
Abstract
Ischemic stroke is a leading cause of disability and is considered now the fourth leading cause of death. Many clinical trials have shown that stroke patients with acute elevation in blood glucose at onset of stroke suffer worse functional outcomes, longer in-hospital stay, and higher mortality rates. The only therapeutic hope for these patients is the rapid restoration of blood flow to the ischemic tissue through intravenous administration of the only currently proven effective therapy, tissue plasminogen activator (tPA). However, even this option is associated with the increased risk of intracerebral hemorrhage. Nonetheless, the underlying mechanisms through which hyperglycemia (HG) and tPA worsen the neurovascular injury after stroke are not fully understood. Accordingly, this review summarizes the latest updates and recommendations about the management of HG and coadministration of tPA in a clinical setting while focusing more on the various experimental models studying (1) the effect of HG on stroke outcomes, (2) the potential mechanisms involved in worsening the neurovascular injury, (3) the different therapeutic strategies employed to ameliorate the injury, and finally, (4) the interaction between HG and tPA. Developing therapeutic strategies to reduce the hemorrhage risk with tPA in hyperglycemic setting is of great clinical importance. This can best be achieved by conducting robust preclinical studies evaluating the interaction between tPA and other therapeutics in order to develop potential therapeutic strategies with high translational impact.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Thrombolytic therapy with tissue plasminogen activator (tPA) to reopen occluded cerebral blood vessels is currently the best chance acute ischemic stroke patients have of recovering normal function. Elevated blood glucose at the time of acute stroke increases the risk of hemorrhagic transformation with tPA treatment, and it is associated with poor clinical outcomes, longer in-hospital stay, increased cost, and mortality. As almost 40 % of stroke patients present with hyperglycemia, this is an important clinical problem. The optimal approach to manage these patients, especially with respect to glucose control and tPA treatment, is not clear, and the various professional guidelines differ in their recommendations. The mechanisms contributing to exacerbated neurovascular injury and poor outcomes are not fully understood. The purposes of this review are to briefly summarize the clinical evidence on hyperglycemia and tPA interactions in acute ischemic stroke and discuss how preclinical studies approach this problem with an emphasis on the experimental models of hyperglycemia and methods of reperfusion used in these studies.
Clinical Evidence
Acute stroke patients who have hyperglycemia on admission or persistent hyperglycemia during the first 3 days of hospitalization have worse functional outcomes than patients without hyperglycemia [1–3]. This finding has been confirmed by many, although not all [4], observational stroke studies. A large proportion of acute stroke patients with hyperglycemia have diabetes mellitus. The complications associated with chronic diabetes mellitus may be contributing to a worse functional outcome in stroke patients compared to those without diabetes. However, many studies show worse clinical outcomes in acute stroke patients with hyperglycemia without a history of diabetes [5, 6]. The interpretation of such findings is complicated by the fact that some acute stroke patients with hyperglycemia have undiagnosed (unknown) diabetes mellitus.
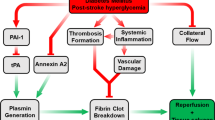
The exact mechanisms by which hyperglycemia (during acute ischemic stroke) leads to worse functional outcome have not been established. It could be that the hyperglycemia during ischemia somehow results in greater brain injury compared to normoglycemia. Hyperglycemia during acute brain ischemia may impair thrombolysis and reperfusion [7, 8]. For example, hyperglycemia (HG) increases coagulation by increasing thrombin production and stimulating the tissue factor pathway [9, 10]. HG also reduces the fibrinolytic activity of tPA by increasing the production of plasminogen activator inhibitor (PAI)-1 [11]. Additionally, hyperglycemia during acute brain ischemia may exacerbate or accelerate some of the pathologic processes involved in ischemic brain injury [12]. In addition, hyperglycemia increases the risk of cerebral hemorrhage in acute stroke patients treated with intravenous tPA [2, 5, 13–15]. A list of major clinical and preclinical studies that reported increased bleeding with tPA and HG is presented in Table 1.
It is not clear whether there is a blood glucose threshold that increases the risk of unfavorable functional outcomes in acute stroke. Some acute stroke studies report a hyperglycemia threshold for worse functional outcomes [16], but others report a linear relation between blood glucose and poor functional outcomes [5]. If hyperglycemia during acute stroke is detrimental, it might be beneficial to lower the blood glucose level swiftly during the initial few hours or days after stroke onset. However, this remains controversial. In traumatic brain injury, intensive insulin therapy was associated with increased risk of hypoglycemia and no improvement in the neurologic outcomes [17, 18]. In ischemic stroke, one limited clinical efficacy trial did not show a benefit of intravenous insulin treatment for mild hyperglycemia [19]. Yet, another small study showed that insulin treatment was associated with a high incidence of hypoglycemia and greater infarct growth in patients with persistent arterial occlusion compared with controls [20]. Another trial of intravenous insulin treatment for patients with greater hyperglycemia and predominantly with diabetes mellitus in acute stroke is ongoing [21].
As might be expected from the paucity of scientific evidence for efficacy, there are limited guideline recommendations for the blood glucose goals during acute stroke. The American Heart Association/American Stroke Association current guidelines [22] recommend maintaining the blood glucose level in the range of 140–180 mg/dL during the acute stroke hospitalization. The European Stroke Organization guideline [23] recommends lowering the blood glucose with insulin to below 180 mg/dL. No specific mention is made in the US or the European guidelines regarding lowering the blood glucose during tPA therapy [22, 23]. Since hyperglycemia increases the risk of cerebral hemorrhage when tPA is used in acute stroke, it might be beneficial to correct the hyperglycemia as soon as possible. Intravenous insulin has an onset of action of approximately 1–2 min and could be administered as soon as tPA therapy is contemplated. As each patient’s insulin needs and reaction to stress are individualized, a general insulin dose recommendation cannot be made. Hypoglycemia (<60 mg/dL) can be detrimental and should be avoided. Nonetheless, if the blood glucose is, for example, 400 mg/dL, an intravenous dose of 10 units regular insulin seems reasonable, as it should lower the blood glucose by a clinically significant amount with little risk for hypoglycemia. Additional doses will likely be needed to maintain the blood glucose <180 mg/dL.
Thrombolytic therapy for acute stroke has been approved in many countries. It should be noted that in Europe, prior stroke with concomitant diabetes is an exclusion criteria from tPA treatment [1–3, 24, 25]. For this reason, the European Cooperative Acute Stroke Study (ECASS) III, which provided the clinical evidence to extend the therapeutic window up to 4.5 h for the administration of tPA, did not include patients with prior stroke and concomitant diabetes or patients with blood glucose over 400 mg/dL [25]. However, whether hyperglycemia shortens the therapeutic window for tPA is not yet determined. Developing strategies to reduce the hemorrhage risk with tPA thrombolysis in hyperglycemic settings is therefore of great importance. Equally important, determining the therapeutic potential and known side effects of tPA under hyperglycemic conditions along with other commonly found comorbidities/risk factors in stroke patients is critical. In this regard, the potential interaction of tPA with experimental therapeutics needs to be carefully evaluated, and experimental studies are critical to provide the much needed preclinical data to move the field forward.
Preclinical Evidence
In this section, we will first summarize different experimental models of hyperglycemia and the effect of hyperglycemia on short-term stroke outcomes. We will then discuss potential mechanisms contributing to exacerbated reperfusion injury and, finally, review the different therapeutic strategies employed to reduce the injury (pretreatment and acute post-stroke treatment) and promote recovery (chronic post-stroke treatment).
Models of Hyperglycemia and Impact on Short-Term Outcomes
Acute hyperglycemia may result from a stress response or due to preexisting diabetes. Early experimental studies in this field used mainly acute changes in blood glucose in which hyperglycemia was induced by glucose injection or by depleting insulin-producing islet cells with streptozotocin (STZ) injection a few days prior to stroke surgery and reported short-term outcomes. As recently reviewed, most, if not all of these studies, used the suture occlusion model of stroke and showed that acute hyperglycemia increases neuronal and vascular injury after ischemic stroke leading to poor outcomes as reported in clinical studies [26, 27].
Studies using different diabetic animal models highlighted the importance of preexisting disease on stroke injury and functional outcomes [28]. Nedergaard et al. reported that compared with normoglycemia, the infarct volume was decreased in hypoglycemic rats, unaltered in acute diabetes induced by single STZ injection 2 days before middle cerebral artery occlusion (MCAO), and increased in chronic diabetes induced by STZ injection 4 months before MCAO [29]. When Zucker Diabetic Fatty Rats (ZDF) (a type 2 diabetic rat model that mimics the chronic metabolic and inflammatory abnormalities observed in humans) with blood glucose of 350–450 mg/dL were subjected to 2-h suture occlusion and 4-h reperfusion, there was a significant increase in neutrophil adhesion and aggregation after reperfusion. This was associated with an increase in the cerebral expression of the inflammatory mediators sICAM, IL-1β, and E-selectin, infarct size, and worse neurological outcomes [30]. Several studies also reported greater gray and white matter injury, higher mortality, increased cerebral edema, and worsened neurological outcomes in type 2 diabetic (db/db) mice after brain ischemia which was associated with upregulation of matrix metalloprotease-9 (MMP-9) [31–35]. Using the lean Goto-Kakizaki (GK) model of type 2 diabetes, we showed that diabetes mediates pathological remodeling and neovascularization of the brain, and ischemia/reperfusion injury superimposed on this preexisting vascular disease causes extensive vascular injury and bleeding into the brain [36–40]. Despite the fact that GK rats developed relatively smaller infarcts compared to control animals at 24 h after stroke, the functional outcome was worse most likely due to increased bleeding and edema.
While discrepancies in infarct size in these studies may result from differences in the duration of ischemia, an important observation that stems out from the review of these preclinical studies is the vulnerability of the vasculature to relatively small changes in blood glucose and how this in turn worsens the functional outcomes. There is no doubt that neuroprotection is very important, but poor outcomes of stroke cannot be solely explained by greater infarcts in hyperglycemic stroke. For example, Xing et al. reported that hyperglycemia increases blood-brain barrier (BBB) permeability and hemorrhagic transformation (HT) and worsens outcome but does not affect the infarct volume [41]. We also showed that a mild elevation in blood glucose (140–200 mg/dL) achieved by intraperitoneal injection of 40 % glucose solution did not increase infarct size yet worsened vascular injury and neurological outcomes [42]. As discussed above, diabetic GK rats develop greater HT and edema compared to their nondiabetic counterparts leading to unfavorable outcomes without a significant increase in infarct size. Therefore, as we move forward in stroke research, the roles of the vasculature and HT on functional outcomes and recovery need to be carefully delineated.
Mechanisms of Hyperglycemic Reperfusion Injury
Although restoration of blood flow to the ischemic tissue is essential to rescuing the penumbral tissue, reperfusion can increase the ischemic injury [43], and hyperglycemia further exacerbates the process. Mechanisms contributing to exacerbated neurovascular injury, HT, and poor outcomes in hyperglycemic stroke are likely to be multifactorial, and different mechanisms may be at play at the neuronal, glial, and/or vascular levels as recently reviewed [26–28]. Augmented oxidative and nitrative stress under hyperglycemic conditions can modify tight junction proteins and structure compromising BBB integrity. For example, occludin levels are decreased to a greater extent under hyperglycemic hypoxic conditions [44]. HG also heightens endothelial mitochondrial damage and decreases BBB transport which can lead to loss of BBB integrity and increased hemorrhage [45, 46]. Since the goal of reperfusion therapies after stroke is to open the occluded artery and reestablish the cerebral blood flow (CBF), in this review, we will focus on cerebrovascular mechanisms and review how hyperglycemic reperfusion affects cerebrovascular function and ultimately CBF. We will also summarize the limited number of studies investigating the effect of tPA on cerebrovascular tone.
Hyperglycemia and Cerebral Blood Flow After Stroke
To study the influence of acute hyperglycemia on CBF during ischemia/reperfusion injury, Kawai et al. occluded the middle cerebral arteries (MCA) for 2 or 4 h followed by 2 h of reperfusion in rats made acutely hyperglycemic by intraperitoneal administration of glucose 20 min before MCA occlusion. They showed that CBF was reduced in the ischemic hemisphere of hyperglycemic rats compared to normoglycemic controls. They also found that poor restoration of CBF in hyperglycemic rats was associated with increased infarct size [47]. These results are in agreement with other studies in rats [48–50] and cats [51–53] which reported reduced CBF in hyperglycemic compared to normoglycemic animals that could limit compensatory blood flow mechanisms and accentuate brain dysfunction after ischemic injury. In contrast, Gisselsson et al. found that acute hyperglycemia had no effect on reperfusion blood flow after 30 min of ischemia and 1 h of reperfusion, refuting the hypothesis that exacerbated injury after acute hyperglycemic stroke is caused by disturbances in CBF [54]. This discrepancy between studies could result from differences in hyperglycemia severity and its duration.
Hyperglycemia and Cerebral Autoregulation
Cerebral autoregulation is critical for maintaining constant blood flow despite changes in perfusion pressure [55–57]. Autoregulation is highly pronounced in the brain, and large cerebral arteries contribute significantly to cerebral autoregulation to protect downstream microvessels [58, 59]. Although the mechanisms of autoregulation in the brain are not fully understood, several studies have shown that the myogenic behavior of the cerebral smooth muscles plays a crucial role in the development of autoregulation. Myogenic response describes the intrinsic ability of the smooth muscle cells to constrict in response to increased pressure and dilate in response to decreased pressure to achieve constant blood flow [56, 60, 61]. To investigate the influence of acute exposure to high glucose levels on cerebral myogenic behavior, rat posterior cerebral arteries were exposed to 44 versus 5.5 mmol/L L-glucose, and the amount of basal tone and the response to transmural pressure were determined. High glucose levels induced vasodilation and loss of basal tone which made the vessels incapable of responding to changes in transmural pressure [62]. These results are consistent with the findings of Sieber et al. which demonstrated that acute hyperglycemia leads to an elevation in CBF and a reduction in cerebrovascular resistance in nondiabetic dogs [63]. Helpern et al. also showed that rats infused with 25 % D-glucose experienced impaired cerebral autoregulation and increased CBF which persisted for 15 min even after normalization of plasma glucose levels [64].
Chronic hyperglycemia has a profound effect on vascular and endothelial function. However, very few studies have focused on the effect of diabetes on the myogenic reactivity of cerebral vessels. Similar to acute hyperglycemia, diabetes can modify cerebrovascular resistance and CBF. Enhanced myogenic tone was reported in cerebral arteries isolated from STZ-induced diabetic rats compared to control [65]. Similar findings were observed in cerebral arteries isolated from type 2 diabetic rats [66–68]. While these findings contradict with a previous report showing a decrease in myogenic tone in cerebral vessels isolated from diabetic rats, differences in the degree of HG may account for this disparity [69]. Taken together, these findings indicate that acute and chronic exposure to high glucose has a deleterious effect on vascular reactivity which may ultimately affect CBF.
Hyperglycemia and Cerebral Myogenic Behavior After Stroke
Little attention has been given to the impact of acute and chronic hyperglycemia on myogenic behavior of cerebral vessels during ischemia/reperfusion injury. Cipolla et al. investigated the effect of acute hyperglycemia on CBF and the reactivity of penetrating arterioles before and after MCAO. The penetrating arterioles are the major vessels involved in lacunar stroke, a type of stroke characterized with favorable outcomes after moderate hyperglycemia [4]. They showed that reperfusion blood flow was not influenced by acute hyperglycemia prior to and after stroke. Basal tone was reduced in both normoglycemic and hyperglycemic animals to the same degree suggesting that it was due to ischemia/reperfusion injury and independent of glucose. They also demonstrated that acute hyperglycemia had no effect on endothelium-dependent vasodilator production in penetrating arterioles which may explain why lacunar strokes are not worsened by hyperglycemia [70]. However, studies done on cortical stroke revealed a deleterious effect of acute hyperglycemia on vascular reactivity. When naïve MCAs were perfused intraluminally with plasma of acutely hyperglycemic rats that underwent 2-h MCAO/2-h reperfusion, they reported increased myogenic tone and endothelial dysfunction, suggesting that circulating factors in plasma due to acute hyperglycemic stroke are vasoactive in nonischemic cerebral vessels [71].
We recently showed that ischemia/reperfusion injury has the global effect of decreasing the myogenic tone of MCAs in both ischemic and nonischemic hemispheres. We also demonstrated that ischemia/reperfusion injury superimposed with acute elevation of blood glucose caused exacerbation of myogenic dysfunction in the nonischemic hemisphere, which was associated with worse stroke outcomes [72]. Similar results were reported by our group showing that myogenic tone of MCAs isolated from control and type 2 diabetic rats was reduced after exposure to 20 min of oxygen-glucose deprivation to mimic ischemic injury, which was greater in the diabetic group [67]. Taken together, these findings suggest that acute or chronic HG may exacerbate myogenic dysfunction and impair effective reperfusion, which ultimately can contribute to the detrimental effect of hyperglycemia on stroke outcomes.
tPA and Cerebrovascular Function
As discussed above, the only effective and FDA-approved treatment for ischemic stroke is to reestablish CBF using recombinant tPA. The most critical adverse effect of tPA administration, hampering its use in stroke patients, is symptomatic intracerebral hemorrhage [73]. To determine whether the hemorrhagic complications associated with tPA are due to a direct deleterious effect on vascular reactivity, MCAs were perfused with tPA and myogenic tone was determined. Intraluminal perfusion of tPA significantly impaired myogenic reactivity in isolated MCAs [65]. This myogenic dysfunction was augmented in arteries exposed to ischemia/reperfusion injury and perfused with tPA. In addition, treatment with tPA caused endothelial dysfunction and diminished vasodilation to acetylcholine and 5HT reactivity, and again, these effects were exacerbated if vessels were exposed to ischemia/reperfusion [74]. In control rats, tPA diminished myogenic tone of MCAs and caused concentration-dependent vasodilation which was prevented by the stabilization of actin cytoskeleton of smooth muscle [75]. These findings suggest that tPA administration has a direct detrimental effect on myogenic reactivity which could contribute to vascular injury after stroke.
Vascular tone is highly regulated by the activity of potassium channels which is a major determinant of membrane potential [59]. A prior study reported that potassium channel activity was impaired after cerebral hypoxia/ischemia, which was aggravated by tPA treatment leading to autoregulation impairment via upregulation of extracellular signal-regulated kinases/mitogen-activated protein kinases [76]. These findings are in agreement with the data which showed that exogenous plasminogen activator administration augmented the hypercapnic and hypotensive cerebrovasodilation impairment in the newborn pig [77, 78]. Taken together, all these studies provide evidence that tPA treatment may adversely affect the cerebrovascular reactivity, especially if superimposed on ischemic injury. However, the impact of tPA administration on vascular function in the presence of hyperglycemia is yet to be determined. Better understanding of how tPA therapy affects vascular reactivity during acute hyperglycemic stroke might be crucial for avoiding its deleterious effects that profoundly constrain its clinical utility.
Treatment Strategies in Experimental Hyperglycemic Acute Ischemic Stroke
This section will summarize different therapeutic strategies employed to reduce ischemic injury (pretreatment and acute post-stroke treatment) and promote recovery (chronic post-stroke treatment).
Preventive Treatment Strategies
Recent clinical and experimental studies suggest that statins play a pivotal role in reducing the incidence of stroke and myocardial infarction in patients with vascular diseases. This was suggested to be due to statins’ pleiotropic rather than lipid-lowering effects [79, 80]. After 4 weeks of diabetes induced by STZ injection, animals were treated with vehicle or simvastatin (1 mg/kg/day) for 14 days. Subsequently, mice were subjected to 90-min MCAO and 24-h reperfusion. Diabetes aggravated the stroke outcome and increased the infarct size compared to nondiabetic mice. Pretreatment with simvastatin for 14 days prior to stroke significantly reduced the infarct volume and improved the neurological outcomes in both diabetic and nondiabetic mice [81].
As discussed above, diabetic GK rats develop greater vascular injury and have poor functional outcomes. These rats also exhibit extensive cerebrovascular remodeling and neovascularization, which was prevented by early glucose control with metformin or inhibition of matrix metalloproteases with minocycline for 5 weeks starting at the onset of diabetes [82]. Prevention of vascular remodeling significantly reduced the vascular injury and improved the functional outcome when compared to nontreated GK rats [38]. This suggests that glycemic control and vasculoprotective treatments may be effective preventive strategies in reducing stroke injury.
Thiazolidinediones (TZD) are peroxisome proliferator-activated receptor agonists that are commonly used to lower blood glucose. One study evaluated the neuroprotective effects of rosiglitazone in type 2 diabetic db/db mice and their nondiabetic db/+ littermates. All animals were subjected to 45 min of focal cerebral ischemia (suture occlusion) followed by 3 days of reperfusion. Rosiglitazone 4 mg/kg (i.p.) treatment 4 h prior to stroke or 2 mg/kg (i.p.) at 2 h reperfusion significantly decreased the infarct volume and induced neuroprotection without affecting blood glucose. This suggests that TZDs have direct neuroprotective effects independent of blood glucose lowering. However, feeding mice with a chow fortified with rosiglitazone for 3 weeks before inducing MCAO significantly decreased blood glucose in db/db mice without affecting the db/+ (normoglycemic genetic control of db/db) blood glucose levels. The long-term oral pretreatment with rosiglitazone induced significantly better neuroprotection and reduction in the infarct size compared to the bolus injection [83]. In an interesting study, Kumari and his group showed that cerebral inflammatory response is needed for recovery after stroke, and this response was delayed and diminished in the brains of diabetic stroked mice [84]. In a recent study, they also showed that the administration of Draglitazone for 7 days before induction of hypoxia/reperfusion significantly reduced the infarct size in diabetic mice. Draglitazone also restored the compromised inflammatory response through increasing the expression of TNF-α, IL-1β, and IL-6 at the early phase of recovery [33].
Acute-Subacute Treatment Strategies
The key question now is whether the detrimental effects of hyperglycemia during acute brain ischemia can be reversed by rapidly correcting the hyperglycemia, consequently improving outcomes. In animals, rapid correction of hyperglycemia during focal brain ischemia resulted in less brain injury than persistent hyperglycemia [85–92] unless there was hypoglycemia [92, 93]. Insulin is the most commonly used agent to regulate blood glucose and has been shown to reduce ischemic brain damage when given immediately before [86, 91, 94, 95] or within minutes after experimental brain ischemia [96–99]. When type 1 diabetic rats were subjected to 2-h MCAO and 24-h reperfusion by the suture occlusion model, acute or chronic administration of low dose of insulin (2 U/kg) did not alter the lesion size or the number of apoptotic cells in the brain. However, the chronic treatment with high dose of insulin (12 U/kg) for 7 days significantly reduced the lesion volume and the apoptotic levels [100].
These observations suggest that the deleterious effects of hyperglycemia during acute brain ischemia may be at least partly reversible by rapid correction of hyperglycemia as recently reviewed [1]. Although underlying mechanisms are not fully understood, it has been suggested that insulin might be neuroprotective independent of its blood glucose-lowering effects. However, there are several key studies that provided strong evidence that regulation of blood glucose is the main factor for reducing ischemic damage with insulin therapy. One study investigated three groups of rats with transient focal brain ischemia: (1) control vehicle treatment, (2) insulin pretreatment (2–3 IU/kg) 60 min prior to ischemia, and (3) insulin pretreatment plus glucose to maintain normal blood glucose the same as in the control group. The resulting mean blood glucose in groups 1, 2, and 3 was 151, 61, and 182 mg/dL, respectively [89]. There was no difference in infarct size between groups 1 and 3, whereas group 2 with lower blood glucose had smaller infarcts. This suggests that the reduction in the infarct size might be due to the blood glucose lowering but not due to direct insulin’s neuroprotective effect. Earlier studies reported a linear relationship between blood glucose and pathological stroke outcomes [87]. When hyperglycemic cats were given insulin after MCAO, blood glucose decreased to hypoglycemic levels, and this caused increased infarct size and early death when compared to cats receiving saline, again suggesting that when glucose levels are not optimal, presence of insulin does not confer neuroprotection [87]. Interestingly, one study showed that the administration of insulin-like growth factor-1 (IGF-1) 30 min before MCAO significantly decreased the lesion volume (infarct size) and decreased the number of apoptotic cells in the central nervous system as indicated by TUNEL staining and caspase-3 immunoreactivity [101].
In addition to glycemic control, other successful tactics have been reported. For example, deferoxamine (DFX) administration, which is an iron chelator, immediately after MCAO attenuated the mortality rate, HT, infarct volume, and brain edema in diabetic rats. These findings suggest that DFX may provide potential means to reduce HT in acute ischemic stroke patients [102]. Another study reported that the inhalation of hydrogen gas during reperfusion reduces hyperglycemia-induced HT and brain infarction resulting in improved neurological function [103]. A recently published study demonstrated the neuroprotective effect of Ginkgo biloba extracts in hyperglycemic rats. Diabetes was induced by single intravenous injection of STZ (50 mg/kg) 4–6 weeks before MCAO. They showed that the Ginkgolide B was able to reduce the levels of malondialdehyde (lipid peroxidation product), reactive oxygen species, and the infarct size leading to better outcome [104], suggesting that oxidative stress might be contributing to hyperglycemic stroke injury.
We showed that therapeutic targets after stroke, especially vasculoprotective approaches, may differ in diabetic models. Atorvastatin (15 mg/kg) administration, one dose directly after reperfusion and the second dose 12 h after a 3-h MCAO, reduced the bleeding rates, hemoglobin content, and infarct volumes in both control and type 2 diabetic GK rats [39]. These effects, however, were independent of changes in plasma and brain tissue lipid peroxides and nitrotyrosine levels. A follow-up study evaluated the effects of acute manipulation of potential targets for vascular protection (i.e., NFκB, peroxynitrite, and matrix metalloproteinases) on vascular injury and functional outcome in GK rats. Animals received a single dose of either FeTPPS (peroxynitrite decomposition catalyst), curcumin (NFκB inhibitor), or minocycline (broad-spectrum MMP inhibitor) at reperfusion. All treatments reduced hemorrhagic transformation in diabetic animals, and this was associated with a reduction in the MMP9 activity. The different treatments improved the neurological outcomes in varying degrees. In control animals, all treatments reduced MMP9 activity yet bleeding was not reduced suggesting that therapeutic targets for neurovascular protection and dosing of potential treatments may differ in control versus diabetic states [36].
Chronic Treatment Strategies for Recovery
Type 1 diabetic rats, subjected to temporary MCAO by the suture model 2 weeks after induction of diabetes, did not show increased lesion volume. However, they exhibited significantly increased brain hemorrhage, BBB disruption, and worsened functional outcomes after 14 days of MCAO. In this model, mortality occurred within the first 3 days after surgery, and all animals that died exhibited hemorrhage. Niaspan (40 mg/kg), a prolonged-release formulation of Niacin which is in current clinical use for increasing HDL cholesterol and also known to improve endothelial function. Niaspan administration did not alter the lesion volume or the mortality rate in diabetic rats when started at 24 h after MCAO for 14 days. However, this approach significantly reduced the hemorrhage volume and the BBB leakage and improved the functional outcomes. Niaspan also promoted cerebrovascular remodeling which was accompanied by reduced expression of angiopoietin-2 (Ang) and increased the expression of Ang 1 in the ischemic brain [105]. The same group recently investigated the long-term therapeutic effects of Niaspan on axonal remodeling after stroke in type 1 diabetic rats. Interestingly, Niaspan (40 mg/kg) treatment for 28 days, starting 24 h after stroke in diabetic rats, significantly increased the axonal density in the ipsilateral motor cortex compared to saline-treated diabetic animals [106]. Previously, the same group also showed that long-term Niaspan (40 or 80 mg/kg) treatment for 14 days after MCAO improved the functional outcomes and promoted angiogenesis in nondiabetic male Wistar rats [107].
We have previously shown that GK rats exhibit dysfunctional cerebral neovascularization [82], and when these rats are subjected to ischemia reperfusion injury, they develop HT and worse neurological outcomes [38]. A follow-up study was conducted to investigate the effect of diabetes and glycemic control on reparative neovascularization and functional recovery in diabetes. While control animals showed angiogenesis in the peri-infarct area and even in the contralateral hemisphere, diabetic animals did not only show impaired angiogenesis but also there was regression of existing vessels and exacerbated astrogliosis. Diabetic animals also exhibited worse neurological outcomes, anxiety-like behavior, and cognitive deficits after stroke when compared to the normoglycemic stroked rats. Glycemic control with metformin (300 mg/kg/day) for 14 days after stroke significantly improved the cerebrovascular repair and the functional outcomes in these animals suggesting that glucose control in the recovery phase may be very important for neurovascular repair [108].
Therapeutic angiogenesis is being pursued as a potential treatment for stroke recovery. The use of bone marrow stromal cell (BMSC) therapy was shown to be promising in promoting and improving functional recovery in nondiabetic rats after stroke [109]. However, this was not the case with diabetic rats. Chen et al. showed that treating type 1 diabetic rats with BMSCs 24 h after stroke worsened the long-term outcomes at 14 days. BMSCs also increased the mortality, BBB leakage, and brain hemorrhage when compared to diabetic untreated animals [110]. Along with our findings in GK rats, these results suggest that promoting new vessel formation after stroke when there is preexisting diabetic vascular disease may not be useful. Stabilization and maintaining the integrity of existing blood vessels may prove more beneficial. Along with our previous study in which we showed vasculoprotective treatment approaches exert differential effects in control versus diabetic rats [36], this study strongly suggests that stroke treatment strategies should be compared in control and disease models.
Hyperglycemia, Stroke, and tPA
In spite of the importance of tPA in the clinical setting, only a few experimental studies were conducted to evaluate the role that tPA plays in either improving or worsening the outcomes in hyperglycemic stroke. In an acute hyperglycemia model, ischemia was induced by occluding both common carotid arteries and the left proximal MCA with microaneurysm clips for 90 min. tPA was infused 10 min before reperfusion and outcome was assessed at day 3. Hyperglycemia exacerbated the brain damage in tPA-treated animals through increasing the infarct size, brain hemorrhage, and edema [111]. It is also worth mentioning that the tPA-induced brain hemorrhage increased with elevated levels of hyperglycemia. In the same study, the administration of the NADPH oxidase inhibitor, apocynin, significantly reduced the tPA-induced brain hemorrhage [111].
In another study, Ning et al. showed that the administration of tPA 2 h after embolic stroke in type 1 diabetic rats significantly increased HT and brain swelling. tPA failed to reduce the brain infarct and improve functional outcomes [112]. However, a similar study conducted by Fan et al. reported decreased infarction with the use of tPA in diabetic rats as compared to untreated diabetic rats. Diabetes increased the infarct volume, edema, and brain hemorrhage when compared to control rats. The administration of tPA slightly but significantly decreased the infarct size in diabetic rats, but it failed to ameliorate the brain swelling. However, tPA significantly increased intracerebral hemorrhage in diabetic rats compared to control animals [113]. In a recent study by the same group using the same diabetic embolic stroke model, they showed that coadministration of minocycline with tPA after stroke significantly reduced the brain infarction, brain swelling, and tPA-induced brain HT, providing evidence that combination therapy with minocycline plus tPA may be beneficial in improving stroke outcomes in diabetes [114]. The same group used again the embolic stroke model to evaluate the effects of early glycemic control by insulin in combination with tPA on stroke outcomes. Insulin was administered at 1 h after stroke, and tPA (10 mg/kg) was given at 1.5 h after stroke. The use of either insulin alone or tPA alone had no effect on the ischemic infarction. However, the early glycemic control with insulin in combination with tPA significantly reduced the brain infarction, brain swelling, and the tPA-induced hemorrhage [115].
Moving Forward: Lessons Learned from Clinical and Experimental Studies
Based on the literature discussed above, there are several important points that should be emphasized in order to advance the field.
-
1.
The severity of hyperglycemia seems to be important for the neuronal injury as mild elevations do not increase the infarct size. However, the vasculature is more susceptible to even small elevations in blood glucose which mediate greater edema and HT leading to poor outcomes in hyperglycemic ischemic brain injury. Preventive and therapeutic strategies that offer vasculoprotection seem to promote neuronal protection and repair.
-
2.
The stroke model with respect to method of occlusion and reperfusion and the duration of ischemia prior to reperfusion may impact the injury and recovery.
-
3.
With no exception, all the above studies were conducted with male and relatively young animals. One study reported that male diabetic mice showed higher mortality rates and larger infarct size than females [34, 116]. Thus, there is a great need for studies involving female and older animals in hyperglycemic stroke research.
-
4.
Therapeutic strategies tested focus on reductions in infarct size and improvement of neurological function. However, there is a need to better understand how these treatment approaches would affect vascular function and CBF.
-
5.
Therapeutic interventions in hyperglycemic stroke should evaluate tPA interactions. This is important as although it was previously demonstrated that single use of erythropoietin (EPO) in mechanical occlusion [117] or embolic [118] models of stroke was protective, EPO failed to improve clinical outcomes and increased the mortality in patients receiving tPA [119]. This was further investigated, and Jia et al. later showed that the late administration of tPA in combination with EPO worsened the outcome in embolic model of stroke [120]. As reviewed above, there is only one study that evaluated the interaction of tPA with another therapeutic agent in experimental hyperglycemic stroke setting, highlighting the need for additional studies of this kind.
References
McCormick MT, Muir KW, Gray CS, Walters MR. Management of hyperglycemia in acute stroke: how, when, and for whom? Stroke. 2008;39(7):2177–85.
Poppe AY, Majumdar SR, Jeerakathil T, Ghali W, Buchan AM, Hill MD, et al. Admission hyperglycemia predicts a worse outcome in stroke patients treated with intravenous thrombolysis. Diabetes Care. 2009;32(4):617–22.
Yong M, Kaste M. Dynamic of hyperglycemia as a predictor of stroke outcome in the ECASS-II trial. Stroke. 2008;39(10):2749–55.
Uyttenboogaart M, Koch MW, Stewart RE, Vroomen PC, Luijckx GJ, De Keyser J. Moderate hyperglycaemia is associated with favourable outcome in acute lacunar stroke. Brain. 2007;130(Pt 6):1626–30.
Bruno A, Levine SR, Frankel MR, Brott TG, Lin Y, Tilley BC, et al. Admission glucose level and clinical outcomes in the NINDS rt-PA stroke trial. Neurology. 2002;59(5):669–74.
Capes SE, Hunt D, Malmberg K, Pathak P, Gerstein HC. Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients: a systematic overview. Stroke. 2001;32(10):2426–32.
Calleja AI, Garcia-Bermejo P, Cortijo E, Bustamante R, Rojo Martinez E, Gonzalez Sarmiento E, et al. Insulin resistance is associated with a poor response to intravenous thrombolysis in acute ischemic stroke. Diabetes Care. 2011;34(11):2413–7.
Cosentino F, Battista R, Scuteri A, De Sensi F, De Siati L, Di Russo C, et al. Impact of fasting glycemia and regional cerebral perfusion in diabetic subjects: a study with technetium-99 m-ethyl cysteinate dimer single photon emission computed tomography. Stroke. 2009;40(1):306–8.
Gentile NT, Vaidyula VR, Kanamalla U, DeAngelis M, Gaughan J, Rao AK. Factor VIIa and tissue factor procoagulant activity in diabetes mellitus after acute ischemic stroke: impact of hyperglycemia. Thromb Haemost. 2007;98(5):1007–13.
Rao AK, Chouhan V, Chen X, Sun L, Boden G. Activation of the tissue factor pathway of blood coagulation during prolonged hyperglycemia in young healthy men. Diabetes. 1999;48(5):1156–61.
Pandolfi A, Giaccari A, Cilli C, Alberta MM, Morviducci L, De Filippis EA, et al. Acute hyperglycemia and acute hyperinsulinemia decrease plasma fibrinolytic activity and increase plasminogen activator inhibitor type 1 in the rat. Acta Diabetol. 2001;38(2):71–6.
Bruno A, Liebeskind D, Hao Q, Raychev R, Investigators US. Diabetes mellitus, acute hyperglycemia, and ischemic stroke. Curr Treat Options Neurol. 2010;12(6):492–503.
Meretoja A, Putaala J, Tatlisumak T, Atula S, Artto V, Curtze S, et al. Off-label thrombolysis is not associated with poor outcome in patients with stroke. Stroke. 2010;41(7):1450–8.
Putaala J, Sairanen T, Meretoja A, Lindsberg PJ, Tiainen M, Liebkind R, et al. Post-thrombolytic hyperglycemia and 3-month outcome in acute ischemic stroke. Cerebrovasc Dis. 2011;31(1):83–92.
Alvarez-Sabin J, Molina CA, Montaner J, Arenillas JF, Huertas R, Ribo M, et al. Effects of admission hyperglycemia on stroke outcome in reperfused tissue plasminogen activator–treated patients. Stroke. 2003;34(5):1235–41.
Fuentes B, Castillo J, San Jose B, Leira R, Serena J, Vivancos J, et al. The prognostic value of capillary glucose levels in acute stroke: the GLycemia in Acute Stroke (GLIAS) study. Stroke. 2009;40(2):562–8.
Coester A, Neumann CR, Schmidt MI. Intensive insulin therapy in severe traumatic brain injury: a randomized trial. J Trauma. 2010;68(4):904–11.
Bilotta F, Caramia R, Cernak I, Paoloni FP, Doronzio A, Cuzzone V, et al. Intensive insulin therapy after severe traumatic brain injury: a randomized clinical trial. Neurocrit Care. 2008;9(2):159–66.
Gray CS, Hildreth AJ, Sandercock PA, O’Connell JE, Johnston DE, Cartlidge NE, et al. Glucose-potassium-insulin infusions in the management of post-stroke hyperglycaemia: the UK Glucose Insulin in Stroke Trial (GIST-UK). Lancet Neurol. 2007;6(5):397–406.
McCormick M, Hadley D, McLean JR, Macfarlane JA, Condon B, Muir KW. Randomized, controlled trial of insulin for acute poststroke hyperglycemia. Ann Neurol. 2010;67(5):570–8.
Bruno A, Durkalski VL, Hall CE, Juneja R, Barsan WG, Janis S, et al. The Stroke Hyperglycemia Insulin Network Effort (SHINE) trial protocol: a randomized, blinded, efficacy trial of standard vs. intensive hyperglycemia management in acute stroke. Int J Stroke. 2013. doi:10.1111/ijs.12045.
Jauch EC, Saver JL, Adams Jr HP, Bruno A, Connors JJ, Demaerschalk BM, et al. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2013;44(3):870–947.
European Stroke Organisation (ESO) Executive Committee, ESO Writing Committee. Guidelines for management of ischaemic stroke and transient ischaemic attack 2008. Cerebrovasc Dis. 2008;25(5):457–507.
Mishra NK, Ahmed N, Davalos A, Iversen HK, Melo T, Soinne L, et al. Thrombolysis outcomes in acute ischemic stroke patients with prior stroke and diabetes mellitus. Neurology. 2011;77(21):1866–72.
Hacke W, Kaste M, Bluhmki E, Brozman M, Davalos A, Guidetti D, et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med. 2008;359(13):1317–29.
Martini SR, Kent TA. Hyperglycemia in acute ischemic stroke: a vascular perspective. J Cereb Blood Flow Metab. 2007;27(3):435–51.
Ergul A, Li W, Elgebaly MM, Bruno A, Fagan SC. Hyperglycemia, diabetes and stroke: focus on the cerebrovasculature. Vasc Pharmacol. 2009;51:44–9.
Ergul A, Kelly-Cobbs A, Abdalla M, Fagan SC. Cerebrovascular complications of diabetes: focus on stroke. Endocr Metab Immune Disord Drug Targets. 2012;12(2):148–58.
Nedergaard M, Diemer NH. Focal ischemia of the rat brain, with special reference to the influence of plasma glucose concentration. Acta Neuropathol. 1987;73(2):131–7.
Ritter L, Davidson L, Henry M, Davis-Gorman G, Morrison H, Frye JB, et al. Exaggerated neutrophil-mediated reperfusion injury after ischemic stroke in a rodent model of type 2 diabetes. Microcirculation. 2011;18(7):552–61.
Kumari R, Willing LB, Patel SD, Baskerville KA, Simpson IA. Increased cerebral matrix metalloprotease-9 activity is associated with compromised recovery in the diabetic db/db mouse following a stroke. J Neurochem. 2011;119(5):1029–40.
Chen J, Cui X, Zacharek A, Cui Y, Roberts C, Chopp M. White matter damage and the effect of matrix metalloproteinases in type 2 diabetic mice after stroke. Stroke. 2011;42(2):445–52.
Kumari R, Willing LB, Patel SD, Krady JK, Zavadoski WJ, Gibbs EM, et al. The PPAR-gamma agonist, darglitazone, restores acute inflammatory responses to cerebral hypoxia-ischemia in the diabetic ob/ob mouse. J Cereb Blood Flow Metab. 2010;30(2):352–60.
Vannucci SJ, Willing LB, Goto S, Alkayed NJ, Brucklacher RM, Wood TL, et al. Experimental stroke in the female diabetic, db/db, mouse. J Cereb Blood Flow Metab. 2001;21(1):52–60.
Tureyen K, Bowen K, Liang J, Dempsey RJ, Vemuganti R. Exacerbated brain damage, edema and inflammation in type-2 diabetic mice subjected to focal ischemia. J Neurochem. 2011;116(4):499–507.
Kelly-Cobbs AI, Prakash R, Li W, Pillai B, Hafez S, Coucha M, et al. Targets of vascular protection in acute ischemic stroke differ in type 2 diabetes. Am J Physiol Heart Circ Physiol. 2013;304(6):806–15.
Li W, Prakash R, Kelly-Cobbs AI, Ogbi S, Kozak A, El-Remessy AB, et al. Adaptive cerebral neovascularization in a model of type 2 diabetes: relevance to focal cerebral ischemia. Diabetes. 2010;59:228–35.
Elgebaly MM, Prakash R, Li W, Ogbi S, Johnson MH, Mezzetti EM, et al. Vascular protection in diabetic stroke: role of matrix metalloprotease-dependent vascular remodeling. J Cereb Blood Flow Metab. 2010;30(12):1928–38.
Elewa HF, Kozak A, El-Remessy AB, Frye RF, Johnson MH, Ergul A, et al. Early atorvastatin reduces hemorrhage after acute cerebral ischemia in diabetic rats. J Pharmacol Exp Ther. 2009;330(2):532–40.
Ergul A, Elgebaly MM, Middlemore ML, Li W, Elewa H, Switzer JA, et al. Increased hemorrhagic transformation and altered infarct size and localization after experimental stroke in a rat model type 2 diabetes. BMC Neurol. 2007;7:33.
Xing Y, Jiang X, Yang Y, Xi G. Hemorrhagic transformation induced by acute hyperglycemia in a rat model of transient focal ischemia. Acta Neurochir Suppl. 2011;111:49–54.
Elgebaly MM, Ogbi S, Li W, Mezzetti EM, Prakash R, Johnson MH, et al. Neurovascular injury in acute hyperglycemia and diabetes: a comparative analysis in experimental stroke. Transl Stroke Res. 2011;2(3):391–8.
McCord JM. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med. 1985;312(3):159–63.
Shao B, Bayraktutan U. Hyperglycaemia promotes cerebral barrier dysfunction through activation of protein kinase C-beta. Diabetes Obes Metab. 2013;15(11):993–9.
Keep RF, Andjelkovic AV, Stamatovic SM, Shakui P, Ennis SR. Ischemia-induced endothelial cell dysfunction. Acta Neurochir Suppl. 2005;95:399–402.
Kawai N, Keep RF, Betz AL, Nagao S. Hyperglycemia induces progressive changes in the cerebral microvasculature and blood–brain barrier transport during focal cerebral ischemia. Acta Neurochir Suppl. 1998;71:219–21.
Kawai N, Keep RF, Betz AL. Hyperglycemia and the vascular effects of cerebral ischemia. Stroke. 1997;28(1):149–54.
Nakai H, Yamamoto YL, Diksic M, Worsley KJ, Takara E. Triple-tracer autoradiography demonstrates effects of hyperglycemia on cerebral blood flow, pH, and glucose utilization in cerebral ischemia of rats. Stroke. 1988;19(6):764–72.
Quast MJ, Wei J, Huang NC, Brunder DG, Sell SL, Gonzalez JM, et al. Perfusion deficit parallels exacerbation of cerebral ischemia/reperfusion injury in hyperglycemic rats. J Cereb Blood Flow Metab. 1997;17(5):553–9.
Duckrow RB, Beard DC, Brennan RW. Regional cerebral blood flow decreases during chronic and acute hyperglycemia. Stroke. 1987;18(1):52–8.
Ginsberg MD, Welsh FA, Budd WW. Deleterious effect of glucose pretreatment on recovery from diffuse cerebral ischemia in the cat. I. Local cerebral blood flow and glucose utilization. Stroke. 1980;11(4):347–54.
Venables GS, Miller SA, Gibson G, Hardy JA, Strong AJ. The effects of hyperglycaemia on changes during reperfusion following focal cerebral ischaemia in the cat. J Neurol Neurosurg Psychiatry. 1985;48(7):663–9.
Wagner KR, Kleinholz M, de Courten-Myers GM, Myers RE. Hyperglycemic versus normoglycemic stroke: topography of brain metabolites, intracellular pH, and infarct size. J Cereb Blood Flow Metab. 1992;12(2):213–22.
Gisselsson L, Smith ML, Siesjo BK. Hyperglycemia and focal brain ischemia. J Cereb Blood Flow Metab. 1999;19(3):288–97.
Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev. 1999;79(2):387–423.
Schubert R, Mulvany MJ. The myogenic response: established facts and attractive hypotheses. Clin Sci (Lond). 1999;96(4):313–26.
Peterson EC, Wang Z, Britz G. Regulation of cerebral blood flow. Int J Vasc Med. 2011;2011:823525.
Faraci FM, Heistad DD. Regulation of large cerebral arteries and cerebral microvascular pressure. Circ Res. 1990;66(1):8–17.
Faraci FM, Heistad DD. Regulation of the cerebral circulation: role of endothelium and potassium channels. Physiol Rev. 1998;78(1):53–97.
Bayliss WM. On the local reactions of the arterial wall to changes of internal pressure. J Physiol. 1902;28(3):220–31.
Palomares SM, Cipolla MJ. Vascular protection following cerebral ischemia and reperfusion. J Neurol Neurophysiol. 2011;2011.
Cipolla MJ, Porter JM, Osol G. High glucose concentrations dilate cerebral arteries and diminish myogenic tone through an endothelial mechanism. Stroke. 1997;28(2):405–10.
Sieber FE, Brown PR, Wu Y, Koehler RC, Traystman RJ. Cerebral blood flow responsivity to CO2 in anesthetized chronically diabetic dogs. Am J Physiol. 1993;264(4 Pt 2):H1069–75.
Helpern JA, Branch CA, Huang N, Hernandez L. Impaired autoregulation with hyperglycemia. Proc Intl Sot Mag Reson Med. 2000;8:1300.
Zimmermann PA, Knot HJ, Stevenson AS, Nelson MT. Increased myogenic tone and diminished responsiveness to ATP-sensitive K+ channel openers in cerebral arteries from diabetic rats. Circ Res. 1997;81(6):996–1004.
Jarajapu YP, Guberski DL, Grant MB, Knot HJ. Myogenic tone and reactivity of cerebral arteries in type II diabetic BBZDR/Wor rat. Eur J Pharmacol. 2008;579(1–3):298–307.
Kelly-Cobbs AI, Prakash R, Coucha M, Knight RA, Li W, Ogbi SN, et al. Cerebral myogenic reactivity and blood flow in type 2 diabetic rats: role of peroxynitrite in hypoxia-mediated loss of myogenic tone. J Pharmacol Exp Ther. 2012;342(2):407–15.
Kelly-Cobbs A, Elgebaly MM, Li W, Ergul A. Pressure-independent cerebrovascular remodelling and changes in myogenic reactivity in diabetic Goto-Kakizaki rat in response to glycaemic control. Acta Physiol (Oxf). 2011;203(1):245–51.
Kold-Petersen H, Brondum E, Nilsson H, Flyvbjerg A, Aalkjaer C. Impaired myogenic tone in isolated cerebral and coronary resistance arteries from the goto-kakizaki rat model of type 2 diabetes. J Vasc Res. 2012;49(3):267–78.
Cipolla MJ, Godfrey JA. Effect of hyperglycemia on brain penetrating arterioles and cerebral blood flow before and after ischemia/reperfusion. Transl Stroke Res. 2010;1(2):127–34.
Palomares SM, Gardner-Morse I, Sweet JG, Cipolla MJ. Peroxynitrite decomposition with FeTMPyP improves plasma-induced vascular dysfunction and infarction during mild but not severe hyperglycemic stroke. J Cereb Blood Flow Metab. 2012;32(6):1035–45.
Coucha M, Li W, Hafez S, Fagan SC, Ergul A. The role of contralateral cerebrovascular myogenic dysfunction in hyperglycemic stroke. Stroke. 2013;44:A153. Abstract.
Tanne D, Kasner SE, Demchuk AM, Koren-Morag N, Hanson S, Grond M, et al. Markers of increased risk of intracerebral hemorrhage after intravenous recombinant tissue plasminogen activator therapy for acute ischemic stroke in clinical practice: the Multicenter rt-PA Stroke Survey. Circulation. 2002;105(14):1679–85.
Cipolla MJ, Lessov N, Clark WM, Haley Jr EC. Postischemic attenuation of cerebral artery reactivity is increased in the presence of tissue plasminogen activator. Stroke. 2000;31(4):940–5.
Cipolla MJ, Curry AB. Dilation to tissue plasminogen activator of middle cerebral arteries is prevented by actin cytoskeletal stabilization. Stroke. 2000;32:360. Abstract.
Armstead WM, Riley J, Cines DB, Higazi AA. tPA contributes to impairment of ATP and Ca sensitive K channel mediated cerebrovasodilation after hypoxia/ischemia through upregulation of ERK MAPK. Brain Res. 2011;1376:88–93.
Armstead WM, Cines DB, Higazi AA. Plasminogen activators contribute to impairment of hypercapnic and hypotensive cerebrovasodilation after cerebral hypoxia/ischemia in the newborn pig. Stroke. 2005;36(10):2265–9.
Armstead WM, Riley J, Kiessling JW, Cines DB, Higazi AA. PAI-1-derived peptide EEIIMD prevents impairment of cerebrovasodilation by augmenting p38 MAPK upregulation after cerebral hypoxia/ischemia. Am J Physiol Heart Circ Physiol. 2010;299(1):H76–80.
Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet. 1994;344(8934):1383–9.
Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360(9326):7–22.
Cakmak A, Yemisci M, Koksoy C, Yazgan U, Dincer D, Dalkara T. Statin pre-treatment protects brain against focal cerebral ischemia in diabetic mice. J Surg Res. 2007;138(2):254–8.
Prakash R, Johnson M, Fagan SC, Ergul A. Cerebral neovascularization and remodeling patterns in two different models of type 2 diabetes. PloS ONE. 2013;8(2):e56264.
Tureyen K, Kapadia R, Bowen KK, Satriotomo I, Liang J, Feinstein DL, et al. Peroxisome proliferator-activated receptor-gamma agonists induce neuroprotection following transient focal ischemia in normotensive, normoglycemic as well as hypertensive and type-2 diabetic rodents. J Neurochem. 2007;101(1):41–56.
Kumari R, Willing LB, Krady JK, Vannucci SJ, Simpson IA. Impaired wound healing after cerebral hypoxia-ischemia in the diabetic mouse. J Cereb Blood Flow Metab. 2007;27(4):710–8.
Anderson RE, Tan WK, Martin HS, Meyer FB. Effects of glucose and PaO2 modulation on cortical intracellular acidosis, NADH redox state, and infarction in the ischemic penumbra. Stroke. 1999;30(1):160–70.
de Courten-Myers G, Myers RE, Schoolfield L. Hyperglycemia enlarges infarct size in cerebrovascular occlusion in cats. Stroke. 1988;19(5):623–30.
de Courten-Myers GM, Kleinholz M, Wagner KR, Myers RE. Normoglycemia (not hypoglycemia) optimizes outcome from middle cerebral artery occlusion. J Cereb Blood Flow Metab. 1994;14(2):227–36.
Bomont L, MacKenzie ET. Neuroprotection after focal cerebral ischaemia in hyperglycaemic and diabetic rats. Neurosci Lett. 1995;197(1):53–6.
Hamilton MG, Tranmer BI, Auer RN. Insulin reduction of cerebral infarction due to transient focal ischemia. J Neurosurg. 1995;82(2):262–8.
Quast MJ, Wei J, Huang NC. Nitric oxide synthase inhibitor NG-nitro-L-arginine methyl ester decreases ischemic damage in reversible focal cerebral ischemia in hyperglycemic rats. Brain Res. 1995;677(2):204–12.
Yip PK, He YY, Hsu CY, Garg N, Marangos P, Hogan EL. Effect of plasma glucose on infarct size in focal cerebral ischemia-reperfusion. Neurology. 1991;41(6):899–905.
Zhu CZ, Auer RN. Optimal blood glucose levels while using insulin to minimize the size of infarction in focal cerebral ischemia. J Neurosurg. 2004;101(4):664–8.
Meden P, Andersen M, Overgaard K, Rasmussen RS, Boysen G. The effects of early insulin treatment combined with thrombolysis in rat embolic stroke. Neurol Res. 2002;24(4):399–404.
Warner DS, Gionet TX, Todd MM, McAllister AM. Insulin-induced normoglycemia improves ischemic outcome in hyperglycemic rats. Stroke. 1992;23(12):1775–80.
Wass CT, Lanier WL. Glucose modulation of ischemic brain injury: review and clinical recommendations. Mayo Clin Proc. 1996;71(8):801–12.
Siemkowicz E, Hansen AJ, Gjedde A. Hyperglycemic ischemia of rat brain: the effect of post-ischemic insulin on metabolic rate. Brain Res. 1982;243(2):386–90.
Voll CL, Auer RN. The effect of postischemic blood glucose levels on ischemic brain damage in the rat. Ann Neurol. 1988;24(5):638–46.
Voll CL, Whishaw IQ, Auer RN. Postischemic insulin reduces spatial learning deficit following transient forebrain ischemia in rats. Stroke. 1989;20(5):646–51.
Tyson R, Peeling J, Sutherland G. Metabolic changes associated with altering blood glucose levels in short duration forebrain ischemia. Brain Res. 1993;608(2):288–98.
Rizk NN, Rafols JA, Dunbar JC. Cerebral ischemia-induced apoptosis and necrosis in normal and diabetic rats: effects of insulin and C-peptide. Brain Res. 2006;1096(1):204–12.
Rizk NN, Myatt-Jones J, Rafols J, Dunbar JC. Insulin like growth factor-1 (IGF-1) decreases ischemia-reperfusion induced apoptosis and necrosis in diabetic rats. Endocrine. 2007;31(1):66–71.
Xing Y, Hua Y, Keep RF, Xi G. Effects of deferoxamine on brain injury after transient focal cerebral ischemia in rats with hyperglycemia. Brain Res. 2009;1291:113–21.
Chen CH, Manaenko A, Zhan Y, Liu WW, Ostrowki RP, Tang J, et al. Hydrogen gas reduced acute hyperglycemia-enhanced hemorrhagic transformation in a focal ischemia rat model. Neuroscience. 2010;169(1):402–14.
Huang M, Qian Y, Guan T, Huang L, Tang X, Li Y. Different neuroprotective responses of Ginkgolide B and bilobalide, the two Ginkgo components, in ischemic rats with hyperglycemia. Eur J Pharmacol. 2012;677(1–3):71–6.
Ye X, Chopp M, Cui X, Zacharek A, Cui Y, Yan T, et al. Niaspan enhances vascular remodeling after stroke in type 1 diabetic rats. Exp Neurol. 2011;232(2):299–308.
Yan T, Chopp M, Ye X, Liu Z, Zacharek A, Cui Y, et al. Niaspan increases axonal remodeling after stroke in type 1 diabetes rats. Neurobiol Dis. 2012;46(1):157–64.
Chen J, Cui X, Zacharek A, Jiang H, Roberts C, Zhang C, et al. Niaspan increases angiogenesis and improves functional recovery after stroke. Ann Neurol. 2007;62(1):49–58.
Prakash R, Li W, Qu Z, Johnson MA, Fagan SC, Ergul A. Vascularization pattern after ischemic stroke is different in control versus diabetic rats: relevance to stroke recovery. Stroke. 2013;44(10):2875–82.
Chen J, Li Y, Wang L, Zhang Z, Lu D, Lu M, et al. Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke. 2001;32(4):1005–11.
Chen J, Ye X, Yan T, Zhang C, Yang XP, Cui X, et al. Adverse effects of bone marrow stromal cell treatment of stroke in diabetic rats. Stroke. 2011;42(12):3551–8.
Won SJ, Tang XN, Suh SW, Yenari MA, Swanson RA. Hyperglycemia promotes tissue plasminogen activator-induced hemorrhage by increasing superoxide production. Ann Neurol. 2011;70(4):583–90.
Ning R, Chopp M, Yan T, Zacharek A, Zhang C, Roberts C, et al. Tissue plasminogen activator treatment of stroke in type-1 diabetes rats. Neuroscience. 2012;222:326–32.
Fan X, Qiu J, Yu Z, Dai H, Singhal AB, Lo EH, et al. A rat model of studying tissue-type plasminogen activator thrombolysis in ischemic stroke with diabetes. Stroke. 2012;43(2):567–70.
Fan X, Lo EH, Wang X. Effects of minocycline plus tissue plasminogen activator combination therapy after focal embolic stroke in type 1 diabetic rats. Stroke. 2013;44(3):745–52.
Fan X, Ning M, Lo EH, Wang X. Early insulin glycemic control combined with tPA thrombolysis reduces acute brain tissue damages in a focal embolic stroke model of diabetic rats. Stroke. 2013;44(1):255–9.
Zhang L, Nair A, Krady K, Corpe C, Bonneau RH, Simpson IA, et al. Estrogen stimulates microglia and brain recovery from hypoxia-ischemia in normoglycemic but not diabetic female mice. J Clin Invest. 2004;113(1):85–95.
Siren AL, Fratelli M, Brines M, Goemans C, Casagrande S, Lewczuk P, et al. Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc Natl Acad Sci U S A. 2001;98(7):4044–9.
Wang Y, Zhang ZG, Rhodes K, Renzi M, Zhang RL, Kapke A, et al. Post-ischemic treatment with erythropoietin or carbamylated erythropoietin reduces infarction and improves neurological outcome in a rat model of focal cerebral ischemia. Br J Pharmacol. 2007;151(8):1377–84.
Ehrenreich H, Weissenborn K, Prange H, Schneider D, Weimar C, Wartenberg K, et al. Recombinant human erythropoietin in the treatment of acute ischemic stroke. Stroke. 2009;40(12):e647–56.
Jia L, Chopp M, Zhang L, Lu M, Zhang Z. Erythropoietin in combination of tissue plasminogen activator exacerbates brain hemorrhage when treatment is initiated 6 hours after stroke. Stroke. 2010;41(9):2071–6.
Acknowledgments
Adviye Ergul is a Research Career Scientist at the Charlie Norwood Veterans Affairs Medical Center in Augusta, GA. This work was supported in part by VA Merit Award (BX000347) and NIH award (NS054688) to Adviye Ergul, VA Merit Award (BX000891) and NIH award (NS063965) to Susan C. Fagan, and American Heart Association Predoctoral Fellowships (12PRE11300001 to Maha Coucha and 13PRE 17090026 to Sherif Hafez).
Conflict of Interest
The authors declare no conflict of interest. The contents do not represent the views of the Department of Veterans Affairs or the United States Government.
Author information
Authors and Affiliations
Corresponding author
Additional information
Sherif Hafez and Maha Coucha equally contributed to this review.
Rights and permissions
About this article
Cite this article
Hafez, S., Coucha, M., Bruno, A. et al. Hyperglycemia, Acute Ischemic Stroke, and Thrombolytic Therapy. Transl. Stroke Res. 5, 442–453 (2014). https://doi.org/10.1007/s12975-014-0336-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12975-014-0336-z