
Abstract
Background
HS1793, a novel analogue of resveratrol, was previously determined to be more potent at lower dosages by improving mitochondrial function and increased mitochondrial biogenesis-related proteins. In this study, we focused on targeting the mitochondria to address muscle wasting with HS-1793.
Method
Dosage screening was performed by evaluating for cytotoxicity and cell proliferation. Mitochondrial mass, mitochondrial membrane potential (Δψm), reactive oxygen species (ROS) level, and mitochondria biogenesis-regulated genes and proteins were analyzed to determine the effects on mitochondrial biogenesis.
Results
HS-1793 reduced ROS generation, but treatment did not interfere with cellular viability at low dosages. HS-1793 also regulated mitochondrial function by increasing cellular and mitochondrial ATP synthesis function, stabilizing Δψm and decreasing ROS. More importantly, these dysfunction in these parameters were ameliorated by HS-1793 in a simulated oxidative stress model with tBHP. We also observed increase in mitochondrial mass and upregulation in vital mitochondrial biogenesis-related gene PGC1-α as a response to HS-1793 treatment. Moreover, phosphorylation of AKT and mTOR proteins, which are considered as regulators of skeletal muscle function were also increased during the treatment. Finally, HS-1793 also demonstrated protective effects against cisplatin-induced skeletal muscle cell injury by increasing expression of mitochondrial biogenesis-relate markers.
Conclusion
Taken altogether, it shows the viability of HS-1793 as a compound that can restore mitochondrial function and render protection in skeletal muscle cells, especially during high oxidative stress levels.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mitochondrial dysfunction has been well attributed to skeletal muscle pathologies, brought about by changes in mitochondrial energetics or oxidative damage (Abrigo et al. 2019). Mitochondria not only appear susceptible to damage mediated by increased stress, but also play significant roles in the regulation of cell function (Ballinger 2005). Cachexia often accompanies skeletal muscle pathology, which increases patient morbidity and mortality. Since skeletal muscle wasting can precede cachexia, preventive strategies mostly focus on muscle mass preservation (Josiak et al. 2014).
Expansive research efforts aimed at bringing new therapeutic agents to address skeletal muscle diseases have produced a plethora of new compounds and drugs, including resveratrol, a naturally-occurring compound found in mulberries, grape, peanuts, pines (Jasinski et al. 2013; Alway 2017). However, high concentration of resveratrol is needed to be a more effective therapeutic agent as it has been found to have low cytotoxicity among different therapeutic agents (Cecchinato 2007). Due to this, more dynamic resveratrol analogs were examined that exhibited more efficiency as therapeutic agents (Um 2010). A newly developed novel analogue of resveratrol called HS-1793 has gained attention. HS-1793 has no unstable double bond, has a different aromatic ring position in two of the three hydroxyl groups, less photosensitive, and is more metabolically stable, all of which contributes to its increased potency (Jeong 2014a).
HS-1793 has been proven to be more potent than resveratrol as it exerts toxic effect in cancer cells (Kim 2014). It has exhibited negative effect against cell proliferation while alternately enhancing apoptosis. HS-1793 was able to inhibit expression of mitochondrial proteins that promote mitochondrial biogenesis that in turn, promote tumorigenesis (Jeong 2012). In addition, it has also been proven to have a protective effect in ischemia/reperfusion system (Jeong 2013). Thus, it is thought that through these, HS-1793 is a potential way of treating not only metabolic diseases but also possibly has a positive effect on muscle cells. This study then aimed to determine how HS-1793 works in relation to improved muscle function by targeting the mitochondria.
Materials and methods
Chemicals
HS-1793 (4-(6-hydroxy-2-naphthyl)-1,3-benzenediol) was kindly provided by Professor Hongsuk Suh, Pusan National University in Busan, South Korea. Cisplatin, tBHP (t-BuOOH), SNP (disodium nitroferricyanide), and Dox were obtained from Sigma-Aldrich. (St. Louis, MO, USA).
Cell culture
Mouse myoblast C2C12 cells (ATCC, Manassas, VA, USA) were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum, 50 U/mL penicillin, and 50 μg/mL streptomycin (Lonza, Walkersville, MD, USA).
Measurement of cell proliferation
Cell viability was measured via quantitative colorimetric assay using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (Sigma-Aldrich, St. Louis, MO, USA). MTT is converted into formazan crystals which are an indicator of mitochondrial activity in living cells. These were quantified by measuring the complex’s optical density at 570 nm using a microplate reader (Molecular Device, Sunnyvale, CA, USA).
Measurement of cytotoxicity
Cytotoxicity was evaluated via quantitative fluorescence assay using CellTox Green cytotoxicity assay (Promega, Madison, WI, USA). The change in membrane integrity that occurs as a result of cell death is measured in terms of fluorescence (excitation/emission = 485 nm/530 nm) using a microplate reader (Molecular Device, Sunnyvale, CA, USA).
Measurement of mitochondrial activity
Cells were incubated with 1 μM Mito-Sox for 20 min/10 µM CM-H2DCF-DA for 30 min at 37 °C for mitochondria ROS and with 5 µM TMRE for 30 min at 37 °C for membrane potential. The levels of the fluorescent probes were measured using a FACSCanto II flow cytometer (BD Biosciences), or with a multi-plate reader (Molecular Device, Sunnyvale, CA, USA).
Measurement of ATP
To measure the basal and mitochondrial ATP levels, we used the Mitochondrial ToxGlo Assay (Promega, Madison, WI) and followed the manufacturer’s protocol. Cells were separated by centrifugation at 200 g for 10 min, and 50 μL fresh medium containing 10 mM glucose (cellular ATP; glycolysis and mitochondrial ATP) or galactose (instead of glucose; only mitochondrial ATP) was added to each well. Plates were incubated at 37 °C in a humidified and CO2-supplemented incubator for 90 min. 100 μL assay solution was added to each well, and plates were then incubated at room temperature for 30 min. Luminescence was measured using a luminometer (Molecular Device, Sunnyvale, CA, USA).
Measurement of mitochondrial mass
To measure the mitochondrial mass, cells were stained with acridine orange 10-nonyl bromide at a final concentration of 2.5 μM in phosphate buffered saline (PBS) (NAO; Invitrogen, Carlsbad, CA, USA). NAO fluorescence for each group was measured in laser confocal microscope (LSM 700, Carl-Zeiss, Oberkochen, Germany). The cells were excited using 488 nm and emission of NAO was measured beyond 585 nm. The mean intensity of the region of interest (ROI) was measured in each cell and analyzed by ZEN2009 software (Carl-Zeiss, Oberkochen, Germany).
To confirm the mitochondrial mass, cells were in 60 mm cell culture plate. After 16 h, the cells were treated with 0, 5, or 10 μM of HS-1793 for 24 h. The cells were stained with 2.5 μM NAO for 30 min at 37 °C. After washing twice with PBS, relative signal intensity of NAO in cells was analyzed using a multi-plate reader (Molecular Device, Sunnyvale, CA, USA).
Western blot
Cell lysates were centrifuged at 14,000 rpm for 15 min at 4 °C. Protein concentrations were determined by Bradford protein assay (Bio-Rad, Hercules, CA, USA), and 30 μg of protein was loaded per lane onto 10% SDS polyacrylamide gels. Gels were transferred onto nitrocellulose membranes (Whatman, Freiburg, Germany) and incubated with specific antibodies PGC-1α (ABCam, Cambridge, MA, CA, USA), PARP, SOD2, mTOR, phosphor-mTOR, AKT, phosphor-AKT, and beta-tubulin (Cell Signaling, Danvers, MA, USA). Western blot analysis was performed using these antibodies and a detection kit, Ab signal™ (AbClon, Seoul, Korea).
Data analysis
All experiments were performed in triplicate unless stated otherwise. Data are presented as means ± standard error of the mean (SEM). Student’s t test and one-way ANOVA was used to compare values between groups wherever applicable, and p ≤ 0.05 was considered significant.
Results
HS-1793 attenuated tBHP-induced mitochondrial dysfunction
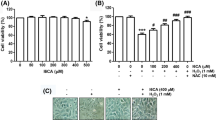
We first confirmed for the effect of HS-1793 at various dosages. HS-1793 greatly affected cell toxicity at high dosages while doses lower than 10 μM did not affect cell viability in C2C12 cells (Fig. 1a). Since lower dosages did not induce toxic effects, 5 and 10 μM were selected and used in the succeeding experiments.

HS-1793 is non-toxic at low dosages and improves mitochondrial function of C2C12 cells. a Cytotoxicity assay shows HS-1793 is highly toxic to C2C12 cells at doses ≤ 50 μM, but less toxic at dosages lower than 10 μM. b ATP assay shows HS-1793 increased cellular and mitochondrial ATP in a dose-dependent manner even in the presence of cardiotoxic tBHP compound. c High ROS production during tBHP treatment was decreased during HS-1793 treatment. d Mitochondrial membrane potential decreased in tBHP-treated cells and recovered upon HS-1793 treatment. All experiments were performed in triplicates. *p < 0.05
We then investigated whether HS-1793 influences energy production and mitochondrial function. Dose-dependent HS-1793 treatment increased cellular and mitochondrial ATP production. Moreover, even with the presence of cardiotoxic tBHP, higher ATP production was observed which can denote a protective effect on the mitochondria (Fig. 1b). HS-1793 reduced cellular reactive oxygen species (ROS), especially in tBHP-treated group (Fig. 1c). Despite observing a decrease in mitochondrial membrane potential (Δψm) during single treatment at 10 μM, HS-1793 treatment increased Δψm which was lowered upon introduction of tBHP (Fig. 1d). These results suggest that HS-1793 has the ability to improve cellular and mitochondrial ATP synthesis function via increased mitochondrial contents stress by preventing ROS generation and mitochondrial membrane potential depolarization. Overall, HS-1793 does not interfere with cell viability and does not contribute to mitochondrial dysfunction in mouse myoblast C2C12 cells at low dosages.
HS-1793 enhanced mitochondrial function and biogenesis marker expression and related pathway
Next, we investigated whether the observed increase in mitochondrial function was due to increased mitochondrial contents. We measured mitochondrial contents using 10-nonylacridine orange (NAO) staining and confocal microscopy. We found that dose-dependent HS-1793 treatment significantly increased mitochondrial mass in a dose-dependent manner (Fig. 2a, b). This result was confirmed by additional NAO measurement using spectrophotometry (Fig. 2c). We then examined for mitochondrial biogenesis-regulated gene expression levels for further analysis of mitochondrial biogenesis. As described in previous studies, mitochondrial biogenesis is regulated by PGC-1α (Fernandez-Marcos and Auwerx 2011). We observed PGC-1α mRNA expression was increased by HS-1793, which was parallel to the increase in PGC-1α protein expression. However, related transcription factors TFAM and NRF1 were not changed (Fig. 3a, b).

HS-1793 increased mitochondrial contents. a Respective confocal images and b relative fluorescence intensity of NAO-stained C2c12 cells. c Quantitative analysis of NAO-stained C2C12 using spectrophotometry shows HS-1793 treatment increased mitochondrial contents in a dose-dependent manner. All experiments were performed in triplicates. *p < 0.05

HS-1793 increased mitochondrial biogenesis regulator PGC-1α but not its transcription factors TFAM and NRF1. a HS-1793 increased expression of PGC-1α mRNA and b PGC-1α protein expression. qPCR was done in triplicates. Values below images indicate relative intensity measurements of PGC-1α to β-tubulin. *p < 0.05
Active forms of mammalian target of rapamycin (mTOR) and protein kinase B (AKT), phosphor-mTOR and phosphor-AKT respectively, also affect skeletal muscle protein expression (Ogasawara 2016). Western blot analysis was performed to determine whether AKT /mTOR signaling is activated during HS-1793 treatment. Treatment of HS-1793 increased both mTOR (by as much as ~ 1.5-fold increase) and AKT phosphorylation (by as much ~ 2.2-fold increase) (Fig. 4).

Activation of AKT/mTOR signaling pathway in HS-1793 treatment. HS-1793 affected mTOR phopshorylation and increased expression of p-AKT were observed. Values below images indicate relative intensity measurements of p-AKT and p-mTOR to AKT and MTOR, respectively
HS-1793 attenuated cisplatin-induced oxidative stress
Cisplatin, a known chemotherapeutic drug, is also known to have muscle wasting side effects (Coletti 2018). We tested whether treatment of HS-1793 will ameliorate effects of cisplatin-induced oxidative stress. Since the effect of HS-1793 treatment was observed even at 5 μM in our previous experiments, we used 5 μM in this particular experiment. There was a notable decrease in cell death expression marker PARP during the treatment of HS-1793, as compared to cisplatin-only treatment. Interestingly, this decrease was accompanied by increased expression of mitochondrial biogenesis marker PGC-1α and oxidative stress marker in mitochondria SOD2 (Fig. 5a, b). These results strengthen earlier claims of the possibility of the ameliorative effect of HS-1793 in oxidative stress-induced muscle wasting through the regulation of mitochondria bioenergetics and biogenesis.

HS-1793 ameliorates cisplatin-induced oxidative stress. a Cisplatin and b HS-1793 co-treatment with cancer drug cisplatin. HS-1793 decreased cell death as evidenced by decreased PARP and exhibited increases similar in PGC-1α, SOD2 ratio. Values below images indicate relative intensity measurements of PARP, PGC-1α, and SOD2 to b-tubulin
Discussion
Over the past two decades, researchers have studied the ability of pharmacological interventions to prevent or reverse the progression of diseases. Popular compounds such as resveratrol have shown great promise (Raj 2014), although more efficient compounds and drugs are being synthesized such as HS-1793, which also holds great potential for mitochondrial targeting. The effects conferred by HS-1793 might have a mitochondrial origin. HS-1793 was observed to induce murine breast cancer cell apoptosis via a mitochondrial pathway through caspase activation or contributions of apoptosis-inducing factor and endonuclease G (Kim 2012). In addition, HS-1793 was also observed to regulate mitochondrial biogenesis gene marker expression (Jeong 2014b). These previous reports indicate that cell survivability heavily relies on mitochondrial function regulation. We therefore proceeded to identify how HS-1793 regulates cell survival and mitochondrial function in a non-cancer cell line such as skeletal muscle cells.
Our current findings are the first to show HS-1793 is able to regulate mitochondrial function in C2C12 skeletal muscle cell lines by increasing ATP production, and stabilizing Δψm and ROS generation. HS-1793 was able to improve ATP production in simulated oxidative stress models, as well as recovering Δψm and lowering high levels of ROS. High levels of ROS cause functional oxidative damages to proteins, lipids, nucleic acids and cell components. These are all considered as etiological factors which contribute to sarcopenia, wasting, and related muscle diseases. With this, it may lower levels of ROS that can then activate signaling molecules such as PGC-1α that control cellular mechanisms for muscle adaptation (Irrcher et al. 2009). Additionally, maintaining high levels of PGC-1α under catabolic conditions was observed to protect muscle mass in pathological states such as heart failure and sarcopenia by inhibiting the transcriptional activity of FoxO3 and NF-κB, but not their protein synthesis (Egerman and Glass 2014). Moreover, PGC-1α can increase protein synthesis in C2C12 cells independent from the AKT/Akt/mTOR pathway is a crucial muscle mass and atrophy regulator in vivo (Chauhan et al. 2013; Bodine 2001).
Activated Akt phosphorylates proteins modulating various cellular processes. Pharmacologic approaches which activate Akt will be useful in regulating muscle synthesis (Lai 2004). Such as in the case of mTOR when targeted by AKT: its activation causes muscle cells to increase protein synthesis (Wang and Proud 2006). Atrophy and hypertrophy of skeletal muscle are associated with Ser2448 phosphorylation, suggesting its importance in protein synthesis (Reynolds et al. 2002). Ser2448 phosphorylation could be brought about by the phosphorylation of AKT at Ser473. In summary, the findings support the hypothesis that HS-1793 plays a pivotal role in regulating mitochondrial activity in in vitro skeletal muscle cells, resulting in mitochondrial biogenesis and muscle synthesis-related pathway activation.
Conclusion
The effects of HS-1793 in vitro were dose-dependent in alleviating mitochondrial dysfunction caused by exogenous oxidative stress. HS-1793 even at low concentrations demonstrated a significant effect in attenuating the detrimental impact of a high oxidative stress. Uncovering the underlying mechanism is a step forward in providing new ideas and strategies on how to address skeletal muscle pathologies through mitochondrial targeting.
References
Abrigo J, Simon F, Cabrera D, Vilos C, Cabello-Verrugio C (2019) Mitochondrial dysfunction in skeletal muscle pathologies. Curr Protein Pept Sci 20:536–546. https://doi.org/10.2174/1389203720666190402100902
Alway SE et al (2017) Resveratrol enhances exercise-induced cellular and functional adaptations of skeletal muscle in older men and women. J Gerontol A Biol Sci Med Sci 72:1595–1606. https://doi.org/10.1093/gerona/glx089
Ballinger SW (2005) Mitochondrial dysfunction in cardiovascular disease. Free Radic Biol Med 38:1278–1295. https://doi.org/10.1016/j.freeradbiomed.2005.02.014
Bodine SC et al (2001) Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol 3:1014–1019. https://doi.org/10.1038/ncb1101-1014
Cecchinato V et al (2007) Resveratrol-induced apoptosis in human T-cell acute lymphoblastic leukaemia MOLT-4 cells. Biochem Pharmacol 74:1568–1574. https://doi.org/10.1016/j.bcp.2007.08.001
Chauhan M, Punga T, Punga AR (2013) Muscle-specific regulation of the mTOR signaling pathway in MuSK antibody seropositive (MuSK+) experimental autoimmune Myasthenia gravis (EAMG). Neurosci Res 77:102–109. https://doi.org/10.1016/j.neures.2013.07.008
Coletti D (2018) Chemotherapy-induced muscle wasting: an update. Eur J Transl Myol 28:7587. https://doi.org/10.4081/ejtm.2018.7587
Egerman MA, Glass DJ (2014) Signaling pathways controlling skeletal muscle mass. Crit Rev Biochem Mol Biol 49:59–68. https://doi.org/10.3109/10409238.2013.857291
Fernandez-Marcos PJ, Auwerx J (2011) Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr 93:884S–890. https://doi.org/10.3945/ajcn.110.001917
Irrcher I, Ljubicic V, Hood DA (2009) Interactions between ROS and AMP kinase activity in the regulation of PGC-1alpha transcription in skeletal muscle cells. Am J Physiol Cell Physiol 296:C116–123. https://doi.org/10.1152/ajpcell.00267.2007
Jasinski M, Jasinska L, Ogrodowczyk M (2013) Resveratrol in prostate diseases—a short review. Cent Eur J Urol 66:144–149. https://doi.org/10.5173/ceju.2013.02.art8
Jeong SH et al (2012) An analogue of resveratrol HS-1793 exhibits anticancer activity against MCF-7 cells via inhibition of mitochondrial biogenesis gene expression. Mol Cells 34:357–365. https://doi.org/10.1007/s10059-012-0081-7
Jeong SH et al (2013) HS-1793, a recently developed resveratrol analogue protects rat heart against hypoxia/reoxygenation injury via attenuating mitochondrial damage. Bioorg Med Chem Lett 23:4225–4229. https://doi.org/10.1016/j.bmcl.2013.05.010
Jeong MH et al (2014a) In vitro genotoxicity assessment of a novel resveratrol analogue, HS-1793. Toxicol Res 30:211–220. https://doi.org/10.5487/TR.2014.30.3.211
Jeong MH et al (2014b) Protective activity of a novel resveratrol analogue, HS-1793, against DNA damage in 137Cs-irradiated CHO-K1 cells. J Radiat Res (Tokyo) 55:464–475. https://doi.org/10.1093/jrr/rrt140
Josiak K, Jankowska EA, Piepoli MF, Banasiak W, Ponikowski P (2014) Skeletal myopathy in patients with chronic heart failure: significance of anabolic-androgenic hormones. J Cachexia Arcopenia Muscle 5:287–296. https://doi.org/10.1007/s13539-014-0152-z
Kim HJ et al (2012) The novel resveratrol analogue HS-1793 induces apoptosis via the mitochondrial pathway in murine breast cancer cells. Int J Oncol 41:1628–1634. https://doi.org/10.3892/ijo.2012.1615
Kim JA et al (2014) HS-1793, a resveratrol analogue, induces cell cycle arrest and apoptotic cell death in human breast cancer cells. Int J Oncol 44:473–480. https://doi.org/10.3892/ijo.2013.2207
Lai KM et al (2004) Conditional activation of akt in adult skeletal muscle induces rapid hypertrophy. Mol Cell Biol 24:9295–9304. https://doi.org/10.1128/MCB.24.21.9295-9304.2004
Ogasawara R et al (2016) The role of mTOR signalling in the regulation of skeletal muscle mass in a rodent model of resistance exercise. Sci Rep 6:31142. https://doi.org/10.1038/srep31142
Raj P et al (2014) Potential of resveratrol in the treatment of heart failure. Life Sci 95:63–71. https://doi.org/10.1016/j.lfs.2013.12.011
Reynolds THT, Bodine SC, Lawrence JC Jr (2002) Control of Ser2448 phosphorylation in the mammalian target of rapamycin by insulin and skeletal muscle load. J Biol Chem 277:17657–17662. https://doi.org/10.1074/jbc.M201142200
Um HJ et al (2010) Differential effects of resveratrol and novel resveratrol derivative, HS-1793, on endoplasmic reticulum stress-mediated apoptosis and Akt inactivation. Int J Oncol 36:1007–1013. https://doi.org/10.3892/ijo_00000581
Wang X, Proud CG (2006) The mTOR pathway in the control of protein synthesis. Physiology 21:362–369. https://doi.org/10.1152/physiol.00024.2006
Acknowledgements
This work was supported by the Ministry of Education of Korea (2010-0020224), and by the Korea Government Ministry of Science and ICT (2018R1A2A3074998).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Human and animal rights
The article does not contain any studies with human and animal and this study was performed following institutional and national guidelines.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Marquez, J., Park, N., Garcia, M.V.F. et al. HS-1793 protects C2C12 cells from oxidative stress via mitochondrial function regulation. Mol. Cell. Toxicol. 16, 359–365 (2020). https://doi.org/10.1007/s13273-020-00090-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13273-020-00090-w