Kumar et al. [24] found significantly higher exposures (AUC) in Indian children aged < 12 years (17.5 µg/ml*h vs 9.4 µg/ml*h, p = 0.030) and that gender predicted higher Cmax values in female patients.
Lee et al. [68] conducted a pharmacokinetic study with prothionamide in 17 patients with MDR-TB. The patients received oral doses of 375 mg or 250 mg twice daily for at least 2 weeks in addition to their other tuberculosis drugs. Blood sampling occurred over 24 h. Mean Cmax was 22.2 µg/ml (± 1.1 µg/ml), time to maximum concentration was 3.6 h (± 1.3 h), elimination half-life ranged from 1.5 to 3.8 h and mean AUC0–12 h was 11.0 ± 3.7 µg*h/ml. No major differences in pharmacokinetic parameters were noted between patients with BMIs above and below 18.5. The key finding from this study is that low body weight did not alter pharmacokinetic data and therefore may not need dosage adjustments in patients with poor nutritional statuses [68].
3.4 Therapeutic Drug Monitoring
The evidence surrounding therapeutic drug monitoring is controversial [8, 69, 70]. Studies, to date, have not demonstrated strong associations between therapeutic drug monitoring and better clinical outcomes. Studies are also limited by inappropriate sampling, lack of long-term outcome data, and lack of drug susceptibility testing [8, 69, 70]. Although collected individualized response information can be useful, positive outcome data are likely necessary before widespread use can be recommended. While therapeutic drug monitoring may provide more insight into dose and exposure targets, its role may currently be reserved for research purposes or perhaps those patients with underlying reasons for alterations in drug exposure. The American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America currently recommend therapeutic drug monitoring for patients with poor response despite adherence and a fully susceptible strain, severe gastrointestinal abnormalities, drug–drug interactions, impaired renal clearance, HIV infection, diabetes mellitus, or treatment using second-line drugs [71]. It must be noted, however, that any recommendation to implement routine therapeutic drug monitoring in practice must be balanced against required resources, especially considering the distribution of MDR-TB across world regions [8]. Nevertheless, studies should continue to explore the role of therapeutic drug monitoring for widespread practice, in pursuit of determining which patients and which drugs may most benefit from its use. Cost analyses should also be conducted to compare the costs of potentially improved therapy with therapeutic drug monitoring versus the high costs of repeated or failed MDR-TB therapy.
4 Summary
We aimed to review current pharmacokinetic and pharmacodynamic topics relating to drugs used to treat MDR-TB that may have important consequences for clinical outcomes. A general conclusion is that the literature in this area is still lacking and much work needs to be carried out, in order to optimize MDR-TB treatment outcomes. Specifically, determining exposure targets, as well as optimal doses, should be priorities for future research. At the same time, researchers should focus on how to achieve pharmacokinetic/pharmacodynamic outcomes in resource limited settings.
References
World Health Organization. Tuberculosis. 2018. https://www.who.int/en/news-room/fact-sheets/detail/tuberculosis. Accessed 5 Dec 2019.
Furin J, Cox H. Tuberculosis. Lancet. 2019;393:1642–56.
Gunther G. Multidrug-resistant and extensively drug-resistant tuberculosis: a review of current concepts and future challenges. Clin Med. 2014;14:279–85.
Dean AS, Cox H, Zignol M. Epidemiology of drug-resistant tuberculosis. Adv Exp Med Biol. 2017;1019:209–20.
World Health Organization. UN general assembly high level meeting on ending TB. 2018. https://www.who.int/news-room/events/un-general-assembly-high-level-meeting-on-ending-tb. Accessed 5 Dec 2019.
WHO consolidated guidelines on drug-resistant tuberculosis treatment. Geneva: World Health Organization; 2019. Licence: CC BY-NC-SA 3.0 IGO.
Rizk ML, Bhavnani SM, Drusano G, Dane A, Eakin AE, Guina T, et al. Considerations for dose selection and clinical pharmacokinetics/pharmacodynamics for the development of antibacterial agents. Antimicrob Agents Chemother. 2019;63:e02309–18.
Technical report on the pharmacokinetics and pharmacodynamics (PK/PD) of medicines used in the treatment of drug-resistant tuberculosis. Geneva: World Health Organization; 2018 (WHO/CDS/TB/2018.6). Licence: CC BY-NC-SA 3.0 IGO.
Srivastava S, Pasipanodya JG, Meek C, Leff R, Gumbo T. Multidrug-resistant tuberculosis not due to noncompliance but to between-patient pharmacokinetic variability. J Infect Dis. 2011;204:1951–9.
Mariandyshev A, Eliseev P. Drug-resistant tuberculosis threatens WHO’s End-TB strategy. Lancet Infect Dis. 2017;17:674–5.
Alffenaar JC, Migliori GB, Gumbo T. Multidrug-resistant tuberculosis: pharmacokinetic and pharmacodynamic science. Lancet Infect Dis. 2017;17:898.
Reynolds J, Heysell SK. Understanding pharmacokinetics to improve tuberculosis treatment outcome. Expert Opin Drug Metab Toxicol. 2014;10:813–23.
Peloquin CA, Phillips PPJ, Mitnick CD, Eisenach K, Patientia RF, Lecca L, et al. Increased doses lead to higher drug exposures of levofloxacin for treatment of tuberculosis. Antimicrob Agents Chemother. 2018;62:e00770-18.
Peloquin CA, Hadad DJ, Molino LPD, Palaci M, Boom WH, Dietze R, et al. Population pharmacokinetics of levofloxacin, gatifloxacin, and moxifloxacin in adults with pulmonary tuberculosis. Antimicrob Agents Chemother. 2008;52:852–7.
Ghimire S, Maharjan B, Jongedijk EM, Kosterink JGW, Ghimire GR, Touw DJ, et al. Levofloxacin pharmacokinetics and pharmacodynamics and outcome in MDR-TB patients. Eur Respir J. 2019;53:1802107.
Thee S, Garcia-Prats AJ, Draper HR, McIlleron HM, Wiesner L, Castel S, et al. Pharmacokinetics and safety of moxifloxacin in children with multidrug-resistant tuberculosis. Clin Infect Dis. 2015;60:549–56.
Mpagama SG, Ndusilo N, Stroup S, Kumburu H, Peloquin CA, Gratz J, et al. Plasma drug activity in patients on treatment for multidrug-resistant tuberculosis. Antimicrob Agents Chemother. 2014;58:782–8.
van’t Boveneind-Vrubleuskaya N, Seuruk T, van Hateren K, van der Laan T, Kosterink JGW, van der Werf TS, et al. Pharmacokinetics of levofloxacin in multidrug- and extensively drug-resistant tuberculosis patients. Antimicrob Agents Chemother. 2017;61:e00343-17.
Denti P, Garcia-Prats AJ, Draper HR, Wiesner L, Winckler J, Thee S, et al. Levofloxacin population pharmacokinetics in South African children treated for multidrug-resistant tuberculosis. Antimicrob Agents Chemother. 2018;62:e01521-17.
Malik AA, Brooks MB, Siddiqui S, Fuad J, Peloquin CA, Amanullah F, et al. Pharmacokinetics of levofloxacin in children treated for exposure to drug-resistant tuberculosis. Antimicrob Agents Chemother. 2019;63(5):e02569-18.
Ghimire S, van’t Boveneind-Vrubleuskaya N, Akkerman OW, de Lange WCM, van Soolingen D, Kosterink JGW, et al. Pharmacokinetic/pharmacodynamic-based optimization of levofloxacin administration in the treatment of MDR-TB. J Antimicrob Chemother. 2016;71:2691–703.
Deshpande D, Pasipanodya JG, Mpagama SG, Bendet P, Srivastava S, Koeuth T, et al. Levofloxacin pharmacokinetics/pharmacodynamics, dosing, susceptibility breakpoints, and artificial intelligence in the treatment of multidrug-resistant tuberculosis. Clin Infect Dis. 2018;67:S293–302.
Mase SR, Jereb JA, Gonzalez D, Martin F, Daley CL, Fred D, et al. Pharmacokinetics and dosing of levofloxacin in children treated for active or latent multidrug-resistant tuberculosis, Federated States of Micronesia and Republic of the Marshall Islands. Pediatr Infect Dis J. 2016;35(4):414–21.
Kumar AKH, Kumar A, Kannan T, Bhatia R, Agarwal D, Kumar S, et al. Pharmacokinetics of second-line antituberculosis drugs in children with multidrug-resistant tuberculosis in India. Antimicrob Agents Chemother. 2018;62(5):e02410–7.
Garcia-Prats AJ, Purchase SE, Osman M, Draper HR, Schaaf HS, Wiesner L, et al. Pharmacokinetics, safety, and dosing of novel pediatric levofloxacin dispersible tablets in children with multidrug-resistant tuberculosis exposure. Antimicrob Agents Chemother. 2019;63(4):e01865-18.
Chang MJ, Jin B, Chae J, Yun H, Kim ES, Lee YJ, et al. Population pharmacokinetics of moxifloxacin, cycloserine, p-aminosalicylic acid and kanamycin for the treatment of multi-drug-resistant tuberculosis. Int J Antimicrob Agents. 2017;49:677–87.
Ganatra SR, Udwadia ZF, Mullerpattan JB, Banka RA, Gandhare AR, Sadani MA, Rodrigues CS. High dose moxifloxacin for drug resistant tuberculosis; is 600 mg the optimal dose in high resistance settings? Am J Resp Crit Care Med. 2016;193:A4570.
Gumbo T, Louie A, Deziel MR, Parsons LM, Salfinger M, Drusano GL. Selection of a moxifloxacin dose that suppresses drug resistance in Mycobacterium tuberculosis, by use of an in vitro pharmacodynamic infection model and mathematical modeling. J Infect Dis. 2004;190:1642–51.
Zvada SP, Denti P, Sirgel FA, Chigutsa E, Hatherill M, Charalambous S, et al. Moxifloxacin population pharmacokinetics and model-based comparison of efficacy between moxifloxacin and ofloxacin in African patients. Antimicrob Agents Chemother. 2014;58:503–10.
Heinrichs MT, Vashakidze S, Nikolaishvili K, Sabulua I, Tukvadze N, Bablishvili N, et al. Moxifloxacin target site concentrations in patients with pulmonary TB utilizing microdialysis: a clinical pharmacokinetic study. J Antimicrob Chemother. 2018;73(2):477–83.
Diacon AH, Donald PR, Pym A, Grobusch M, Patientia RF, Mahanyele R, et al. Randomized pilot trial of eight weeks of bedaquiline (TMC207) treatment for multidrug-resistant tuberculosis: long-term outcome, tolerability, and effect on emergence of drug resistance. Antimicrob Agents Chemother. 2012;56:3271–6.
van Heeswijk RPG, Dannemann B, Hoetelmans RMW. Bedaquiline: a review of human pharmacokinetics and drug-drug interactions. J Antimicrob Chemother. 2014;69:2310–8.
Akkerman OW, Odish OF, Bolhuis MS, de Lange WC, Kremer HP, Luijckx GJ, et al. Pharmacokinetics of bedaquiline in cerebrospinal fluid and serum in multidrug-resistant tuberculous meningitis. Clin Infect Dis. 2016;62(4):523–4.
Svensson EM, Dosne AG, Karlsson MO. Population pharmacokinetics of bedaquiline and metabolite M2 in patients with drug-resistant tuberculosis: the effect of time-varying weight and albumin. CPT Pharmacometrics Syst Pharmacol. 2016;5(12):682–91.
McLeay SC, Vis P, van Heeswijk RPG, Green B. Population pharmacokinetics of bedaquiline (TMC207), a novel antituberculosis drug. Antimicrob Agents Chemother. 2014;58:5315–24.
Perrineau S, Lachâtre M, Lê MP, Rioux C, Loubet P, Fréchet-Jachym M, et al. Long-term plasma pharmacokinetics of bedaquiline for multidrug- and extensively drug-resistant tuberculosis. Int J Tuberc Lung Dis. 2019;23(1):99–104.
Svensson EM, Karlsson MO. Modelling of mycobacterial load reveals bedaquiline’s exposure-response relationship in patients with drug-resistant TB. J Antimicrob Chemother. 2017;72:3398–405.
Rouan M, Lounis N, Gevers T, Dillen L, Gilissen R, Raoof A, Andries K. Pharmacokinetics and pharmacodynamics of TMC207 and its N-desmethyl metabolite in a murine model of tuberculosis. Antimicrob Agents Chemother. 2012;56:1444–51.
Doan TN, Cao P, Emeto TI, McCaw JM, McBryde ES. Predicting the outcomes of new short-course regimens for multidrug-resistant tuberculosis using intrahost and pharmacokinetic-pharmacodynamic modeling. Antimicrob Agents Chemother. 2018;62(12):e01487-18.
Tsuyuguchi K, Sasaki Y, Mitarai S, Kurosawa K, Saito Y, Koh T, et al. Safety, efficacy, and pharmacokinetics of bedaquiline in Japanese patients with pulmonary multidrug-resistant tuberculosis: an interim analysis of an open-label, phase 2 study. Respir Investig. 2019;57(4):345–53.
Wasserman S, Meintjes G, Maartens G. Linezolid in the treatment of drug-resistant tuberculosis: the challenge of its narrow therapeutic index. Exp Rev Anti Infect Ther. 2016;14:901–15.
Garcia-Prats AJ, Schaaf HS, Draper HS, Garcia-Cremades M, Winckler J, Wiesner L, et al. Pharmacokinetics, optimal dosing, and safety of linezolid in children with multidrug-resistant tuberculosis: combined data from two prospective observational studies. PLoS Med. 2019;16(4):e1002789.
Song T, Lee M, Jeon HS, Park Y, Dodd LE, Dartois V, et al. Linezolid trough concentrations correlate with mitochondrial toxicity-related adverse events in the treatment of chronic extensively drug-resistant tuberculosis. EBioMedicine. 2015;2(11):1627–33.
Srivastava S, Magombedze G, Koeuth T, Sherman C, Pasipanodya JG, Raj P, et al. Linezolid dose that maximizes sterilizing effect while minimizing toxicity and resistance emergence for tuberculosis. Antimicrob Agents Chemother. 2017;61:e00751-17.
Bolhuis MS, Akkerman OW, Sturkenboom MGG, Ghimire S, Srivastava S, Gumbo T, Alffenaar JC. Linezolid-based regimens for multidrug-resistant tuberculosis (TB): a systematic review to establish or revise the current recommended dose for TB treatment. Clin Infect Dis. 2018;67:S327–35.
Brown AN, Drusano GL, Adams JR, Rodriquez JL, Jambunathan K, Baluya DL, et al. Preclinical evaluations to identify optimal linezolid regimens for tuberculosis therapy. mBio. 2015;6:e01741-15.
Alffenaar JC, van Altena R, Harmelink IM, Filguera P, Molenaar E, Wessels AMA, et al. Comparison of the pharmacokinetics of two dosage regimens of linezolid in multidrug-resistance and extensively drug-resistant tuberculosis patients. Clin Pharmacokinet. 2010;49:559–65.
Millard J, Pertinez H, Bonnett L, Hodel EM, Dartois V, Johnson JL, et al. Linezolid pharmacokinetics in MDR-TB: a systematic review, meta-analysis and Monte Carlo simulation. J Antimicrob Chemother. 2018;73:1755–62.
Kempker RR, Heinrichs MT, Nikolaishvili K, Sabulua I, Bablishvili N, Gogishvili S. A comparison of linezolid lung tissue concentrations among patients with drug-resistant tuberculosis. Eur Respir J. 2018;51(2):1702166.
Gopal M, Padayatchi N, Metcalfe JZ, O’Donnell MR. Systematic review of clofazimine for the treatment of drug-resistant tuberculosis. Int J Tuberc Lung Dis. 2013;17:1001–7.
Court R, Wiesner L, Stewart A, de Vries N, Harding J, Maartens G, et al. Steady state pharmacokinetics of cycloserine in patients on terizidone for multidrug-resistant tuberculosis. Int J Tuberc Lung Dis. 2018;22(1):30–3.
Deshpande D, Alffenaar JC, Koser CU, Dheda K, Chapagain ML, Simbar N, et al. D-Cycloserine pharmacokinetics/pharmacodynamics, susceptibility, and dosing implications in multidrug-resistant tuberculosis: a Faustian deal. Clin Infect Dis. 2018;67:S308–16.
Mulubwa M, Mugabo P. Analysis of terizidone in plasma using HPLC-UV method and its application in a pharmacokinetic study of patients with drug-resistant tuberculosis. Biomed Chromatogr. 2018;32:e4325.
Stinson K, Kurepina N, Venter A, Fujiwara M, Kawasaki M, Timm J, et al. MIC of delamanid (OPC-67683) against Mycobacterium tuberculosis clinical isolates and a proposed critical concentration. Antimicrob Agents Chemother. 2016;60:3316–22.
Gler MT, Skripconoka V, Sanchez-Garavito E, Xiao H, Cabrera-Rivero JL, Vargas-Vasquez DE, et al. Delamanid for multidrug-resistant pulmonary tuberculosis. N Engl J Med. 2012;366:2151–60.
Sturkenboom MGG, Simbar N, Akkerman OW, Ghimire S, Bolhuis MS, Alffenaar JC. Amikacin dosing for MDR tuberculosis: a systematic review to establish or revise the current recommended dose for tuberculosis treatment. Clin Infect Dis. 2018;67:S303–7.
Modongo C, Pasipanodya JG, Magazi BT, Srivastava S, Zetola NM, Williams SM, et al. Artificial intelligence and amikacin exposures predictive of outcomes in multidrug-resistant tuberculosis patients. Antimicrob Agents Chemother. 2016;60:5928–32.
Srivastava S, Modongo C, Siyambalapitiyage Dona CW, Pasipanodya JG, Deshpande D, Gumbo T. Amikacin optimal exposure targets in the hollow-fiber system model of tuberculosis. Antimicrob Agents Chemother. 2016;60:5922–7.
Modongo C, Pasipanodya JG, Zetola NM, Williams SM, Sirugo G, Gumbo T. Amikacin concentrations predictive of ototoxicity in multidrug-resistant tuberculosis patients. Antimicrob Agents Chemother. 2015;59(10):6337–43.
Ahmad N, Ajuha SD, Akkerman OW, Alffenaar JC, Anderson LF, Baghaei P, et al. Treatment correlates of successful outcomes in pulmonary multidrug-resistant tuberculosis: an individual patient data meta-analysis. Lancet. 2018;392:821–34.
Sotgiu G, D’Ambrosio L, Centis R, Tiberi S, Esposito S, Dore S, et al. Carbapenems to treat multidrug and extensively drug-resistant tuberculosis: a systematic review. Int J Mol Sci. 2016;17:373.
van Rijn SP, Zuur MA, Anthony R, Willfert B, van Altena R, Akkerman OW, et al. Evaluation of cabapenems for treatment of multi- and extensively drug-resistant Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2019;63:e01489-18.
van Rijn SP, van Altena R, Akkerman OW, van Soolingen D, van der Laan T, de Lange WCM. Pharmacokinetics of ertapenem in patients with multidrug-resistant tuberculosis. Eur Respir J. 2016;47:1229–34.
Chirehwa MT, McIlleron H, Rustomjee R, Mthiyane T, Onyebujoh P, Smith P, Denti P. Pharmacokinetics of pyrazinamide and optimal dosing regimens for drug-sensitive and -resistant tuberculosis. Antimicob Agents Chemother. 2017;61:e00490-17.
Mugabo P, Mulubwa M. Population pharmacokinetic modelling of pyrazinamide and pyrazinoic acid in patients with multi-drug resistant tuberculosis. Eur J Drug Metab Pharmacokinet. 2019;44(4):519–30.
Sy SK, de Kock L, Diacon AH, Werely CJ, Xia H, Rosenkranz B, et al. N-acetyltransferase genotypes and the pharmacokinetics and tolerability of para-aminosalicylic acid in patients with drug-resistant pulmonary tuberculosis. Antimicrob Agents Chemother. 2015;59(7):4129–38.
Deshpande D, Pasipanodya JG, Mpagama SG, Srivastava S, Bendet P, Koeuth T, et al. Ethionamide pharmacokinetics/pharmacodynamics-derived dose, the role of MICs in clinical outcome, and the resistance arrow of time in multidrug-resistant tuberculosis. Clin Infect Dis. 2018;67:S317–26.
Lee HW, Kim DW, Park JH, Kim S, Lim M, Phapale PB, et al. Pharmacokinetics of prothionamide in patients with multidrug resistant tuberculosis. Int J Tuberc Lung Dis. 2009;13:1161–6.
Mota L, Al-Efraij K, Campbell JR, Cook VJ, Marra F, Johnson J. Therapeutic drug monitoring in anti-tuberculosis treatment: a systematic review and meta-analysis. Int J Tuberulosis Lung Dis. 2016;20:819–26.
Wilby KJ, Ensom MH, Marra F. Review of evidence for measuring drug concentrations of first-line antitubercular agents in adults. Clin Pharmacokinet. 2014;53:873–90.
Nahid P, Dorman SE, Alipanah N, Barry PM, Brozek JL, Cattamanchi A, et al. Official American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America clinical practice guidelines: treatment of drug susceptible tuberculosis. Clin Infect Dis. 2016;63:e147–95.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was provided for this manuscript.
Conflict of interest
Dr. Kyle John Wilby and Mrs. Farhat Naz Hussain declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Wilby, K.J., Hussain, F.N. A Review of Clinical Pharmacokinetic and Pharmacodynamic Relationships and Clinical Implications for Drugs Used to Treat Multi-drug Resistant Tuberculosis. Eur J Drug Metab Pharmacokinet 45, 305–313 (2020). https://doi.org/10.1007/s13318-019-00604-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-019-00604-5