
Abstract
Background
Indole-6-carboxaldehyde (I6CA), a natural indole derivative derived from the brown algae Sargassum thunbergii (Mertens) Kuntze, is known to have several pharmacological activities. However, the antioxidant effects of I6CA have not been identified.
Objective
The study aimed to investigate the protective effect of I6CA and its underlying mechanism against oxidative stress-induced damage in C2C12 mouse skeletal myoblasts.
Results
The findings revealed that pretreatment with I6CA protected hydrogen peroxide (H2O2)-induced cytotoxicity and DNA damage through blockage of intracellular reactive oxygen species (ROS) generation. I6CA also significantly suppressed C2C12 cells against H2O2-induced apoptosis by preventing loss of mitochondrial membrane potential and cytosolic release of cytochrome c, decreasing the rate of Bax/Bcl-2 expression and reducing the activity of caspases. In addition, I6CA markedly attenuated the decrease in ATP content induced by H2O2 and restored H2O2-induced activation of AMP-activated protein kinase (AMPK). However, the cytoprotective effects of I6CA against H2O2 were eliminated by compound C, a specific AMPK signaling blocker.
Conclusion
The current results indicate that I6CA was able to protect C2C12 myoblast DNA damage and apoptosis from oxidative stress by at least preserving mitochondrial homeostasis mediated through the ROS-AMPK signaling pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Muscles require a large amount of oxygen due to the high energy demand for contractile activity and are one of the most vulnerable organs for oxidative stress, characterized by the overproduction of reactive oxygen species (ROS). Myoblasts are embryonic precursors of muscle cells produced by tissue-resident stem cells called satellite or muscle stem cells. They differentiate into muscle cells through myogenesis, a process that is fused to multi-nucleated myotubes (Sambasivan and Tajbakhsh 2015; Chang and Rudnicki 2014). The role of ROS in myogenic differentiation is complicated by a wide range of cellular responses depending on the level of ROS. At an appropriate level, ROS regulate many cellular signaling pathways, including myogenic differentiation and biosynthetic metabolism, but high levels of ROS are closely related to impaired myogenesis (Sandiford et al. 2014; Sestili et al. 2009; Hansen et al. 2007). In addition, excess ROS in myoblasts, as well as in many other cell types, could increase chromosomal aberration and DNA strand breakage, leading to DNA damage and apoptosis (Santa-Gonzalez et al. 2016; del Río et al. 2006; Caporossi et al. 2003). These observations indicate that ROS levels should be tightly regulated, because high levels of ROS contribute to the loss of myoblast function, promotion of myoblast death and further exacerbating muscle function.
Indole and its derivatives having an aromatic heterocyclic structure are biologically active molecules formed from naturally occurring glucosinolates. These compounds are found not only in cruciferous vegetables, but also in a number of natural resources, including actinomycetes, fungi, algae and marine sponges (Prieto et al. 2019; Sánchez-Pujante et al. 2017). Many studies have shown that indole derivatives have received considerable attention due to their potential pharmacological properties, including anti-inflammatory (Garg et al. 2019; Ampofo et al. 2018), antioxidant (Wang et al. 2019, 2020; Garg et al. 2019; Hasan et al. 2018), and anti-cancer activities (Wan et al. 2019; Sidhu et al. 2015) as well as their inhibitory effects against various cardiovascular and metabolic diseases (Hendrikx and Schnabl 2019; Zhang et al. 2018; Konopelski and Ufnal 2018). In particular, several previous studies have shown that indole derivatives can activate antioxidant defense systems and improve damage against oxidative stress (Wei et al. 2019; Hajra et al. 2013, 2018). However, whether they can weaken oxidative stress-induced injury in myoblasts has not been well studied. Recently, in the process of exploring novel indole derivatives with anti-obesity effects, Kang et al. (2017) isolated indole‑6‑carboxaldehyde (I6CA) from the marine brown algae Sargassum thunbergii (Mertens) Kuntze and found that I6CA was effective in suppressing adipogenesis. In addition, Kim et al. (2019) reported that I6CA suppressed tumor invasion and metastasis. However, to date, there is a lack of evidence to describe the underlying mechanisms and limited information available about whether I6CA can reduce oxidative stress-induced damage. Therefore, in this study, the antioxidant potential of I6CA against oxidative stress (H2O2)-induced cytotoxicity in immortalized mouse myoblast C2C12 cells was evaluated.
Materials and methods
Reagents and antibodies
Dulbecco's modified Eagle’s medium (DMEM), fetal calf serum (FCS), antibiotics mixtures and other cell culture reagents were obtained from WelGENE Inc. (Gyeongsan, Republic of Korea). I6CA, H2O2, N-acetyl-l-cysteine (NAC), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetra-zolium bromide (MTT), DNase-free RNase A, ethidium bromide (EtBr), 4,6-diamidino-2-phenylindole (DAPI), 5,5′,6,6′-tetrachloro-1,1′3,3′-tetraethyl-imidacarbocyanune iodide (JC-1), compound C and AICAR were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). 2′,7′-Dichlorofluorescein diacetate (DCF-DA) and a firefly luciferase-based ATP Bioluminescence assay kit were obtained from Molecular Probes (Eugene, OR, USA) and Roche Applied Science (Indianapolis, IN, USA), respectively. MitoSOX™ red dye was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit and colorimetric activity assay kits were purchased from R&D Systems Inc. (Minneapolis, MN, USA). Bradford assay reagent and the mitochondria and cytoplasmic protein extraction kit were obtained from Bio-Rad Laboratories (Hercules, CA, USA) and Active Motif, Inc. (Carlsbad, CA, USA), respectively. Polyvinylidene difluoride (PVDF) membranes were purchased from Merck Millipore (Bedford, MA, USA). HT 8-oxo-dG enzyme-linked immunosorbent assay (ELISA) Kit II and Genomic DNA purification kit were supplied by Trevigen (Gaithersburg, MD, USA) and Promega Corporation (Madison, WI, USA), respectively. Primary antibodies were purchased from Abcam, Inc. (Cambridge, MA, UK), Cell Signaling Technology (Danvers, MA, USA), and Santa Cruz Biotechnology, Inc. Horseradish peroxidase (HRP)-conjugated secondary antibodies and enhanced chemiluminescence (ECL) detection system were obtained from Amersham Life Science (Arlington Heights, IL, USA). All other chemicals not specifically cited here were supplied by Sigma-Aldrich Chemical Co.
Cell culture and MTT assay
C2C12 cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in DMEM containing 10% heat-inactivated FCS and antibiotics mixture in a water-saturated humidified incubator at 5% CO2 and 37 °C. I6CA was dissolved in dimethyl sulfoxide (DMSO), and diluted with cell culture medium to adjust the final treatment concentrations before use in experiments. To measure cell viability using an MTT assay as described by Hasan et al. (2019), C2C12 cells were treated with the indicated concentrations of I6CA for 24 h or pretreated with I6CA, NAC or compound C for 1 h and then incubated with or without H2O2 for 24 h. Then, MTT solution was added to a final concentration of 0.5 mg/ml and incubated at 37 °C for 3 h. At the end of the incubation, the culture supernatants were discarded, and the formed formazan crystals were dissolved in DMSO. Finally, the optical density values were acquired with an ELISA reader (Dynatech Laboratories, Chantilly, VA, USA) at 450 nm. The optical density of the formazan crystals formed in untreated control cells was used to represent 100% viability. In a parallel experiment, changes in cell images were captured by a phase-contrast microscope (Carl Zeiss, Oberkochen, Germany).
Measurement of ROS generation
The production of intracellular and mitochondrial ROS was evaluated using DCF-DA and MitoSOX™ red dye, respectively. To measure the amount of ROS generated in cells, cells were treated with or without I6CA or NAC for 1 h, before another 1 h culture with the addition of H2O2. The cells were washed with phosphate-buffered saline (PBS), and lysed with PBS containing 1% Triton X-100 for 10 min at 37 °C. The cells were then stained with 10 µM DCF-DA or 1 μM MitoSOX™ red for 30 min at room temperature (RT) in the dark, and washed with PBS. Intracellular ROS generation was immediately recorded at 515 nm by a flow cytometer (Becton Dickinson, San Jose, CA, USA). The results were expressed as the percentage increase relative to untreated cells (Yoon et al. 2019). It was also analyzed the levels in ROS by fluorescence microscopy. Stained cells were washed twice with PBS and observed with a fluorescence microscope (Carl Zeiss).
Comet assay
A comet assay was used to analyze the migration of the DNA from individual cells in the gel as previously described (Aristizabal-Pachon and Castillo 2019). After the cells were exposed to H2O2 with or without I6CA, the cells were suspended in 0.5% low melting point agarose (LMA) at 37 °C, aliquoted and then spread onto a fully frosted microscope slide precoated with 1% normal melting agarose. After the agarose solidified in the dark, the slide was covered with 0.5% LMA and submerged in lysis solution for 1 h at 4 °C. The slides were then incubated in a gel electrophoresis device containing 300 mM NaOH and 10 mM Na-EDTA (pH 13) for 30 min and then were subjected to electrophoresis for 30 min at 300 mA for 20 min to draw negatively charged DNA toward the anode. When electrophoresis was complete, the slides were washed with neutralizing buffer (0.4 M Tris, pH 7.5) at 4 °C and stained with 20 µg/ml propidium iodide (PI). The nuclear images were visualized and captured using a fluorescence microscope.
Western blot analysis
At the end of the treatment period, the cells were collected and lysed on ice for 30 min in lysis buffer as previously described (Park et al. 2020). The mitochondrial and cytoplasmic protein fractions were obtained using a commercial mitochondrial fractionation kit according to the manufacturer’s instructions. The protein concentration of the collected supernatants was measured using the Bradford assay reagent according to the manufacturer’s protocol. Subsequently, the same amount of protein from each sample was separated by sodium-dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis and transferred to PVDF membranes. The membranes were blocked with Tris-buffered saline (10 mM Tris–Cl, pH 7.4) containing 5% skim milk and 0.5% Tween-20 for 1 h at RT and then incubated over-night at 4 °C with primary antibodies. After washing with PBS, the membranes were incubated with the appropriate HRP-conjugated secondary antibodies for 2 h at RT. The protein bands were detected using an ECL detection system, and the signals were visualized using a chemiluminescence imager (Azure Biosystems, Inc., Dublin, CA, USA).
Determination of 8-hydroxy-2′-deoxyguanosine (8-OHdG) concentration
After experimental treatment, the levels of intracellular 8-OHdG were quantitated using the 8-OHdG ELISA kit II, according to the manufacturer’s protocol. In brief, DNA was extracted using the Genomic DNA purification kit and was quantitated, and the DNA concentration of each sample was adjusted to a final concentration of 200 µg/ml. Then, the DNA was digested by DNase I and alkaline phosphatase sequentially for 1 h at 37 °C. To determine the levels of 8-OHdG in the culture supernatants, the cell culture medium was clarified by centrifugation of the cell debris, and the amount of 8-OHdG was measured at 450 nm using an ELISA plate reader based on the manufacturer's instructions. Subsequently, the concentration of 8-OHdG for each sample was quantified from the standard curve.
Nuclear staining
To determine apoptosis, changes in nuclear morphology were examined using DAPI staining. Briefly, cells were harvested after treatment with H2O2 in the absence or presence of I6CA, washed with PBS, and fixed with 3.7% paraformaldehyde in PBS for 10 min at RT. The cells were washed with PBS again and stained with 2.5 µg/ml DAPI solution for 10 min at RT. The cells were observed via a fluorescence microscope (Carl Zeiss).
Detection of apoptosis by annexin V staining
The degree of apoptosis was detected using an annexin V-FITC apoptosis detection kit according to the manufacturer’s instructions. In brief, after treatment with H2O2 in the absence or presence of I6CA, the cells were washed with cold PBS, fixed in 75% ethanol at 4 °C for 30 min and then stained with annexin V-FITC and propidium iodide (PI) for 20 min at RT in the dark. Using a flow cytometer, the fluorescence intensities of the cells were quantified as percentages of annexin V-positive and PI-negative (annexin V+/PI−) cells in the total cell populations as indicators of apoptotic cells, whereas the V−/PI− cells in the total cell population was considered normal (Zhang et al. 2020).
DNA fragmentation assay
Following treatment, cells were washed with PBS and lysed in a lysis buffer followed by the addition of RNase A (0.5 µg/ml) for an additional 1 h at 37 °C as previously described (Park et al. 2019). Subsequently, nuclear DNA in the supernatant was extracted with an equal volume of neutral phenol–chloroform–isoamyl alcohol mixture (25:24:1) and mixed with DNA loading buffer. DNA samples were analyzed by electrophoresis on a 1.5% agarose gel for 5 h at 60 V. The gels were stained with EtBr (0.1 µg/ml) to show the DNA fragments and photographed using a gel documentation system (VilberLourmat, Torcy, France).
Measurement of mitochondrial membrane potential (MMP, Δψm)
Mitochondrial function was determined by membrane potential-specific fluorescence staining using a sensor of mitochondrial potential, JC-1. Briefly, after treatment with H2O2 in the absence or presence of I6CA or NAC, the cells were trypsinized. The collected cell pellets were suspended in PBS and incubated with 10 µM JC-1 at 37 °C for 20 min. The cells were then washed with cold PBS and analyzed using a flow cytometer. We also analyzed the changes in the MMP by fluorescence microscopy. To this end, the cells cultured on glass cover slips were treated with H2O2 in the absence or presence of I6CA. After 24 h of treatment, the cells were incubated in a medium containing 10 µM JC-1 at 37 °C for 20 min. The stained cells were washed twice with PBS and observed with a fluorescence microscope.
Determination of caspase-3 and caspase-9 activity
The enzymatic activities of caspase-3 and caspase-9 were determined using colorimetric activity assay kits according to the manufacturer’s instructions. Briefly, the cells were incubated in a supplied lysis buffer on ice for 15 min. The supernatants were collected, and a total of 200 μg protein lysate was incubated with 5 μL of caspase-3 or caspase-9 substrates in the dark for 2 h at 37 °C, according to the kit protocol. The concentrations of p-nitroanilide released from the substrate by caspase-3 and caspase-9 were calculated from the absorbance values at 405 nm. According to their concentration curve, the results of at least three independent experiments were expressed as fold change, compared with the untreated control cells.
Determination of ATP levels
Intracellular ATP levels were measured by an ATP Bioluminescence assay according to the kit instructions. In brief, the cells were lysed with the lysis buffer provided in the kit and the supernatants were collected. An equal amount of supernatant and luciferaserere agent, which catalyzed the light production from ATP and luciferin, were mixed. The firefly luciferase activity using the emitted light was immediately measured using a luminometer (Turner Designs, Inc. San Jose, CA, USA) and ATP levels were determined according to the ATP standard curve. To compare intracellular ATP concentrations between treatment groups, ATP levels were expressed as nmole per mg cell protein.
Statistical analysis
The results were expressed as the mean ± standard deviation (SD) of at least three independent experiments. Statistical analyses were performed using the SPSS software, version 16.0 (SPSS Inc., Chicago, IL, USA). The statistical significance was analyzed by one-way ANOVA. A value of P < 0.05 was considered to indicate a statistically significant difference.
Results
I6CA inhibited H2O2-induced cytotoxicity in C2C12 cells
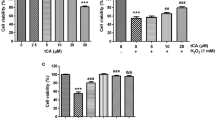
To investigate the protective effect of I6CA against oxidative stress using H2O2, the effect of I6CA on the survival rate of C2C12 cells was first investigated. As shown in Fig. 1a, there were no significant differences in cell viability in cells treated with less than 400 μM of I6CA compared with the control. However, the cell viability of C2C12 cells treated with 500 μΜ I6CA was slightly reduced. Therefore, to study the cytoprotective effects of I6CA against H2O2-induced oxidative stress, a concentration of I6CA was chosen to be less than 400 μM. Next, to achieve the optimized oxidative stress conditions, the concentration of H2O2 was selected as 1 mM, with a survival rate of about 60% (Fig. 1b). Subsequently, the ability of I6CA to counteract H2O2-induced cytotoxicity was investigated, and it was found that I6CA significantly inhibited the H2O2-mediated loss of C2C12 cell viability in a concentration-dependent manner (Fig. 1b). It was also found that pretreatment of NAC, a well-established ROS scavenger, had a complete inhibitory effect on H2O2-induced cytotoxicity when compared with the control (Fig. 1b). In addition, the morphological changes in the C2C12 cells treated with H2O2 alone were alleviated by pretreatment with I6CA (Fig. 1c).

Protective effect of I6CA on H2O2-induced cytotoxicity in C2C12 cells. The cells were treated with the various concentrations of I6CA for 24 h (a), or pretreated with or without the indicated concentrations of I6CA or 10 mM NAC for 1 h, and then cultured in the presence of 1 mM H2O2 for 24 h (b, c). a, b The cell viability was determined by an MTT reduction assay. The results are expressed as the mean ± SD obtained from three independent experiments (*p < 0.05, and ***p < 0.001 compared with the untreated cells; #p < 0.05 ###p < 0.01 and ###p < 0.001 compared with the H2O2-treated cells). c Representative images of the cells were captured by a phase-contrast microscope (original magnification, × 200)
I6CA suppressed H2O2-induced ROS generation in C2C12 cells
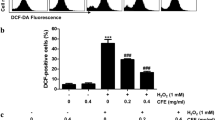
It was next examined whether I6CA abolished H2O2-induced ROS generation, and found that the generation of ROS in H2O2-treated C2C12 cells peaked within 1 h and gradually decreased over time (data not shown). However, pretreatment with I6CA significantly reduced the effect of H2O2 on ROS overproduction, and NAC as a ROS scavenger also almost completely eliminated the production of ROS (Fig. 2a, b). Similarly, I6CA markedly suppressed fluorescence intensity of both DCF-DA and MitoSox in H2O2-stimulated cells (Fig. 2c, d), which is demonstrating that I6CA has potent scavenging activity for ROS derived from mitochondria and intracellular.

Attenuation of H2O2-induced ROS generation by I6CA in C2C12 cells. The cells were pretreated with 400 μM I6CA or 10 mM NAC for 1 h and then stimulated with or without 1 mM H2O2 for an additional 1 h. The medium was removed, and the cells were incubated with medium containing 10 μM DCF-DA (a–c) or 1 μM MitoSOX™ red (d) for 30 min. a Intracellular ROS production was measured using a flow cytometer in DCF-DA stained cells, and representative profiles are shown. b The measurements were made in triplicate, and the values are expressed as the mean ± SD (***p < 0.001 compared with the untreated cells; ###p < 0.001 compared with the H2O2-treated cells). DCF (c) and MitoSOX™ red (d) fluorescence images of cells cultured under the same conditions were captured by a phase-contrast microscope. Each image is representative of at least three independent experiments
I6CA attenuated H2O2-induced DNA damage in C2C12 cells
To validate whether I6CA could protect H2O2-induced DNA damage was performed. As shown in Fig. 3a, increased comet tail moment (DNA migration) was observed in H2O2-treated cells, but not in cells treated with I6CA alone or control cells. However, pretreatment with I6CA clearly reduced the H2O2-induced the DNA migration to levels similar to those observed in the control cells. The DNA damage blocking effect of I6CA was also confirmed by analyzing the phosphorylation of γH2AX (p-γH2AX) and the production of 8-OHdG. As presented in Fig. 3b–d, although no significant change was observed in the total protein expression of γH2AX in the cells treated with H2O2 alone, the expression of p-γH2AX obviously increased, and higher levels of 8-OHdG were also observed in the H2O2-treated cells compared with the control cells. However, pretreatment with I6CA clearly attenuated the H2O2-induced phosphorylation of γH2AX and significantly suppressed the production of 8-OhdG, indicating that I6CA effectively blocked the DNA damage caused by oxidative stress.

Protection of H2O2-induced DNA damage by I6CA in C2C12 cells. The cells were treated with or without 400 μM I6CA for 1 h before treatment with 1 mM H2O2 for 24 h. a A comet assay was performed, and representative images were captured using a fluorescence microscope (original magnification, × 200). b The cell lysates were prepared, and p-γH2AX and γH2AX expression was identified by Western blot analysis. The equivalent loading of proteins in each well was confirmed by actin. c Bar graphs indicate the relative band density of the phosphorylated protein/non phosphorylated protein ration in western blot analysis. d The DNA samples of cells were subjected to assessment of the 8-OHdG levels. c, d The measurements were made in triplicate, and the results are expressed as the mean ± SD (***p < 0.001 compared to untreated cells; #p < 0.05 compared with the H2O2-treated cells)
I6CA reduced H2O2-induced apoptosis in C2C12 cells
To examine the ability of I6CA to prevent H2O2-induced apoptosis, DAPI staining, annexin V-FITC/PI double staining and DNA fragmentation assays were performed. As shown in Fig. 4a, the nuclei were round-shaped with homogeneous fluorescence intensity in the control group and I6CA alone treated group. H2O2-treated cells exhibited typical apoptotic morphology characterized by nuclear condensation and fragmentation, whereas cells pretreated with I6CA prior to H2O2 exposure had significantly decreased the apoptotic features. Consistent with DAPI staining, the results of flow cytometry analysis using annexin V/PI staining showed that H2O2 triggered a higher magnitude of apoptosis compared with controls; however, after supplementation with I6CA, the percentage of apoptotic cells significantly decreased (Fig. 4b, c). In addition, H2O2 alone notably increased DNA fragmentation as detected by agarose gel electrophoresis, which was markedly attenuated in cells pretreated with I6CA (Fig. 4d).

Inhibitory effect of I6CA on H2O2-induced apoptosis in C2C12 cells. The cells were treated with or without 200 or 400 μM I6CA for 1 h before treatment with 1 mM H2O2 for 24 h. a The cells were stained with DAPI solution and stained nuclei were observed using a fluorescence microscope (original magnification, × 200). Each image is representative of at least three independent experiments. b, c The cells were fixed and stained with annexin V-FITC and PI for flow cytometry analysis. b The results show early apoptosis, defined as annexin V+ and PI− cells (lower right quadrant), and late apoptosis, defined as annexin V+ and PI+ (upper right quadrant) cells, and representative profiles are shown. c The percentages of apoptotic cells were determined by expressing the numbers of annexin V+ cells as percentages of all the present cells. The data represent the mean ± SD of three independent experiments (***p < 0.001 compared with the untreated cells; ###p < 0.001 compared with the H2O2-treated cells). d DNA fragmentation was analyzed by extracting genomic DNA, electrophoresis in 1.5% agarose gel, and then visualizing by EtBr staining
I6CA protected H2O2-induced mitochondrial dysfunction in C2C12 cells
To analyze whether inhibition of mitochondrial impairment is a mechanism involved in the protective effect of I6CA, JC-1 dye was used to estimate the MMP. The JC-1 monomer ratio of C2C12 cells treated with H2O2 was higher than that of untreated control cells or cells treated with I6CA alone (Fig. 5a, b), indicating that H2O2 reduced the MMP. The ratio of red/green fluorescence was also significantly decreased after H2O2 treatment compared with untreated cells, but this reduction was reversed in cells pretreated with I6CA prior to H2O2 treatment (Fig. 5c).

Inhibition of H2O2-induced mitochondrial dysfunction by I6CA in C2C12 cells. The cells were treated with 400 μM I6CA or 10 mM NAC for 1 h and then exposed to 1 mM H2O2 for 24 h. a The cells were collected and stained with JC-1. The JC-1 fluorescence intensity was detected to evaluate the changes in the MMP using a flow cytometer. b The percentage of cells with JC-1 monomers is indicated by bars, and the data represent the mean ± SD of triplicate determinations (***p < 0.001 compared with the untreated cells; ###p < 0.001 compared with the H2O2-treated cells). c JC-1 fluorescence images of the cells treated with 1 mM H2O2 in the presence or absence of 400 μM I6CA are shown. Red fluorescence indicates high membrane potential, and green fluorescence represents low membrane potential. Representative images were captured using a fluorescence microscope (original magnification, × 400)
I6CA abolished the change in expression of apoptosis regulatory factors caused by H2O2 in C2C12 cells
We next investigated the effect of I6CA on H2O2-induced changes on apoptosis regulatory factors including cytochrome c, Bax, Bcl-2 and caspases by Western blotting. As shown in Fig. 6a–c, the expression of cytochrome c in the H2O2-treated cells increased in the cytoplasmic fraction and decreased in the mitochondrial fraction, indicating that cytochrome c was released from the mitochondria to the cytosol. It was also found that after H2O2 treatment, the protein expression of anti-apoptotic Bcl-2 in cells was remarkably decreased, while pro-apoptotic Bax expression was increased (Fig. 6d–f). In addition, the expression of pro-caspase-9 and pro-caspase-3 was markedly decreased in cells treated with H2O2 alone compared with the control group (Fig. 6d, g and h), and their activity was significantly increased in the results using the colorimetric assay kits (Fig. 6j, k). The degradation of poly (ADP-ribose) polymerase (PARP) was also observed in H2O2-treated cells. However, pretreatment with I6CA reversed these changes (Fig. 6d, i).

Effects of I6CA on the expression of apoptosis regulators in H2O2-treated C2C12 cells. The cells were treated with or without 400 μM I6CA for 1 h before treatment with 1 mM H2O2 for 24 h. a Cytochrome c levels were analyzed by Western blotting on mitochondrial and cytoplasmic fractions isolated from cells. Cytochrome oxidase subunit VI (COX IV) and actin serve as protein loading controls for the mitochondria and cytosol, respectively. d Whole cell lysates were prepared, and Bax, Bcl-2, caspase-9, caspase-3 and PARP expressions were identified by Western blot analysis. The equivalent loading of proteins in each well was confirmed by actin. b, c, e–i) Bar graphs indicate the relative band density in western blot. j, k The activities of caspase-9 (j) and caspase-3 (k) in cell lysates were measured using the respective substrate peptides. The measurements were made in triplicate. b, c, e–k The results are expressed as the mean ± SD (*p < 0.05, **p < 0.01 and ***p < 0.001 compared to untreated cells; #p < 0.05, ##p < 0.01 and ###p < 0.001 compared with the H2O2-treated cells)
AMP‑activated protein kinase (AMPK) was involved in the mitigation of H2O2-mediated cytotoxicity by I6CA in C2C12 cells
To investigate whether the protective effect of I6CA against H2O2-induced mitochondrial dysfunction was related to the improvement of cellular metabolic activity, the changes in intracellular ATP content were investigated. The results showed that C2C12 cells treated with H2O2 alone displayed a significant decrease in ATP production compared with the untreated control (Fig. 7a). However, pretreatment with I6CA was able to maintain ATP synthesis significantly, and although there was no statistical significance, the combination of I6CA and NAC was more effective than I6CA alone. We further examined whether the AMPK signaling pathway mediated the protective effect of I6CA against H2O2-induced reduction of ATP production. The levels of phosphorylated AMPK and its downstream effector, acetyl-CoA carboxylase (ACC), were greatly increased in the H2O2-stimulated cells compared with the control group without changing the expression of their total proteins (Fig. 6b–d). Since reduced ATP production can activate AMPK, the results are evidence that AMPK was activated in response to a decrease in ATP production in H2O2-treated cells. However, I6CA and NAC markedly attenuated their phosphorylation levels in cells cultured under oxidative stress conditions. Finally, we investigated the role of AMPK in the protective effect of I6CA against H2O2-induced cytotoxicity using the AMPK inhibitor compound C and the AMPK activator AICAR. As shown in Fig. 7e, compound C treatment significantly suppressed H2O2-induced cytotoxicity, and its protective effect is similar to that of I6CA. In addition, compound c substantially improved the effect of I6CA in the presence of H2O2. Meanwhile, AMPK activator AICAR markedly induced cytotoxicity in C2C12 cells, and the cell viability was more suppressed by combination treatment of H2O2 and AICAR. AICAR treatment significantly eliminated the protection tendency of I6CA against the H2O2-induced cytotoxicity compared to AICAR non-treated cells in the presence of H2O2 in I6CA pretreated cells. These results suggested that I6CA attenuated H2O2-induced cytotoxicity, which is mediated by down-regulation of AMPK signaling pathway.

Relevance of the AMPK signaling system in the inhibition of H2O2-induced cytotoxicity by I6CA in C2C12 cells. The cells were treated with 400 μM I6CA, 10 mM NAC or 5 μM compound C for 1 h, and then exposed to 1 mM H2O2 for 24 h. a The cells were collected and the cellular ATP concentrations were measured using an ATP determination kit. b Equal amounts of proteins were subjected to Western blot analysis of the listed proteins. Actin was used as an internal control. c, d Bar graphs indicate the relative band density of the phosphorylated protein/ non phosphorylated protein ration in western blot analysis. e The cells were treated with 400 μM I6CA, 1 mM AICAR or 5 μM compound C for 1 h, and then exposed to 1 mM H2O2 for 24 h. The cell viability was determined by MTT assay. a, c–e The results are expressed as the mean ± SD of three independent experiments (*p < 0.05, **p < 0.01 and ***p < 0.001 compared with the control group; #p < 0.05, ##p < 0.01 and ###p < 0.001 compared with the H2O2-treated group; &p < 0.05 and &&p < 0.01 compared with the I6CA and H2O2-treated group)
Discussion
In the present study, we investigated whether I6CA can protect C2C12 myoblasts from oxidative stress. For this purpose, we utilized H2O2, which is widely used as a representative ROS for establishing various oxidative stress models, to induce oxidative damage, and found that H2O2 reduced C2C12 cell viability by triggering DNA damage and apoptosis by promoting ROS production. However, I6CA has been found to have the ability to inhibit H2O2-induced cytotoxicity and provide ROS scavenging activity.
It is well known that caspase-dependent apoptosis can generally be divided into the extrinsic and intrinsic pathways. The extrinsic pathway is initiated by extracellular ligands that bind to death receptors on the cell surface, while activation of intrinsic pathway is associated with intracellular apoptotic signals that cause mitochondrial dysfunction (Bock and Tait 2020; Popgeorgiev et al. 2018). In particular, ROS overload by mitochondrial dysfunction causes free radical attack of the phospholipid bilayer of the mitochondria, which leads to the depolarization of the mitochondrial membrane, resulting in the loss of MMP (Bock and Tait 2020; Xiong et al. 2014). During this process, the permeability of the mitochondrial membranes increases, allowing apoptogenic factors in the mitochondrial intermembrane space, especially cytochrome c, to be released into the cytoplasm (Bock and Tait 2020; Er et al. 2006). Therefore, the loss of MMP and the release of mitochondrial cytochrome c are indicative of impaired mitochondrial function and are evident early phenomena in the onset of intrinsic apoptosis In this study, to evaluate the preventive effect of I6CA on mitochondrial dysfunction, the MMP values and cytochrome c expression were examined, and it was found that the loss of the MMP and cytosolic cytochrome c expression were markedly increased in H2O2-treated cells. However, I6CA pretreatment protected the reduction of the MMP induced by H2O2 and maintained the expression of cytochrome c in mitochondria during H2O2 exposure. The findings indicated that I6CA was able to reverse mitochondrial damage caused by oxidative stress.
Cytochrome c released into the cytoplasm interacts with and activates caspase-9, which in turn triggers activation of effector caspases, such as caspase-3 and caspase-7, eventually completing cell death. This process is accompanied by the degradation of substrate proteins of effector caspases, including PARP, as evidenced that caspase-dependent apoptosis was induced (Kiraz et al. 2016; Hassan et al. 2014). The activation of this caspase cascade is also tightly regulated by the expression of a variety of regulators. Among them, the Bcl-2 family proteins, which are consists of members that either promote or inhibit apoptosis, play an important role in regulating apoptosis by governing mitochondrial outer membrane permeabilization. Pro-apoptotic proteins such as Bax, located on the outer mitochondrial membrane, promote mitochondrial permeability transition or attenuate the barrier function of the mitochondrial outer membrane, leading to release of apoptotic factors. However, anti-apoptotic proteins such as Bcl-2 are essential for maintaining mitochondrial permeability and membrane barrier stabilization to inhibit the release of apoptotic factors (Bock and Tait 2020; Popgeorgiev et al. 2018). In the present study, H2O2 enhanced the expression level of Bax and reduced the level of Bcl-2, which indicates that modifications in Bcl-2 family protein expression contributed to the loss of MMP and resulted in the cytosolic release of cytochrome c. We also found that H2O2 activated caspase-9 and caspase-3, resulting in the degradation of PARP, a representative substrate protein degraded by activated effector caspases (Kiraz et al. 2016; Hassan et al. 2014), which are consistent with previous studies (Park et al. 2019; Choi 2018; Yin et al. 2015; Siu et al. 2009). However, these changes were markedly attenuated in the presence of I6CA, indicating that I6CA can protect C2C12 cells from apoptosis by inhibiting the mitochondrial-related apoptosis pathway activated by H2O2.
Mitochondria are essential organelles that produce most of the energy needed by cells in the form of ATP. Energy deficiency stimulates AMPK to facilitate muscle mitochondrial biosynthesis to produce more ATP, while simultaneously inhibiting multiple anabolic pathways (Kjøbsted et al. 2018; Hardie 2011). AMPK also blocks energy consumption pathways and activates ATP production pathways, including fatty acid β-oxidation and glycolysis through phosphorylation-mediated inhibition of ACC, leading to down-regulation of malonyl-CoA levels (Bullon et al. 2016; Hardie and Pan 2002). Therefore, AMPK is considered a key energy-sensing kinase that activates various catabolic processes while controlling the ratio of intracellular ATP to AMP (Herzig and Shaw 2018; Shirwany and Zou 2014). However, the role of AMPK under oxidative stress conditions in myoblasts is still unclear. Based on our finding that H2O2 diminished ATP production and activated AMPK in C2C12 myoblasts, we investigated whether the AMPK signaling pathway was involved in the protective effect of I6CA against oxidative stress. Our results demonstrated that I6CA was able to effectively improve the energy supply of cells by preserving mitochondrial homeostasis under oxidative conditions, which was associated with inhibition of phosphorylation of ACC as well as AMPK. Therefore, we used compound C, a well-known AMPK inhibitor, and found that the increased cell viability by pretreatment of I6CA in the presence of H2O2 was significantly up-regulated in compound C–co-treated cells. Meanwhile, AMPK activator AICAR markedly eliminated the protection tendency of I6CA against the H2O2-induced cytotoxicity compared to AICAR non-treated cells in the presence of H2O2 in I6CA pretreated cells. The results indicate that inactivation of AMPK signaling in C2C12 cells is, at least, involved as a protective mechanism for initiating an intrinsic apoptosis pathway following H2O2-mediated mitochondrial dysfunction. In this respect, our finding is especially meaningful as I6CA attenuated H2O2-induced cytotoxicity through by down-regulation of AMPK signaling pathway. These results provide evidence that I6CA may have high applicability as a therapeutic for maintaining myoblast function during oxidative stress. However, more detailed studies are needed on the role of other signaling pathways involved in the beneficial effects of I6CA upon inhibition of AMPK activation.
In conclusion, in the present study, we elucidated the protective effect of I6CA against H2O2-induced oxidative damage and explored its mechanism of action in C2C12 myoblasts. According to our results, I6CA significantly reversed the increased intracellular ROS production and mitochondrial damage caused by H2O2, eventually inhibiting DNA damage and apoptosis. In addition, I6CA weakened the activation of AMPK and abolished the reduction of ATP production by H2O2 (Fig. 8). Although this is the first study to demonstrate that I6CA can relieve H2O2-induced oxidative stress in skeletal muscle myoblasts, the underlying mechanisms involved in this process require further study.

Schematic summary for the action mechanism of I6CA on oxidative stress-induced cellular damages in C2C12 skeletal myoblasts. I6CA prevents H2O2-induced mitochondrial dysfunction, DNA damage and apoptosis by regulating the ROS-AMPK signaling pathway
References
Ampofo E, Schmitt BM, Menger MD, Laschke MW (2018) Targeting the microcirculation by indole-3-carbinol and its main derivate 3,3',-diindolylmethane: effects on angiogenesis, thrombosis and inflammation. Mini Rev Med Chem 18:962–968
Aristizabal-Pachon AF, Castillo WO (2019) Genotoxic evaluation of occupational exposure to antineoplastic drugs. Toxicol Res 36:29–36
Ates-Alagoz Z (2013) Antioxidant activities of retinoidal benzimidazole or indole derivatives in in vitro model systems. Curr Med Chem 20:4633–4639
Bock FJ, Tait SWG (2020) Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol 21:85–100
Bullon P, Marin-Aguilar F, Roman-Malo L (2016) AMPK/mitochondria in metabolic diseases. Exp Suppl 107:129–152
Caporossi D, Ciafrè SA, Pittaluga M, Savini I, Farace MG (2003) Cellular responses to H2O2 and bleomycin-induced oxidative stress in L6C5 rat myoblasts. Free Radic Biol Med 35:1355–1364
Chang NC, Rudnicki MA (2014) Satellite cells: the architects of skeletal muscle. Curr Top Dev Biol 107:161–181
Choi YH (2018) Schisandrin A prevents oxidative stress-induced DNA damage and apoptosis by attenuating ROS generation in C2C12 cells. Biomed Pharmacother 106:902–909
del Río LA, Sandalio LM, Corpas FJ, Palma JM, Barroso JB (2006) Reactive oxygen species and reactive nitrogen species in peroxisomes. Production, scavenging, and role in cell signaling. Plant Physiol 141:330–335
Er E, Oliver L, Cartron PF, Juin P, Manon S, Vallette FM (2006) Mitochondria as the target of the pro-apoptotic protein Bax. Biochim Biophys Acta 1757:1301–1311
Garg V, Maurya RK, Thanikachalam PV, Bansal G, Monga V (2019) An insight into the medicinal perspective of synthetic analogs of indole: a review. Eur J Med Chem 180:562–612
Hajra S, Patra AR, Basu A, Bhattacharya S (2018) Prevention of doxorubicin (DOX)-induced genotoxicity and cardiotoxicity: Effect of plant derived small molecule indole-3-carbinol (I3C) on oxidative stress and inflammation. Biomed Pharmacother 101:228–243
Hansen JM, Klass M, Harris C, Csete M (2007) A reducing redox environment promotes C2C12 myogenesis: implications for regeneration in aged muscle. Cell Biol Int 31:546–553
Hardie DG (2011) Sensing of energy and nutrients by AMP-activated protein kinase. Am J Clin Nutr 93:891S–896
Hardie DG, Pan DA (2002) Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem Soc Trans 30:1064–1070
Hasan H, Ismail H, El-Orfali Y, Khawaja G (2018) Therapeutic benefits of indole-3-carbinol in adjuvant-induced arthritis and its protective effect against methotrexate induced-hepatic toxicity. BMC Complement Altern Med 18:337
Hasan MM, Islam MS, Hoque KMF, Haque A, Reza MA (2019) Effect of Citrus macroptera fruit pulp juice on alteration of caspase pathway rendering anti-proliferative activity against Ehrlich's ascites carcinoma in mice. Toxicol Res 35:271–277
Hassan M, Watari H, AbuAlmaaty A, Ohba Y, Sakuragi N (2014) Apoptosis and molecular targeting therapy in cancer. Biomed Res Int 2014:150845
Hendrikx T, Schnabl B (2019) Indoles: metabolites produced by intestinal bacteria capable of controlling liver disease manifestation. J Intern Med 286:32–40
Herzig S, Shaw RJ (2018) AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 19:121–135
Kang MC, Ding Y, Kim EA, Choi YK, de Araujo T, Heo SJ, Lee SH (2017) Indole derivatives isolated from brown alga Sargassum thunbergii inhibit adipogenesis through AMPK activation in 3T3-L1 preadipocytes. Mar Drugs 15:119
Kim TH et al (2019) Indole-6-carboxaldehyde isolated from Sargassum thunbergii inhibits the expression and secretion of matrix metalloproteinase-9. Int J Mol Med 44:1979–1987
Kiraz Y, Adan A, KartalYandim M, Baran Y (2016) Major apoptotic mechanisms and genes involved in apoptosis. Tumour Biol 37:8471–8486
Kjøbsted R et al (2018) AMPK in skeletal muscle function and metabolism. FASEB J 32:1741–1777
Konopelski P, Ufnal M (2018) Indoles—Gut bacteria metabolites of tryptophan with pharmacotherapeutic potential. Curr Drug Metab 19:883–890
Park C et al (2019) Honokiol ameliorates oxidative stress-induced DNA damage and apoptosis of c2c12 myoblasts by ROS generation and mitochondrial pathway. Anim Cells Syst (Seoul) 24:60–68
Park S et al (2020) Myristoylated TMEM39AS41, a cell-permeable peptide, causes lung cancer cell death. Toxicol Res 36:123–130
Popgeorgiev N, Jabbour L, Gillet G (2018) Subcellular localization and dynamics of the Bcl-2 family of proteins. Front Cell Dev Biol 6:13
Prieto MA, López CJ, Simal-Gandara J (2019) Glucosinolates: molecular structure, breakdown, genetic, bioavailability, properties and healthy and adverse effects. Adv Food Nutr Res 90:305–350
Sambasivan R, Tajbakhsh S (2015) Adult skeletal muscle stem cells. Results Probl Cell Differ 56:191–213
Sánchez-Pujante PJ, Borja-Martínez M, Pedreño MÁ, Almagro L (2017) Biosynthesis and bioactivity of glucosinolates and their production in plant in vitro cultures. Planta 246:19–32
Sandiford SD, Kennedy KA, Xie X, Pickering JG, Li SS (2014) Dual oxidase maturation factor 1 (DUOXA1) overexpression increases reactive oxygen species production and inhibits murine muscle satellite cell differentiation. Cell Commun Signal 12:5
Santa-Gonzalez GA, Gomez-Molina A, Arcos-Burgos M, Meyer JN, Camargo M (2016) Distinctive adaptive response to repeated exposure to hydrogen peroxide associated with upregulation of DNA repair genes and cell cycle arrest. Redox Biol 9:124–133
Sestili P et al (2009) Creatine supplementation prevents the inhibition of myogenic differentiation in oxidatively injured C2C12 murine myoblasts. Mol Nutr Food Res 53:1187–1204
Shirwany NA, Zou MH (2014) AMPK: a cellular metabolic and redox sensor. A minireview. Front Biosci (Landmark Ed) 19:447–474
Sidhu JS, Singla R, Mayank JV (2015) Indole derivatives as anticancer agents for breast cancer therapy: a review. Anticancer Agents Med Chem 16:160–173
Siu PM, Wang Y, Alway SE (2009) Apoptotic signaling induced by H2O2-mediated oxidative stress in differentiated C2C12 myotubes. Life Sci 84:468–481
Wan Y, Li Y, Yan C, Yan M, Tang Z (2019) Indole: a privileged scaffold for the design of anti-cancer agents. Eur J Med Chem 183:111691
Wang N et al (2019) Structural modifications of nature-inspired indoloquinolines: a mini review of their potential antiproliferative activity. Molecules 24:E2121
Wang SY, Shi XC, Laborda P (2020) Indole-based melatonin analogues: synthetic approaches and biological activity. Eur J Med Chem 185:111847
Wei PC et al (2019) Neuroprotection of indole-derivative compound NC001-8 by the regulation of the NRF2 pathway in Parkinson's disease cell models. Oxid Med Cell Longev 2019:5074367
Xiong S, Mu T, Wang G, Jiang X (2014) Mitochondria-mediated apoptosis in mammals. Protein Cell 5:737–749
Yin Y et al (2015) Astragalus polysaccharide inhibits autophagy and apoptosis from peroxide-induced injury in C2C12 myoblasts. Cell BiochemBiophys 73:433–439
Yoon HJ, Chay KO, Yang SY (2019) Native low density lipoprotein increases the production of both nitric oxide and reactive oxygen species in the human umbilical vein endothelial cells. Genes Genom 41:373–379
Zhang Y, Li M, Li X, Zhang T, Qin M, Ren L (2018) Isoquinoline alkaloids and indole alkaloids attenuate aortic atherosclerosis in apolipoprotein E deficient mice: a systematic review and meta-analysis. Front Pharmacol 9:602
Zhang N, Li F, Gao J, Zhang S, Wang Q (2020) Osteopontin accelerates the development and metastasis of bladder cancer via activating JAK1/STAT1 pathway. Genes Genom 42:467–475
Acknowledgements
This research was a part of the project titled ‘Omics based on fishery disease control technology development and industrialization (20150242)’, funded by the Ministry of Oceans and Fisheries, Republic of Korea.
Author information
Authors and Affiliations
Contributions
CP, HL and SHP performed most of the experiments, analyzed the results, interpreted the data, wrote and revised the whole manuscript; SHH, KSS and HJC performed the experiments, analyzed the results; SK and HSK revised the manuscript; GYK and YCC suggested the plan and revised the manuscript; YHC planned, supervised and supported the whole study, and revised the whole manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no commercial or other associations that might pose a conflict of interest.
Ethical approval
The article does not contain any studies with human and animal and this study was performed following institutional and national guidelines.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Park, C., Lee, H., Park, SH. et al. Indole-6-carboxaldehyde prevents oxidative stress-induced mitochondrial dysfunction, DNA damage and apoptosis in C2C12 skeletal myoblasts by regulating the ROS-AMPK signaling pathway. Mol. Cell. Toxicol. 16, 455–467 (2020). https://doi.org/10.1007/s13273-020-00102-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13273-020-00102-9