
Abstract
The gut microbiome of earthworms has a complex interdependence with the host. When the soil minerals pass through earthworm’s gut, they may affect the gut microbiota. To gain insight into the response of gut microbiota to the passed minerals, we fed earthworm (Eisenia fetida) on nutrient-poor soil and ore powder, and used high throughput sequencing to characterize the earthworm intestinal microbial community to find evidence for a core bacterial community of the E. fetida. The results showed that earthworms’ gut maintained a core microbiome that appeared in all samples. These core microbiota may play a significant role in a species’ environmental interactions. The composition of intestinal microbiomes varied with substrates. The earthworm guts from two nutrient-poor substrates had similar microbial communities and they were different from nutrient-rich substrate. Proteobacteria and Bacteroidetes were more abundant in the gut of earthworms kept on a nutrient-poor substrate such as ore powder or mineral soil than in the gut of earthworms kept in organic-rich compost soil; some of these microorganisms may help earthworms survive in nutrient-poor substrates.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Earthworms are an important component of soil macrofauna, dominating the biomass of soil invertebrates (Kodama et al. 2014; Plum and Filser 2005). They are known as the “engineers of ecosystem”, and have a significant impact on biological, chemical and physical processes of the soil ecosystem through their activities (Edwards 2004; Zhang et al. 2007). Animal digestive systems host an abundance of microbes; The gut microbiome forms a complex interdependence with the host, and plays an important regulatory role in host nutrient metabolism, immune system and other physiological functions (Byzov et al. 2009; Dillon and Dillon 2004). Like other animals, earthworms host a large number of microorganisms, including the complex microbial communities of their gastrointestinal tracts (Kim et al. 2004; Knapp et al. 2009). The intestines of worms provide appropriate niches for microorganisms, and earthworms mainly utilize the gut bacteria for provision of metabolic capacities and protection against pathogens (Rudi et al. 2009). The earthworms together with the microbiota are responsible for decomposition and turnover of substances in nature (Byzov et al. 2015). Their interactions regulate the biogeochemistry of terrestrial soils (Thakuria et al. 2009).
As a special earthworm “organ”, gut microbes play an important role in biogeochemical processess of soil elements (Edwards and Fletcher 1988; Sruthy et al. 2013). Earthworms swallow great quantities of soil. Previous studies also have found that earthworms can survive not only in nutrient-poor soils but even in ore powder substrates (Liu et al. 2011; Liu et al. 2016). When soil minerals pass through earthworm’s gut, gut microbes may degrade them to some extent besides mechanical grinding action of guts, corrosion of digestive juice, etc. (Basker et al. 1994; Carpenter et al. 2007; Needham et al. 2004). However, there is poor knowledge about the response of gut microbiota to the ingested minerals. In this paper, high-throughput sequencing of 16S rRNA gene amplicons was employed to analyze intestinal microbiota of earthworms reared respectively in the compost soil, mineral soil, and ore powder substrates. The objectives are to characterize the response of the earthworm gut microbiota to the passed nutrient-poor minerals and to provide an insight into the shifting gut microbial population responsible for helping earthworms adapt to different substrate types.
2 Materials and methods
2.1 Materials
Experimental earthworms Eisenia fetida were purchased from the market. These earthworms were reared in compost soil at the time of purchase. The mineral soil used to feed earthworms in the experiments was collected from Weihe Road North, Puyang City, Henan, China. The mineral soil samples were taken from 40 cm below the surface. Potassium ore was purchased from Banqiao Town of Biyang County, Zhumadian,China. The soil and the potassium ore powder were sieved to pass through a 0.074 mm mesh before using.
Organic matter contents in substrates were determined by potassium dichromate titration with the method described in China’s Agricultural Industry Standards NY/T 1121.6–2006. The organic matter contents of the compost soil and mineral soil samples were 18.1% and 0.9%, respectively. No organic matter was found in the potassium ore powder.
The element compositions (Table S1) and mineral constituents (Table S2) of mineral soil and potassium ore were determined with X-ray fluorescence spectrometry (XRF, Axios, PW4400) and X- ray diffraction (XRD, Rigaku, D/MAx-2200), respectively.
2.2 Feeding trial
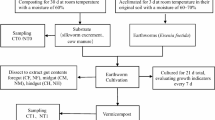
The feeding trial was conducted in Key Lab of Agricultural Biotechnology of Puyang, Henan Province, China. Earthworms were transferred from the compost soil containing 30% moisture to the following two substrates: 1) 1300 g potassium ore powder containing 22% moisture; 2) 1300 g mineral soil containing 30% moisture. These two substrates were placed in 1-L flasks. Twenty earthworms were placed in flask and maintained in the dark at 26 °C for 30 days; substrates without earthworm served as controls. Each treatment was performed in triplicate. The substrates were complemented with appropriate amounts of water during the treatment process to compensate for evaporation.
2.3 Determination of available potassium content
The mineral soil sample after earthworm treatment and its control sample (i.e. no earthworms treatment), potassium ore powder after earthworm treatment and its control sample (i. e. no earthworms treatment) were dried at 50 °C to constant weight in an oven, and available potassium contents were measured using methods described in the People’s Republic of China Forestry Industry Standard “Determination of forest soil K” (LY/T 1236–1999). The method is briefly described as follows: Five grams of dried samples were placed in a 200 mL conical flask, and mixed with 50 mL of 1 M ammonium acetate solution. Contents were tightly sealed and shaken at 150 RPM for 30 min at room temperature, and then centrifuged at 3000 RPM. Supernatant was filtered through a 0.45 μm filter paper, and potassium content of the filtrate was measured using a flame atomic absorption spectrophotometer (TAS-990SUPER F, Beijing Purkinje General Ltd. Co.).
2.4 DNA extraction from intestinal contents
Ten individual earthworms for each biological replicate were randomly chosen, rinsed with tap water, anesthetized with 70% alcohol solution, and then washed with sterile deionized water; they were immediately dissected with sterile scissors, and the intestinal contents were collected for DNA extraction.
Total DNA was extracted using soil DNA extraction kit (Omega, USA) according to the manufacturer’s instructions. For each treatment, the DNA samples from the three biological replicates were then pooled together in equal amounts to generate one mixed sample. The mixed DNA samples were respectively designated CEG, SEG and MEG for earthworms in compost soil, mineral soil, and potassium ore powder, and were examined by 1% agarose gel electrophoresis.
2.5 Amplification of 16S rRNA (V3 + V4) gene fragment
PCR was performed from each sample to produce a fragment of the 16S rRNA gene (covering the V3 + V4 hypervariable regions). To capturing more of the earthworm microbiota 16S rRNA genes, a pair of degenerate primers were used for PCR amplification. The primers were designed and synthesized by Biomarker Biotechnology Co., Ltd. (Beijing, China). Primers were: 5′- ARACTYCTACGGRAGGCWG -3 ‘and 5’- GACTACNVGGGTATCTAATCC -3′. PCR amplification procedure was as follows: Initial 5 min denaturation at 95 °C; 25 cycles of 1 min denaturation at 95 °C, 1 min annealing at 50 °C, 1 min extension at 72 °C; and a final extension at 72 °C for 7 min.
The second step PCR amplification was performed with indexed primers, using the initial amplification product as a template. After sequencing, each sample may be distinguished by an index sequence of second PCR primers. The products of the second round PCR amplification were electrophoretically resolved on a 2% agarose gel to ensure the fragment size were consistent with theoretical value.
2.6 Library construction and MiSeq sequencing
PCR products were purified using AMPure beads (BeckMan, USA), quantified with a Nanodrop 2000 spectrophotometer (Thermo, USA), and then mixed in equal quantity. Mixed samples were recovered from the gel. After quality examination, the samples were sequenced at Biomarker Biotechnology Co., Ltd. (Beijing, China) with the Miseq equipment for paired-end sequencing.
2.7 Bioinformatics analysis
Initially, FLASH software was used to merge paired reads in each sample (Magoč and Salzberg 2011). Followed by filteringthe data with Trimmomatic software to obtain raw tags (Bolger et al. 2014); UCHIME software was then used to compare clean tags with the Gold database, identify and remove chimera sequence (Edgar et al. 2011); these filtered and annotated reads were used for the final analysis.
Sequences were clustered using UCLUST in the QIIME package, with a 97% similarity cutoff for operational taxonomic units (OTUs) (Caporaso et al. 2010; Edgar 2010). Each OTU must represent at least 0.005% of all sequences to be considered valid (Bokulich et al. 2013). According to Silva taxonomy database taxonomy annotation, all the 16S rRNA gene sequences were assigned at an 80% confidence interval to phyla, classes, orders, families and genera.
According to the OTU abundances information in each sample library, Mothur was used to complete α diversity and rarefaction analysis (Schloss et al. 2009). QIIME was used to complete β diversity analysis, obtaining a distance matrix using binary jaccard, bray curtis, unweighted UniFrac and weighted UniFrac algorithms (Grice et al. 2009; Lozupone and Knight 2005). R language tools were used to create heat maps and PCoA curves.
3 Results
3.1 Earthworm effects on mineral weathering
The effect of earthworms on mineral weatheing was observed through changes in the concentration of available potassium (Fig. S1). The average content of available potassium of the mineral soils, treated with earthworms, was 89.56 mg/kg. The average content of their controls, incubated under the same experimental conditions but without earthworms, was 61.82 mg/kg. Treatments with earthworms was 44.87% more than their controls. The average content of available potassium of the potassium-bearing rock powder, treated with earthworms, was 85.96 mg/kg. The average content of their controls, incubated under the same experimental conditions but without earthworms, was 46.98 mg/kg. Treatments with earthworms was 82.97% more than their controls. Statistical analysis indicated that potassium content of the samples treated with earthworms was significantly (p < 0.05) higher than their controls in both mineral soil and potassium ore. There is a little of organic matter in mineral soil, so it is not certain that the increasing available potassium is from the minerals or from the organic matter. However, there is no organic matter in rock powder, its increasing available potassium is only from the weathered minerals. Mineral K is the major proportion of total K in soil and potassium ore, but it cannot be absorbed directly by plants (Liu et al. 2016). Mineral K can only become available for plant through weathering. The above results showed that earthworms promoted the weathering of both soil and ore, and released some potassium from soil and ore.
3.2 Sequencing data analysis
Three samples were sequenced, obtaining a total of 70 Mb filtered data. After merging paired end reads, a total of 131,428 raw spliced tags were obtained. Each sample produced an average of 43,809 raw tags, with the lowest producing 40,741 raw tags. After filtration and removal of the chimeric sequences, 91,187clean tags were obtained, with an average of 30,396 clean tags per sample; the lowest producing sample had 29,816 clean tags (Table S3).
Clean tags were clustered, and 999 OTUs were obtained from earthworm intestinal content samples. Among them, CEG produced 539 OTUs, SEG produced 629 OTUs, and MEG produced 650 OTUs.
Sampling depth of each sample is reflected by the Shannon curve and dilution curve in Fig. 1. Each sample dilution curve and Shannon curve tended to be smooth; the Chao l index estimates number of OTUs in communities (Table 1). Samples CEG, SEG and MEG’s Chao l indices were 619, 791 and 692; meanwhile, detected OTUs accounted for 87.08%, 79.52% and 93.93% of their respective Chao l indices. These data suggest that the sequencing depth was reasonable, and truly reflects the diversity of earthworm gut bacterial community.

Rarefaction curves and Shannon curves of different samples.CEG, the gut of earthworms fed in compost soil; SEG, the gut of earthworms fed in mineral soil; MEG, the gut of earthworms fed in mineral powders
3.3 Earthworm gut flora richness and diversity
The α-diversity indices of earthworm gut bacterial communities were shown in Table 1. As seen from the table, just in terms of gut bacterial species richness, earthworms in mineral soil had the highest Chao 1 abundance index, followed by earthworms in ore powder. Earthworms in compost had the lowest Chao 1. Chao 1 indices simply predict the number of species in a community, without considering the abundance of each species. Therefore, Chao 1 indices cannot fully reflect the diversity of earthworm’s gut flora. Shannon index considers richness and abundance, so it can truly reflect the diversity of earthworm gut flora. Shannon indices of the three samples indicated that earthworm gut microbial diversity was highest in the compost soil treatment, followed by that of mineral soil. The lowest diversity was was observed in the sample of ore powder treatment. Among the three substrates, compost contained the highest organic matter, followed by mineral soil. The ore powder had no organic matter. Therefore, earthworm gut bacterial community diversity positively correlates with to organic matter content of the feeding substrate.
3.4 Gut microbiota composition
The experimental results indicated that earthworm intestinal flora were abundant. Table 2 shows the number of taxa at the phylum, class, order, family, genus levels in the three samples. There were 24 phyla and 338 genera of bacteria found in the gut samples. In addition, there were 111 unidentified OTUs at the phylum level (involving 4086 tags); among these, 14 unidentified OTUs were in CEG (involving 189 tags), 69 OTUs were in SEG (involving 2309 tags), and 28 unidentified OTUs were in MEG (involving 1588 tags). Molecular methods dramatically broadened the diversity and the tree of life as we know it has dramatically expanded due to new genomic sampling of previously enigmatic or unknown microbial lineages (Hug et al. 2016). Our above data suggest that there still exists a large number of unknown new groups of high taxonomic levels.
Identified earthworm gut bacteria include 24 phyla: Proteobacteria, Firmicutes, Actinobacteria, Chloroflexi, Bacteroidetes, Candidate division TM7, Acidobacteria, Cyanobacteria, Nitrospirae, Planctomycetes, Gemmatimonadetes, Verrucomicrobia, Chlamydiae, Deinococcus-Thermus, TM6, MVP-21, Tenericutes, Candidate division WS6, Candidate division BRC1, Candidate division OD1, Chlorobi, Elusimicrobia, Fusobacteria, SM2F11. There were 17, 18 and 21 phyla found in CEG, SEG and MEG, respectively.
Although many species of microorganisms are hosted in the earthworm gut, the dominant microorganisms in the communities include Proteobacteria, Firmicutes, Actinobacteria, Chloroflexi and Bacteroidetes (Fig. 2a), among which, Proteobacteria were the most abundant.

The relative abundance of the dominant bacterial phyla a and genera b in different samples.Both phyla and genera representing more than 1% at least in one gut bacterial community are summarized. Remaining taxa have been grouped into “Others”
At the genus level, the highly abundant, dominant genera in the community were mainly Acinetobacter, Ramlibacter, Enterococcus, Betaproteobacterium Npb-03, Flexibacter, Aeromonas, Bacillus, Mesorhizobium, Ensifer, Flavobacterium, Ilumatobacter, Pedobacter, and Solitalea (Fig. 2b).
3.5 Gut microbiota in different treatments
The number of OTUs shared among the three samples are shown in Fig. 3. As observed, all three samples had a high proportion of OTUs unique to a single sample. CEG had 122 unique OTUs, accounting for 22.63% of its total OTUs; SEG had 155 unique OTUs, accounting 24.64% of its total OTUs; MEG had 115 unique OTUs, accounting for 17.69% of its total OTUs.

Venn diagrams showing the numbers of OTUs shared among three sampling guts of earthworms
Although the total OTUs composition is different between the substrates, there is a core OTUs maintained in the earthworms’ gut. Shared OTUs accounted for a large proportion of the total. The OTUs in common among the three samples accounted for 21.22% of OTUs found in all three samples, 39.33% of OTUs in CEG, 33.70% of OTUs in SEG and 32.62% of OTUs in MEG. The proportions of OTUs in common between two samples were even higher: the shared OTUs between the two samples accounted for a total of between 45.15% and 74.58% of the total OTUs in each single sample.
Proteobacteria dominated the guts of the earthworms, and their OTUs accounted for 49.42%, 62.66% and 62.69% in CEG, SEG and MEG, respectively. Proteobacteria were significantly more abundant in SEG and MEG than in CEG; among these, most were Gammaproteobacteria and Betaproteobacteria. Gammaproteobacteria accounted for 46.96%, 24.67% and 25.36% and Betaproteobacteria accounted for 0.49%, 35.58% and 28.35% of all CEG, SEG and MEG OTUs, respectively.
Bacteroidetes bacteria accounted for 11.35% and 16.61% in SEG and MEG respectively, whereas they accounted for only 1.22% in CEG. Both Bacteroidetes and Betaproteobacteria were also more abundant in SEG and MEG than they were in CEG.
Although Proteobacteria and Bacteroidetes were less abundant in CEG than they were in SEG and MEG, on the whole, CEG had higher microbial diversity. Firmicutes, Actinobacteria, Chloroflexi, Bacteroidetes and Candidate division TM7 were significantly more abundant in CEG than they were in SEG and MEG.
At the genus level, four genera, i. e. Acinetobacter, Enterococcus, Bacillus and Ilumatobacter, were significantly more abundant in CEG than they were in SEG and MEG. Betaproteobacterium Npb-03 and Flavobacterium were significantly more abundant in SEG than they were in CEG and MEG, and Solitalea was found only in SEG. The reduction in substrate organic nutrients and the increase in degree of mineral weathering was accompanied by the increase in the abundances of Ramlibacter, Flexibacter, Mesorhizobium, Ensifer, and Pedobacteria. These five genera were most abundant in the MEG samples, and least abundant in the CEG sample.
Using Fast UniFrac analysis, both weighted and unweighted principal coordinate analysis (PCoA) showed the similarity among the microbial communities (Fig. 4). Both weighted and unweighted PCoA showed that MEG and SEG had similar microbial communities, but they were more different from CEG. UniFrac heat map of samples showed the same results with unweighted and weighted PCoA (Fig. 5).

Unweighted and weighted UniFrac PCoA of bacterial communities in earthworms’ guts. Values on axes indicate percentages of total variation explained by each axis

Heatmaps based on unweighted and weighted UniFrac distance matrices. Colors from red to blue represent the difference of the composition and structure of the two samples from statistically significant to not significant
4 Discussion
4.1 Effects of feeding substrate on earthworm intestinal microbiota
The earthworm gut is colonized by diverse microbial communities (Kim et al. 2004; Knapp et al. 2008). The study presented here tested the influence of different substrates on these communities, and found diverse microbial communities that changed with substrates. Of the 24 phyla and 338 genera of bacteria found among the three earthworm gut samples, they did share a common set of phyla that made up more than 30% of the taxa in each.
Due to differences in the three feeding substrates, microbial diversity indeces were significantly different among the three feeding substrates. Though the number of OUT in CEG is lowest, its Shannon index is highest. The number of OTUs simply reflects the number of species in a community, without considering the abundance of each species. Therefore, it cannot fully reflect the diversity of earthworm’s gut flora. Shannon index considers richness and abundance, so it can truly reflect the diversity of earthworm gut flora. Compost soil are the most nutrient-rich. Mineral soil is poor in organic matter, but is highly weathered, contains more secondary minerals and easily absorbable mineral elements. The highly unweathered potassium ore powder contained no organic matter, consisted mainly of primary minerals, and was poor in easily absorbable mineral elements. As a result, the Shannon index were the highest in earthworms feeding on compost soil, followed by that of mineral soil, and the sample of ore powder treatment has the lowest Shannon index. The feeding substrate greatly influences the intestinal microbial community structure (Koubová et al. 2015). Different substrate components lead to a different microbial community structure (Engel and Moran 2013; Knapp et al. 2009). OTUs unique to each of the three samples accounted for about 20% of all OTUs in each sample, which was a high proportion.
Although high proportions of microbial species were unique to earthworms fed a particular substrates, microbial species common to all three treatments were also highly represented. Such high proportions of shared microbial taxa may be due to the stable microenvironment in earthworms. Although the feeding substrate affects this microenvironment, similarities exist in the gut of earthworms fed on different substrates, and therefore the intestinal microbial species remain partly identical. Despite vast diet-related differences in the composition of the gut microbiome between individuals, there is compelling evidence for the existence of a core gut microbiome in animals (Aira et al. 2015). Many studies found that bacterial communities of E. fetida guts were dominated by Proteobacteria, Actinobacteria, Firmicutes and Bacteroidetes (Ma et al. 2017; Singh et al. 2015), which were consistent with our results. These core microbiota play a significant role in a species’ environmental interactions, often performing functions unachievable by the eukaryotic host (Pass 2015). Food and environment are important factors affecting the structure of the animal gut microbial community (Thakuria et al. 2010). The more similar the food and the environment are, the more similar the animal gut microbial community structure will be (Colman et al. 2012; Maslowski and Mackay 2011).
PCoA and UniFrac distance heat maps showed that microbial communities were similar between MEG and SEG, whose substrates were dominated by mineral components; both communities were quite different from that of the organic-rich CEG. The substrate is not only the microbial habitat, but is also ingested by earthworms into the gut. Thus, the substrate is the key factor that determines the structure of the earthworm gut microbial community. If other factors are similar, similar feeding substrates produce similar gut microbial community structures. The greater the differences among rearing substrates, the greater the structural differences in earthworm gut microbiota.
4.2 Dominant gut microbes in earthworms
Our experimental results showed that Proteobacteria were most abundant in gut microbiota of the earthworms Pheretima carnosa in both mineral soil and ore substrates (Liu et al. 2011). In addition, other scholars analyzed the bacterial 16S rRNA gene fragments from the earthworm Lumbricus rubellus reared in climatic chamber with of three different foods and showed Proteobacteria were dominant in earthworm gut (Knapp et al. 2009). Numerous other studies have demonstrated that Proteobacteria is the most abundant gut microbe not only in earthworms, but also in other animals (Engel and Moran 2013; Ley et al. 2008). For example, using 16S rRNA pyrosequencing, a study of 21 insect orders, 218 species, 305 individuals found that Proteobacteria accounted for 62.1% of all gut microbe reads. Proteobacteria microbes are important for carbon, sulfur and nitrogen cycling on Earth (Kersters et al. 2006). Gut Proteobacteria microbes can ferment food ingredients to aid the host in digestion and absorption of nutrients, augmenting nutrition and energy metabolism in the host (Flint et al. 2012).
Besides Proteobacteria, some phyla such as Actinobacteria, Firmicutes and Bacteroidetes are abundant in earthworms gut, which were consistent with other researchers’ results (Aira et al. 2015; Aira et al. 2016; Ma et al. 2017). For example, after earthworms were fed with cow manure, dwarf shrub or grass litter for six weeks, the intestinal content was dominated by Proteobacteria, especially from the ammasubclass, followed by members of the phyla Bacteroidetes, Actinobacteria and Firmicutes (Knapp et al. 2009). When coconut leaf was vermicomposted using an earthworm, Eudrilus sp., the worm gut contents had Actinobacteria, Firmicutes and Alphaproteobacteria in predominance.
CEG sample was obtained from organic rich compost soil, SEG from highly weathered soil which contained low organic matter, whereas MEG was from poorly weathered rocks containing no organic matter. The substrates for SEG and MEG samples lacked organic matter, and were mainly composed of minerals. Some microbes have significantly higher abundance in SEG and MEG, compared with the CEG sample. Among the dominant phyla, Proteobacteria and Bacteroidetes were more abundant in SEG and MEG than in CEG. In particular, the class Betaproteobacteria in phylum Proteobacteria was highly abundant in SEG and MEG and less abundant in CEG. The five dominant genera that were significantly more abundant in SEG and MEG than in CEG all belong to phyla Proteobacteria and Bacteroidetes. These genera are worthy of more attention because their higher abundances correlated with reductions in substrate organic matter and mineral weathering. Some members of these five genera may play role for helping earthworms survive in extremely nutrient-poor substrate.
5 Conclusions
We assessed abundant microbial community in earthworm gut, where dominated microbial taxa of Proteobacteria, Firmicutes, Actinobacteria, Chloroflexi and Bacteroidetes. Though there is a core microbial community in the earthworms’gut, the total gut microbial composition is different between the substrates. Proteobacteria and Bacteroidetes were more abundant in the gut of earthworms kept on organic-poor substrates than that of earthworms kept in organic-rich compost soil, thus some of them may help earthworms survive in nutrient-poor substrates.
References
Aira M, Bybee S, Pérezlosada M, Domínguez J (2015) Feeding on microbiomes: effects of detritivory on the taxonomic and phylogenetic bacterial composition of animal manures. FEMS Microbiol Ecol 91:817–822
Aira M, Olcina J, Pérez-Losada M, Domínguez J (2016) Characterization of the bacterial communities of casts from Eisenia Andrei fed with different substrates. Appl Soil Ecol 98:103–111
Basker A, Kirkman JH, Macgregor AN (1994) Changes in potassium availability and other soil properties due to soil ingestion by earthworms. Biol Fertil Soils 17:154–158
Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, Mills DA, Caporaso JG (2013) Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods 10:57–59. doi:10.1038/nmeth.2276
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120
Byzov BA, Nechitaylo TY, Bumazhkin BK, Kurakov AV, Golyshin PN, Zvyagintsev DG (2009) Culturable microorganisms from the earthworm digestive tract. Microbiology 78:360–368
Byzov BA, Tikhonov VV, Nechitailo TY, Demin VV (2015) Taxonomic composition and physiological and biochemical properties of bacteria in the digestive tracts of earthworms. Eurasian Soil Science 48:268–275
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336
Carpenter D, Hodson ME, Eggleton P, Kirk C (2007) Earthworm induced mineral weathering: preliminary results. Eur J Soil Biol 43:S176–S183
Colman DR, Toolson EC, Takacs-Vesbach CD (2012) Do diet and taxonomy influence insect gut bacterial communities? Mol Ecol 21:5124–5137
Dillon RJ, Dillon VM (2004) The gut bacteria of insects: nonpathogenic interactions. Entomology 49:71–92
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200
Edwards CA (2004) The importance of earthworms as key representatives of the soil fauna. In: Edwards CA (ed) Earthworm ecology, second edn. CRC Press, Boca Raton, pp 3–11
Edwards CA, Fletcher KE (1988) Interactions between earthworms and microorganisms in organic-matter breakdown. Agric Ecosyst Environ 24:235–247. doi:10.1016/0167-8809(88)90069-2
Engel P, Moran NA (2013) The gut microbiota of insects - diversity in structure and function. FEMS Microbiol Rev 37:699–735
Flint HJ, Scott KP, Louis P, Duncan SH (2012) The role of the gut microbiota in nutrition and health. Nat Rev Gastroenterol Hepatol 9:577–589
Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, Bouffard GG, Blakesley RW, Murray PR, Green ED (2009) Topographical and temporal diversity of the human skin microbiome. Science 324:1190–1192
Hug LA, Baker BJ, Anantharaman K, Brown CT, Probst AJ, Castelle CJ, Butterfield CN, Hernsdorf AW, Amano Y, Ise K (2016) A new view of the tree of life. Nat Microbiol 1:16048
Kersters K, De Vos P, Gillis M, Swings J, Vandamme P, Stackebrandt E (2006) Introduction to the Proteobacteria. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (eds) The prokaryotes. Springer, New York, pp 3–37. doi:10.1007/0-387-30745-1_1
Kim HJ, Shin KH, Hur HG, Cha CJ (2004) Analysis of aerobic and Culturable bacterial community structures in earthworm (Eisenia fetida) intestine. J Appl Biol Chem 47:137–142
Knapp BA, Seeber J, Podmirseg SM, Meyer E, Insam H (2008) Application of denaturing gradient gel electrophoresis for analysing the gut microflora of Lumbricus rubellus Hoffmeister under different feeding conditions. Bull Entomol Res 98:271–279
Knapp BA, Podmirseg SM, Seeber J, Meyer E, Insam H (2009) Diet-related composition of the gut microbiota of Lumbricus rubellus as revealed by a molecular fingerprinting technique and cloning. Soil Biol Biochem 41:2299–2307
Kodama N, Kimura T, Yonemura S, Kaneda S, Ohashi M, Ikeno H (2014) Automated analysis of two-dimensional positions and body lengths of earthworms (Oligochaeta); MimizuTrack. PLoS One 9:e97986
Koubová A, Chroňáková A, Pižl V, Sánchez-Monedero MA, Elhottová D (2015) The effects of earthworms Eisenia spp. on microbial community are habitat dependent. Eur J Soil Biol 68:42–55
Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R (2008) Evolution of mammals and their gut microbes. Science 320:1647–1651
Liu D, Lian B, Wang B, Jiang G (2011) Degradation of potassium rock by earthworms and responses of bacterial communities in its gut and surrounding substrates after being fed with mineral. PLoS One 6:e28803
Liu DF, Lian B, Wang B (2016) Solubilization of potassium containing minerals by high temperature resistant Streptomyces sp.isolated from earthworm's gut. Acta Geochimica 35:262–270
Lozupone C, Knight R (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228–8235
Ma L, Xie Y, Han Z, Giesy JP, Zhang X (2017) Responses of earthworms and microbial communities in their guts to Triclosan. Chemosphere 168:1194–1202. doi:10.1016/j.chemosphere.2016.10.079
Magoč T, Salzberg SL (2011) FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963
Maslowski KM, Mackay CR (2011) Diet, gut microbiota and immune responses. Nat Immunol 12:5–9
Needham SJ, Worden RH, McIlroy D (2004) Animal-sediment interactions: the effect of ingestion and excretion by worms on mineralogy. Biogeosciences 1:113–121
Pass DA (2015) The earthworm microbiome. Dissertation, Cardiff University
Plum NM, Filser J (2005) Floods and drought: response of earthworms and potworms (Oligochaeta: Lumbricidae, Enchytraeidae) to hydrological extremes in wet grassland. Pedobiologia 49:443–453. doi:10.1016/j.pedobi.2005.05.004
Rudi K, Odegård K, Løkken TT, Wilson R (2009) A feeding induced switch from a variable to a homogenous state of the earthworm gut microbiota within a host population. PLoS One 4:e7528
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541
Singh A, Singh DP, Tiwari R, Kumar K, Singh RV, Singh S, Prasanna R, Saxena AK, Nain L (2015) Taxonomic and functional annotation of gut bacterial communities of Eisenia Foetida and Perionyx excavatus. Microbiol Res 175:48–56
Sruthy PB, Anjana JC, Rathinamala J, Jayashree S (2013) Screening of earthworm (Eudrilus eugeniae) gut as a transient microbial habitat. Adv Zool Bot 1:53–56
Thakuria D, Schmidt O, Finan D, Egan D, Doohan FM (2009) Gut wall bacteria of earthworms: a natural selection process. ISME J 4:357–366 http://www.nature.com/ismej/journal/v4/n3/suppinfo/ismej2009124s1.html
Thakuria D, Schmidt O, Finan D, Egan D, Doohan FM (2010) Gut wall bacteria of earthworms: a natural selection process. ISME J 4:357–366
Zhang W, Chen D, Zhao C (2007) Functions of earthworm in ecosystem biodiversity. Science 15:142–153
Acknowledgements
This work was supported by National Natural Science Foundation of China (U1204405 and 41373078) and Natural Science Foundation of Henan Educational Committee, China (14B180010).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Liu, D., Lian, B., Wu, C. et al. A comparative study of gut microbiota profiles of earthworms fed in three different substrates. Symbiosis 74, 21–29 (2018). https://doi.org/10.1007/s13199-017-0491-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13199-017-0491-6