
Abstract
Pt/C and Pt9Bi1/C catalysts are synthesized by wet chemistry, characterized by physicochemical and electrochemical methods, and evaluated towards glucose and methyl-glucoside electrooxidation in alkaline medium. Pt9Bi1/C leads to onset potentials 150 to 350 mV lower than those recorded on Pt/C for glucose and methyl-glucoside oxidation, respectively. From in situ infrared spectroscopy, main reaction products of glucose and methyl-glucoside oxidation are gluconate and methyl-glucuronate, respectively. Chronoamperometry is performed for 6 h in a 25-cm2 electrolysis cell fitted with a Pt9Bi1/C anode to oxidize 18.0 g L−1 glucose and 19.4 g L-1 methyl-glucoside at cell voltages of 0.30 V and 0.50 V, respectively, and a Pt/C cathode to produce hydrogen. Analyses of the reaction products by high-performance liquid chromatography, by 13C nuclear magnetic resonance, and by mass spectroscopy indicate that gluconate and methyl-glucuronate are formed with 100% faradaic efficiency and 100% selectivity at 40% glucose and 37% methyl-glucoside conversions, respectively.

Pt9Bi1/ electrocatalyst is the most outstanding one for the electrooxidation of glucose and methyl-glucoside in terms of activity, selectivity, and faradaic yield towards high value-added gluconate and methyl-glucuronate compounds, respectively.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Biomass represents the most abundant and the only renewable carbon reservoir [1] that can be an alternative to fossil resources for platform chemicals [2]. But, to avoid competition with human feeding, nonedible biomass, such as cellulose, must be preferred to edible biomass, such as starch or vegetal oils, for energy and fine chemistry applications. Cellulose can be depolymerized using highly concentrated solutions of acids or bases [3], but more recently, other cleaner processes have been developed to yield glucose and glucose derivatives [4,5,6].
Oxidation of bio-sourced molecules is a very interesting method for producing valuable chemicals. Classical oxidation methods of organic molecules based on the use of stoichiometric oxidants (permanganate, chromic acid, or nitric acid [7]) are not convenient due to their low environmental quotient [8, 9]. Greener processes, such as catalytic methods, have then been proposed as alternatives for the oxidation of organics in general, and in particular of aldose [10,11,12]. The selective (electro)oxidation of glucose leads generally to gluconic acid because the oxidation of the primary alcohol into carboxylic acid is very difficult to perform, and this reaction remains one of the important problems of organic chemistry [13,14,15], and because the anomeric group of glucose is a reductive function. However, d-glucuronolactone and d-glucaric acid are very interesting compounds for, as examples, hepatoprotective effects and as a common ingredient in energy drinks for the former one [16], and as a precursor for plastics and textile industries for the latter one [10, 17]. But, to obtain these compounds, the primary alcohol group must be oxidized into glucuronic acid.
Amaniampong et al. [18] reported the oxidation of glucose into glucuronic acid with 95% selectivity at 93% conversion using high-frequency ultrasounds (550 kHz) at 80 °C on a copper oxide catalyst. However, the general strategy consists in protecting the reductive anomeric function of glucose by a methyl group and to perform the oxidation of the primary alcohol group. The oxidation of methyl-glucoside has then been carried out by heterogeneous catalysis. Hua et al. [15, 19] developed Pd/La0.5Pb0.5Mn0.9Sn0.1O3 and Pd/CuO–Ce0.5Mn0.5O2 catalysts and performed the reaction with a catalyst/methyl-glucoside mass ratio of 1/6 at 70 °C under stirring and oxygen bubbling. They reported 60% yield over Pd/La0.5Pb0.5Mn0.9Sn0.1O3 and 90% yield over Pd/CuO–Ce0.5Mn0.5O2 in desired products (methyl-glucuronic acid and methyl-glucuronolactone) after acidic hydrolysis. Omri et al. [20] obtained 90% yield with catalysts consisting of TEMPO grafted on nanosized TiO2, AlO2, and CeO2 supports, with a catalyst/methyl-glucoside molar ratio of 1/10 at 35 °C by using the synthetic method developed by Zhao [21] for oxidation of primary alcohols into carboxylic acid. But these methods are both relatively complex to implement in terms of catalyst fabrication and reaction conditions.
Electrochemical reactors are already used in industries for several organic electrosynthesis reactions [22], one of the most famous applications being the Monsanto process for the electrochemical formation of adiponitrile from acrylonitrile. The main advantages of electrochemical methods consist first in only using the molecule to be transformed, water and electrons as reactants, and second in working under mild conditions (room temperature and pressure), which contribute to make electrocatalysis a sustainable nonthermal activation method. For oxidation reactions, electrochemical methods allow activating water molecules at the solid electrode material surface, which will serve as an oxygen source to provide extra oxygen atoms for the oxidation of organic compounds without the use of oxidative agents in the reaction medium (O2 or H2O2, as it is the case in many processes from heterogeneous catalysis). In addition, the activity for an organic reaction can be controlled by modifying the catalyst structure/composition and the selectivity of the reaction by accurately tuning the anode potential (cell voltage), which controls both the water and the organic molecule adsorption/activation at the catalyst surface [23, 24]. For example, Pd3Au7/C catalysts have been shown to be very active [25] for glucose electrooxidation at room temperature and very selective towards gluconic acid formation at a cell voltage of 0.4 V.
In heterogeneous catalysis, the high catalytic activity of platinum-bismuth (PtBi) and palladium-bismuth (PdBi) materials towards controlled oxidation of alcohols, polyols, and carbohydrates was recognized for several decennia [24,25,26,27,28]. More recently, non-alloyed Pt9Bi1/C structure has shown unique catalytic behavior towards the electrooxidation of glycerol [29]: very high activity from very low electrode potentials, prevention of the C–C bond breaking, and very high selectivity towards low-oxidized species (dihydroxyacetone and glyceraldehyde) at low potentials [27, 28].
In this context, the present contribution intends to shed light on the electrooxidation mechanism of glucose and methyl-glucoside on Pt/C and Pt9Bi1/C catalysts by comparing the activity and selectivity of catalysts and the reactivity of molecules. The activity of catalysts will be determined by linear scan voltammetry, whereas the chemical function formed by oxidation of molecules will be monitored with respect to the electrode potential using in situ Fourier transform infrared spectroscopy. At last, chronoamperometry measurements at a fixed cell voltage will be performed in a 25-cm2 surface area electrochemical reactor (electrolysis cell) to accumulate reaction products at the Pt9Bi1/C anode, which could be further analyzed by high-performance liquid chromatography, 13C nuclear magnetic resonance, and mass spectroscopy. Results will show a 100% faradaic yield and a 100% selectivity towards the production of gluconate from glucose and methyl-glucuronate from methyl-glucoside with very low energy consumption that makes Pt9Bi1/C the outstanding electrocatalyst for such reactions.
Experimental
Synthesis of Catalysts by the “Water-in-Oil” Microemulsion Route
The protocols for the synthesis of Pt/C and Pt9Bi1/C are explained elsewhere [25, 27, 28, 30]. Briefly, appropriate amounts of metal salts (H2PtCl6•3 H2O and BiCl3 99.99% purity, Alfa Aesar) are dissolved in ultra-high purity water (Milli-Q, 18.2 MΩ cm, Millipore) to obtain a total metal concentration of 0.10 mol L−1 with the desired Pt/Bi atomic ratios. A 1.6 mL aliquot of aqueous solution containing dissolved metal salts is added to a homogeneous solution of 37 g n-heptane (99% purity, Acros Organics) and 16.1 g polyethylene glycol dodecyl ether (Brij® L4, Sigma-Aldrich). The mixture is stirred until a translucent and stable microemulsion is obtained. Then, 100 mg of sodium borohydride are added as a reducing agent. After reduction of metal salts, a given amount of carbon powder (Vulcan XC72, Cabot Corp.) is added to the colloidal solution to reach a metal loading of 40 wt%. The mixture is then filtered and washed several times with acetone and ultrapure water. The catalytic powder is dried under air overnight in an oven at 343 K before undergoing a thermal treatment for 2 h at 473 K under air to eliminate remaining traces of surfactant.
Catalysts are characterized by thermogravimetric analysis (TGA) to check the metal loading on carbon (TA Instrument model SDT Q600), by transmission electron microscopy (TEM) to evaluate the mean size of particles (JEOL JEM 2100 UHR microscope with a resolution of 0.19 nm), by X-ray diffraction (XRD) to explore the microstructure of materials (PANalytical Empyrean X-ray diffractometer), and by atomic absorption spectroscopy (AAS) after complete dissolution of the catalyst to determine the bulk composition (Perkin Elmer AA200 atomic absorption spectrometer).
Electrochemical Measurements
Cyclic voltammograms (CV) and linear sweep voltammograms (LSV) are recorded at 293 K in a thermostated three-electrode cell using a Voltalab PGZ402 potentiostat (Radiometer Analytical). The reference electrode is a reversible hydrogen electrode (RHE), the counter electrode is a 6-cm2 surface area glassy carbon plate, and the working electrode consists in the catalytic layer deposited on a 0.0707-cm2 geometric surface area glassy carbon disc as substrate. The electrolytes consist in a N2-purged 0.10 mol L−1 NaOH (semiconductor grade 99.99%, Sigma-Aldrich) solution for catalyst characterization, with addition of 18.0 g L−1 of glucose or 19.4 g L−1 methyl-glucoside (99%, Sigma-Aldrich) for electrocatalytic activity evaluations. The working electrode preparation is described elsewhere [25, 27,28,29,30]. Briefly, an aliquot of 3 μL of a catalytic ink (containing 17.7 mg of catalytic powder, 2.646 mL of ultra-high purity water, and 354 μL of 5 wt% Nafion suspension in aliphatic alcohols from Sigma-Aldrich) is dispensed onto the glassy carbon disc, which corresponds to a metal loading of 100 μgmetal cm−2 on the working electrode.
The scheme of the setup for chronoamperometry measurements (CA) at 293 K is presented in supporting information (SI 1). A 25-cm2 single filter press–type electrolysis cell is fitted with a Pt9Bi1/C anode (2.0 mgmetal cm−2) and a Pt/C cathode (0.5 mgPt cm−2) separated by a blotting paper. A 30 mL solution containing 0.10 mol L−1 NaOH and 18.0 g L−1 glucose or 19.4 g L−1 methyl-glucoside (0.10 mol L−1 each) feeds in recirculation at 25 mL min−1 both compartments of the cell. The icell(t) curves are recorded at the constant cell voltages (Ucell = 0.30 V for glucose oxidation and Ucell = 0.50 V for methyl-glucoside oxidation) by using interfaced Agilent E3614 generator to fix the voltage and two Agilent 34405A multimeters to record the applied cell voltage and the current.
Every hour, a 0.6 mL aliquot of the solution is analyzed by high-performance liquid chromatography (HPLC) using a Knauer Azura HPLC equipped with a Transgenomic ICSep COREGEL 107H column for organic acids, aldehydes, alcohols, and ketone molecule separation. Because no standard is available for the expected product from methyl-glucoside oxidation, the comparison of the HPLC peak positions is made with respect to those recorded with standards from glucose and xylose oxidation products at 0.10 M, neutralized with NaOH (when necessary): sodium gluconate, lithium xylonate, glucuronic acid, and glucaric acid (purity ≥ 97%, except for lithium xylonate with purity ≥ 95%). Chromatography analyses are performed with 0.007 mol L−1 H2SO4 aqueous solution as eluent at 0.6 mL min−1 flow rate with a UV detector set at λ = 210 nm. After a 6-h electrolysis experiment, the solutions are analyzed by 13C nuclear magnetic resonance (NMR) using a Bruker Ultrashield 400 plus with a Bruker AD-XT pump and a Bruker B-ACS 60 automatic sample changer. The samples are first neutralized by HCl, then lyophilized and put in D2O (deuterium oxide, 99.9 atom % D, Sigma-Aldrich) for analysis. The exact mass of compounds is determined by mass spectroscopy (MS). A 3-μL sample is injected in direct mode using an autosampler SIL 20AC HT. Analysis is performed in a negative mode (M − 1) with an interface temperature of 623 K, a DL temperature of 523 K, a nebulizer gas flow of 1.5 L min−1, a heat block temperature of 473 K, and a drying gas flow of 10 L min−1. Samples are eluted using a LC-20AD pump by a solution of water/acetonitrile (70/30) containing 5 mM ammonium acetate and 0.1 wt% acetic acid (flow rate of 1 mL min−1). Scan measurement is operated over the m/z range from 100 to 1000. The remaining methyl-glucoside concentrations after the 6-h electrolysis are analyzed by a HPLC equipped with a refractometer (SI 2). Analysis is done at 343 K using a HPLC Shimadzu apparatus composed of a Zorbax NH2-column (250 × 4.6 mm), a Shimadzu LC-20AD pump, and a Shimadzu Waters 2014 detector. The mobile phase (0.4 mL min−1) is an acetonitrile/water 20/80 v/v mixture. The calibration curves are given in SI 2.
In Situ Fourier Transform Infrared Spectroscopy Measurements
In situ Fourier transform infrared spectroscopy (in situ FTIRS) experiments are performed with a Bruker IFS 66 FTIR spectrometer modified for beam reflection on the electrode surface at a 65° incident angle. To remove interferences from atmospheric water and CO2, the beam path is evacuated. An Infrared Associates liquid nitrogen–cooled HgCdTe detector is used. The spectral resolution is 4 cm−1, and each spectrum is obtained by averaging 512 spectra recorded for 35 s. Spectra are recorded every 0.05 V during the linear voltammetry carried out in a 0.10 mol L−1 NaOH and 18.0 g L−1 glucose (19.4 g L−1 methyl-glucoside) electrolyte at 1 mV s−1 (i.e., each spectrum represents the average signal recorded for 0.035 V range every 0.050 V) from 0.050 to 1.200 V vs RHE. The experimental details of the electrochemical setup for SPAIRS (single potential–alteration IR spectroscopy) are described elsewhere [31, 32]. Spectra were normalized as follows:
where RE is the reflectivity recorded at the electrode potential E during the positive potential scan and RERef is the reflectivity recorded at 0.050 V vs RHE. Therefore, negative absorption bands correspond to the formation of species at the electrode surface, and positive absorption bands correspond to the consumption of species at the electrode surface.
Results and Discussion
Physicochemical Characterization of Catalysts
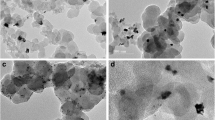
The morphology and the mean size of carbon-supported materials are observed by TEM, whereas the microstructure is observed by XRD. Figure 1a,b shows typical micrographs of Pt/C and Pt9Bi1/C catalysts, respectively, and as the corresponding size distribution histograms are presented in Fig. 1c,d, respectively. The metal particles are quasi-spherical in shape and relatively well-dispersed on the carbon support, although some agglomerates could be found. From the counting of at least 300 isolated particles, the metal particle mean sizes are found to be ca. 5.2 nm and ca. 4.7 nm for Pt/C and Pt9Bi1/C, respectively.

Typical TEM images and size distribution histograms for Pt/C (a, c) and Pt9Bi1/C (b, d)
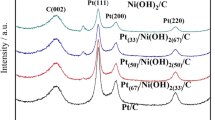
XRD patterns of the Pt/C and Pt9Bi1/C catalysts are depicted in Fig. 2. Both patterns exhibit diffraction peaks of the different crystallographic planes located at the characteristic positions for a face-centered cubic (fcc) structure, typical of metallic Pt. A small diffraction peak located at ca. 25° is attributed to the (002) plane of turbostratic graphite. The apparent mean crystallite sizes (Lv) are calculated using the Scherrer equation from the (111) diffraction peak located close to 2θ = 40°, leading to mean crystallite size values of 3.5 nm and 4.3 nm for Pt/C and Pt9Bi1/C, respectively. Due to size and strain effects, these values of crystallite sizes are underestimated but still consistent with those obtained by TEM. The positions of diffraction peaks related to (111), (200), (220), and (311) planes allow calculating for all materials the average values of the lattice parameters (a) using the Bragg equation [33]. These values are 0.392 nm for both Pt/C and Pt9Bi1/C, indicating the formation of non-alloyed Pt9Bi1 structure using the “water-in-oil” microemulsion method, in agreement with previous works [29, 34]. Previous studies by molecular dynamics simulations and deep physicochemical characterization [29] have shown that Pt9Bi1 nanoparticles prepared by the low-temperature microemulsion method consisted in Bi shell structures surrounding and interacting with well-crystallized Pt cores.

XRD patterns of Pt/C and Pt9Bi1/C nanomaterials
The bulk compositions of binary catalysts as determined by AAS give a Pt/Bi atomic ratio of 89/11, close to the expected ones. At last, the metal loadings on carbon as determined by TGA are 39 wt% and 37 wt% for Pt/C and Pt9Bi1/C, respectively, again close to the expected loadings of 40 wt%. All these results also demonstrate that the “water-in-oil” microemulsion synthesis method is quantitative, meaning that all metal atoms are incorporated in the catalytic particles.
Electrocatalytic Behavior of Catalysts
The surfaces of the catalysts are first characterized by cyclic voltammetry. Figure 3 displays the typical CVs of Pt/C (black line) and Pt9Bi1/C (gray line) recorded in a N2-purged 0.10 M NaOH electrolyte. The current densities are reported versus the geometric surface area of the electrode (i.e., 0.0707 cm2) loaded with the same mass of metals. The Pt/C catalyst presents redox features in the potential region from 0.05 to 0.45 V vs RHE corresponding to hydrogen adsorption on (negative currents) and desorption from (positive currents) the Pt surface, and in the potential range from 0.50 to 1.20 V vs RHE corresponding to Pt surface oxidation into PtO (positive currents) and the reduction of PtO into Pt (negative currents). After modification of Pt by 10 at% of bismuth, the redox features in the 0.05 to 0.45 V vs RHE range have disappeared, which indicates inhibition of the hydrogen adsorption process on Pt. This behavior was already observed by different authors [34, 35], although still not completely understood. It suggests either a (quasi) full coverage of Pt surface by Bi structures or an electronic effect of Bi on Pt inhibiting the hydrogen adsorption process. In the potential range from 0.50 to 1.20 V vs RHE, the positive current peaks at ca. 0.62 V and 0.82 V vs RHE have been proposed to correspond to the oxidation of bismuth atoms from Bi(0) to Bi (II) [36] and Bi (III) [37] in low and strong interactions with platinum surface, respectively [38]. The reduction peak at ca. 0.72 V vs RHE corresponds to the simultaneous reduction of Pt and Bi oxides. The electroactive surface area could be calculated for the Pt/C catalyst from the integration of the hydrogen desorption current densities in the 0.05 to 0.45 V vs RHE region after correction of the double-layer capacitive current (41 m2 gPt−1); but unfortunately, it cannot be calculated for the Pt9Bi1/C catalyst for which the hydrogen adsorption process is inhibited.

Cyclic voltammograms of Pt/C (black line) and Pt9Bi1/C (gray line) nanomaterials, (scan rate = 5 mV s−1, N2-purged 0.10 M NaOH electrolyte, and T = 293 K)
The linear scan voltammetry (LSV) curves for the electrooxidation of glucose and methyl-glucoside over Pt/C (black lines) and Pt9Bi1/C (gray lines) are compared in Fig. 4. The onset potentials Eonset and maximum current density in the catalytically interesting potential region (below 0.8 V vs RHE) jmax are given in Table 1. Regarding the LSVs recorded for glucose electrooxidation in Fig. 4a, Pt9Bi1/C displays much higher activity than Pt/C as lower onset potential and higher current densities are recorded over the whole potential range studied. It is worth to notice the very low onset potential between 0.00 and 0.06 V vs RHE, which makes this catalyst the outstanding one for glucose electrooxidation. Indeed, Ghoshs et al. [39] prepared Au-based NPs on graphene nanosheets active for glucose electrooxidation from the onset potential of ca. 0.30 V vs RHE; Yan et al. [40] obtained an onset potential of ca. 0.40 V vs RHE on non-alloyed PdxAuy/C, whereas Rafaïdeen et al. [25] developed an active-alloyed Pd3Au7/C catalyst presenting an onset potential of ca. 0.1 V vs RHE.

Polarization curves of 0.10 M glucose oxidation (a) of 0.10 M methyl-glucoside oxidation (b) recorded on Pt/C (black lines), on Pt9Bi1/C (gray lines). Scan rate = 5 mV s−1, N2-purged 0.10 M NaOH electrolyte, and T = 293 K
Regarding the LSVs recorded for methyl-glucoside electrooxidation in Fig. 4b, the same trends as for glucose electrooxidation are observed: Pt9Bi1/C is the most active catalyst from ca. .22 V vs RHE. Pt/C displays lower activity for this reaction. But the onset potentials for methyl-glucoside oxidation are shifted towards higher values than those obtained for glucose oxidation, by more than 150 mV and ca. 350 mV on Pt9Bi1/C and Pt/C, respectively. The second remark is that over Pt9Bi1/C, the general shape of methyl-glucoside oxidation differs from that of glucose oxidation, showing only one well-defined oxidation peak rather than an oxidation wave and a kind of current plateau at higher potentials. The shape of the LSVs for methyl-glucoside oxidation resembles a lot those obtained for the oxidation of full polyols, such as glycerol [22, 27] and ethylene glycol [41], which seems to indicate that other functions than the anomeric one are involved in the oxidation reaction.
In Situ Infrared Spectroscopy Measurements
To shed light on the chemical functions formed for the oxidation reactions of glucose and methyl-glucoside at Pt/C and Pt9Bi1/C electrodes, infrared spectra are recorded over the 0.050 to 1.200 V vs RHE potential range every 50 mV with the very low potential scan rate of 1 mV s−1 (Fig. 5).

Infrared spectra recorded for the oxidation of 0.10 M glucose (a, b) and 0.10 M methyl-glucoside in 0.10 M NaOH (c, d) electrolyte on Pt/C (a, c) and Pt9Bi1/C (b, d) catalysts (scan rate = 1 mV s−1, resolution 4 cm−1, and T = 293 K; certical scale: ΔR/R). For the interpretation of the references to colors in this figure legend, the reader is referred to the web version of this article
Figure 5a,b presents the spectra recorded for the oxidation of glucose at Pt/C and Pt9Bi1/C electrodes, respectively. The spectra on both catalysts display the same main infrared absorption bands, but they appear at lower potentials and show higher intensities for Pt9Bi1/C than for Pt/C, confirming the higher activity of the former catalyst as evidenced by voltammetry study. Based on literature data [42,43,44], the infrared absorption bands located at ca. 1044, 1092, and 1136 cm−1 are assigned to the ν(C–O) stretching mode of CH2–OH functions, whereas the medium infrared absorption band at 1414 cm−1 and the strong one at 1581 cm−1 are attributed to the formation of gluconate. The infrared absorption band at ca. 1360 cm−1 was assigned by Beden et al. [42] to δ(CH2) mode of gluconate or gluconolactone. On Pt/C catalysts, a strong absorption band arise in the 2000 to 2100 cm−1 wavenumber range from 0.10 V vs RHE that is assigned to the formation of linearly adsorbed CO [45]. This indicates that Pt/C material can break the C–C bond of the glucose molecules from relatively low electrode potentials. This is confirmed by the appearance of the absorption band centered at 2343 cm−1 for potentials higher than 0.60 V vs RHE. This infrared absorption band is typical of the formation of interfacial CO2 [46,47,48] and indicates that degradation products from glucose, involving C–C bond breaking, are formed. In the case of Pt9Bi1/C catalyst, no absorption band in the 2000 to 2100 cm−1 wavenumber region is visible, but the band at 2343 cm−1 appears for potentials higher than 0.70 V vs RHE. These observations show that the Pt9Bi1/C material has much lower ability to break the C–C bond at low potentials than Pt/C. The high similarity of the spectra in the 1000 to 1800 cm−1 wavenumber range recorded on Pt/C and Pt9Bi1/C materials indicates that gluconate is the main reaction product formed on Pt9Bi1/C catalyst according to the chemical equation presented in the following electrochemical semi-equation (Eq. 2):

Figure 5c,d presents the spectra recorded for the oxidation of methyl-glucoside at Pt/C and Pt9Bi1/C electrodes, respectively. Again, the spectra on both catalysts display the same main infrared absorption bands, but again, they appear at lower potentials and show higher intensities for Pt9Bi1/C than for Pt/C, confirming the higher activity of the former catalyst as evidenced by the voltammetry study. The comparison with the spectra recorded for glucose oxidation shows that if the same infrared absorption bands located at 1414 cm−1 and 1581 cm−1 are present in the spectra of methyl-glucoside oxidation, the band at 1360 cm−1 is no more present and a band at ca. 1305 cm−1 appears. The peaks at 1305, 1414, and 1581 cm−1 are assigned to the formation of carboxylate species. The absence of the peak at 1360 cm−1 assigned to δ(CH2) vibration mode of gluconate or gluconolactone translates into a different reaction pathway without formation of this CH2 group (typical of the gluconate/gluconolactone) in the reaction products of methyl-glucoside oxidation. It is also worth to note the absence, in the case of Pt9Bi1/C, of the absorption band at ca. 2343 cm−1 related to the formation of interfacial CO2 over the whole studied potential range. This demonstrates that the anomeric function is protected, which avoids C–C bond cleavage. It also indicates that in the case of glucose oxidation, the C–C bond-breaking mechanism operates through a successive transformation of the carboxylate group into carbonate or CO2, creating a carboxylate with one less carbon and shortening the molecule one carbon at a time. Indeed, Rafaïdeen et al. [25] showed the presence of xylonate and traces of threonate after the 6-h electrolysis of 0.1 M glucose on a Pd3Au7/C catalyst. This means that the –CHOH–COO− group is less stable than the primary or secondary alcohol groups. From all these observations, it appears that the product formed from methyl-glucoside oxidation presents a carboxylate group which is different to that of gluconate from glucose oxidation, without C–C bond breaking. It is then reasonable to propose that methyl-glucuronate is formed according to the electrochemical semi-reaction presented in Eq. (3):

LSVs (Fig. 4a, b) show that Pt9Bi1/C catalyst displays higher activity (higher current densities) than Pt/C catalyst towards glucose and methyl-glucoside oxidation reactions over the whole potential range studied. In situ FTIRS measurements (Fig. 5) show that gluconate and methyl-glucuronate are obtained as main products on both catalysts for glucose and methyl-glucoside oxidation reactions, respectively. The modification of a catalytic metal, namely platinum, by foreign atoms can affect the amount and composition of chemisorbed species and further the course of their electrooxidation [49]. Actually, three main effects of surface modification have been proposed [50]: the ligand effect (electronic effect) that stipulates changes in surface electronic properties induced by the foreign atoms altering both adsorption and intramolecular bond energies, the ensemble effect (third body or geometric effect) that stipulates that blocking particular adsorption sites by foreign atoms favors a given reaction pathway without decreasing the activity of the catalytic surface, and the bifunctional mechanism that stipulates that OH species adsorbed on the foreign atoms can be provided to the adsorbed organic species on the catalytic surface to complete the oxidation reaction.
Concerning the electrooxidation of glucose on noble metals such as Au and Pt, it is admitted that the first step involves the adsorption of the anomeric carbon and that the formation of gluconolactone occurs [42, 51,52,53]. In low potential region and intermediate potential region on pure Pt surface, gluconolactone can desorb and be converted into gluconate in presence of hydroxyl ions in the electrolyte [54]. For potentials higher than 0.6 V vs RHE, Pt surface can activate water molecules and adsorb OH species, allowing a Langmuir-Hinshelwood mechanism to occur between adsorbed gluconolactone and adsorbed OH species leading to direct carboxylate formation. The increase of activity in the low potential region on the Bi-modified Pt nanoparticles cannot be explained in terms of the bifunctional mechanism, because Bi atoms are expected to be at the zero valent state in the potential range below 0.25 V vs RHE [35, 55]. Therefore, the activity enhancement induced by the presence of bismuth at low electrode potentials is due either to the ligand or to the ensemble effects.
In situ FTIRS measurements for glucose oxidation show the formation of adsorbed CO on Pt/C, but not on Pt9Bi1/C. This observation can be explained by the ensemble effect: the dilution of Pt surface atoms by Bi atoms avoids the multi-bonded adsorption mode of glucose, favors linearly bonded intermediates due to steric effect [56], and further prevents the formation of COads on the Pt surface. This effect will enhance not only the selectivity of the reaction but also the activity of the catalyst by limiting its poisoning by strongly adsorbed CO species. However, the ligand effect cannot be discarded, as it can also explain the total inhibition of hydrogen adsorption on the Pt surface modified by Bi atoms and the simultaneous reduction of Pt and Bi surface oxide observed in the Pt9Bi1/C CV of Fig. 3.
In the case of methyl-glucoside oxidation, the anomeric function is protected, and according to in situ FTIRS measurements and chronoamperometry studies (see below), methyl-glucuronate is formed. At low overpotentials, i.e., E ≤ 0.40 V vs RHE, Pt surface is not expected to activate water and to adsorb OH species necessary to complete the oxidation reaction into carboxylate. This fact explains the onset potential of ca. 0.42 V vs RHE for methyl-glucoside oxidation into methyl-glucuronate through a Langmuir-Hinshelwood mechanism on Pt/C. In this case, the addition of Bi leads to shift the onset potential down to ca. 0.22 V vs RHE (Fig. 4b), where the redox transition from Bi(0) to Bi (II) (Bi (OH)2) is expected to occur [29] allowing the bifunctional effect to take place. This mechanism can also be invoked for enhanced activity of the glucose electrooxidation on Pt9Bi1/C catalyst for electrode potentials higher than 0.25 V vs RHE.
Chronoamperometry Measurements (CA)
Previous voltammetric studies and infrared measurements under electrode potential control allow determining the anode potential range of interest to accumulate products with the selectivity as high as possible. Because methyl-glucoside is less electro-reactive than glucose, the cell voltage has to be increased in this case to obtain enough conversion for further analysis of reaction products; chronoamperometry measurements of glucose and methyl-glucoside oxidations at fixed constant cell voltages of 0.3 V and 0.5 V, respectively, are performed at 293 K for 6 h in a 25-cm2 surface area filter press–like cell fitted with a Pt/C catalyst at the cathode (40 wt% Pt, 0.5 mgPt cm−2) and a Pt9Bi1/C catalyst at the anode (40 wt% metal, 2.0 mgmetal cm−2). In the cathodic compartment, the Pt/C catalyst served for the hydrogen evolution reaction (HER) according to the following reaction:
The hydrogen evolution counter-reaction at a Pt/C cathode has a reversible potential of 0.00 V vs RHE, and this reaction occurs with very high kinetics. This means that the anode potential value is close to the cell voltage value (slightly lower due to the small negative overpotential at the cathode), because the cell voltage corresponds to the difference of electrode potentials (Ucell = Ea – Ec, where Ea is the anode potential, Ec is the cathode potential, and Ucell is the cell voltage). It can then be assumed that under actual operational conditions, the anode potentials remain between 0.25 and 0.30 V vs RHE for glucose electrooxidation and between 0.45 V and 0.50 V vs RHE for methyl-glucoside electrooxidation. In a previous paper, Rafaïdeen et al. [25] showed indeed that the Pt/C cathode potential remained between − 0.03 and − 0.01 V vs RHE for the 6-h electrolysis experiment of glucose electro-conversion at a cell voltage of 0.4 V.
Figure 6 shows the i(t) curves recorded for 6 h electro-reforming of glucose (Fig. 6a) and methyl-glucoside (Fig. 6b) at cell voltages of 0.30 V and 0.50 V, respectively. The initial currents for both glucose and methyl-glucoside electrolysis measurements reach values between 0.05 and 0.06 A. For both compounds, the current decreases monotonously over the first hours of experiments and then reach plateaus of ca. 0.006 A and 0.012 A for glucose and methyl-glucoside, respectively. Pt9Bi1/C catalyst could undergo a first rearrangement of the surface atoms or some poisoning of the surface by adsorbed species from glucose and methyl-glucoside. For the electrooxidation of methyl-glucoside, the bifunctional mechanism [57, 58] is required in order to bring the extra oxygen atom necessary to complete the oxidation reaction towards 1-methyl-glucuronate according to reaction (3). Therefore, the poisoning tolerance of the catalyst surface will depend on the ability of the surface to adsorb OH groups from water and species from methyl-glucoside (and therefore on the coverage balance between both kinds of adsorbates at the surface) and will rapidly reach a steady state. At so low anode potential (ca. 0.50 V vs RHE), it is likely that the balance between both adsorbed species at the surface shifts slightly towards higher coverage of methyl-glucoside species rather than towards OH groups, which explains the rapid decay in current. In contrary, in the case of glucose electrooxidation, at a so low anode potential (close to 0.30 V vs RHE), the mechanism involves the electrochemical formation and desorption of gluconolactone [42, 51, 54], which does not need extra oxygen atoms to be formed. Gluconolactone can be further hydrolyzed chemically into gluconate in the alkaline medium, and the decay of the current is only due to the difference in charge transfer kinetics and desorption rate of gluconolactone species. The difference between methyl-glucoside and glucose oxidation mechanisms can be responsible of the different rates of the current decay observed. The continuous slow current decay for longer electrolysis times corresponds to the depletion of reactants in the medium, as it was previously observed for glucose and xylose electrooxidation on a Pd7Au3/C catalyst [25].

i(t) curves recorded at 293 K in the 25-cm2 electrolysis cell for the oxidation of 0.10 M glucose (a) in 0.10 M NaOH aqueous solution at 0.3 V and 0.10 M methyl-glucoside (b) in 0.10 M NaOH aqueous solution at 0.5 V
Every hour, an aliquot of 0.6 mL of solution is sampled and analyzed by HPLC. Figure 7a shows the liquid chromatograms recorded as a function of time for glucose electrolysis. The retention time of the main peak at ca. 7.9 min corresponds to that of gluconic acid in Fig. 7b. Figure 7c shows the chromatograms recorded as a function of time for methyl-glucoside electrolysis. The retention time of the main peak in this case does not correspond to any standard in Fig. 7b (no commercial standard of methyl-glucuronate is available). But its position is between that of glucuronic acid (ca. 7.4 min) and that of gluconic acid (ca. 7.9 min). In both cases, glucose and methyl-glucoside oxidation products, a peak at a retention time of ca. 6.7 min is observed after 1-h electrolysis and does not increase significantly for longer times. Such a peak was also already observed in the case of xylose electrooxidation [25], and we propose that it could correspond to the dead time peak or to some rapidly oxidized impurity present in commercial samples. In the case of glucose (Fig. 7a), the small peak at ca. 7.6 min disappears after 2-h electrolysis, and therefore, we propose that it is due to the oxidation of an impurity present as a trace. In the case of methyl-glucoside, two very small peaks are present at ca. 7.25 min and 8.6 min; the small intensities of which indicate that these products are present as traces. At last, another peak at the higher retention time of ca. 9.1 min appears and increases with time.

Chromatograms recorded as a function of time for the electrolysis of 0.10 M glucose (a) and 0.10 M methyl-glucoside (c) in 0.10 M NaOH aqueous solutions. The chromatograms for standard solutions at 0.10 M of gluconic acid, glucaric acid, glucuronic acid, and xylonic acid are given in (b). For the interpretation of the references to colors in this figure legend, the reader is referred to the web version of this article
Concerning glucose oxidation, the experimental concentrations of gluconic acid as determined by HPLC are compared in Fig. 8 to the theoretical one determined from the integration of the current along the electrolysis measurement time, considering that only gluconate is formed according to Eq. (2) (two electrons involved for the oxidation of one glucose molecule). Both curves in Fig. 8 present a parallel growth and are very close to each other, with that determined from current integrations slightly above that determined from HPLC measurements. The analysis by 13C NMR of the reaction products after 6-h CA of 0.10 M glucose solution at 0.30 V was performed at the end of electrolysis measurements to confirm the nature of the products formed at the Pt9Bi1/C anode, and the experimental 13C NMR spectrum has been compared with those of standard 0.10 M glucose and 0.10 M gluconic acid solutions (SI 3). In addition to signals related to remaining glucose at δ = 92.1 ppm (α-glucose) and δ = 95.9 ppm (β-glucose), signals at δ = 62.6 ppm and δ = 177 ppm are also present in the spectrum of standard gluconic acid. Owing to the chemical shift value of ca. 177 ppm, this last signal is rather assigned to gluconic acid than to gluconolactone [53]. Zhang et al. [59] explained that the formation of δ- and γ-gluconolactone through a lactonization reaction occurred in acidic media. However, in the present case, the reaction mixture was not acidified before measurements, only neutralized to pH 7, which explains the absence of peaks related to lactones in NMR spectra of reaction mixture.

Comparison of the theoretical concentration of gluconate assuming a 100% selectivity (black line) with the experimental one determined by HPLC (red line). For the interpretation of the references to colors in this figure legend, the reader is referred to the web version of this article
Moreover, mass spectrometry measurements performed on the sample after 6-h CA experiment (SI 4) showed a unique peak at m/z = 195, which indicated that only one product was present with the molecular mass of 196 g corresponding to gluconic acid. Therefore, it can be concluded that the difference between both straight lines in Fig. 8 is due to a systematic error (either slight overestimation of the current density or slight underestimation of the HPLC peak surface area), and that the selectivity towards gluconic acid on Pt9Bi1/C catalyst is 100% for ca. 40% glucose conversion. Holade et al. [53] announced a 100% selectivity towards gluconate at 65% conversion for the electrooxidation of glucose on Au/C catalyst at a cell voltage of 0.8 V, although two HPLIC (high performance liquid ionic chromatography) peaks were obtained (the second one at slightly higher retention time than that assigned to gluconate). Rafaïdeen et al. [25] obtained 87% selectivity for 67% glucose conversion at a cell voltage of 0.4 V after 6-h electrolysis, the main coproduct revealed by a second HPLC peak at a slightly higher retention time than that for gluconate being xylonate. In the present contribution, the selective oxidation of glucose into gluconate was performed at a cell voltage of 0.3 V. Because the electrical energy consumption is only dependent on the cell voltage [60], energy savings of 25% with respect to results of Rafaïdeen et al. [25] and of 62.5% with respect to results from Holade et al. [53] are achieved using Pt9Bi1/C anode catalyst.
For methyl-glucoside oxidation, the integration of the current along the electrolysis measurement time, considering that only methyl-glucuronate is formed according to Eq. (3) (four electrons involved for the oxidation of one methyl-glucoside molecule), allows calculating a final methyl-glucoside conversion of 36%. Analysis by 13C NMR of the reaction products is performed after 6-h electrolysis at the cell voltage of 0.50 V. The reaction mixture is first neutralized with HCl, then lyophilized, and at last, dissolved into D2O. The comparison of the experimental 13C NMR spectrum obtained after 6-h electrolysis with that of a 0.10 M methyl-glucoside standard and that predicted (TopSpin 4.0.6 software) from the structure presented in Fig. 9 is given in supporting information (SI 5). Both spectra contain the typical signal for primary carbon bearing a carboxyl group at δ = 173.2 ppm, whereas that at δ = 55.8 ppm signifies the presence of a methyl group. Hence, the species formed after 6 h of electrolysis of methyl-glucoside may correspond to the structure of methyl-glucuronic acid.

Structure of the compound used to predict the 13C NMR spectrum for comparison with the experimental one from 6-h methyl-glucoside electro-reforming at 0.50 V (see SI 5)
In addition, and because several peaks are obtained in the chromatograms of Fig. 7b, mass spectrometry measurements after 6-h electrolysis point out the presence of two compounds (Fig. 10a): the main one at m/z = 207 corresponds to the negatively ionized methyl-glucuronic acid (molecular mass of 208 g mol−1) and the second one at m/z = 253 is already present in the spectra of a fresh methyl-glucoside solution (Fig. 10b) and therefore does not correspond to a reaction product after electrolysis. This is an important result indicating that according to MS, only methyl-glucuronate is formed at the Pt9Bi1/C anode under our experimental conditions. The small peak at ca. 9.1 min in chromatograms of Fig. 7b, which increases slightly with time, does not give any MS peak, and therefore corresponds likely to a dimer which is further dissociated during acidification and/or ionization processes. The final methyl-glucoside conversion, as determined from the HPLC calibration curve (SI 2), reaches 38%. Both conversions, determined by current integration and HPLC analysis, are very consistent, the difference likely lying in the experimental standard deviations, which confirms the formation of only one product: methyl-glucuronate. Those analytical results evidenced then a 100% selectivity towards methyl-glucuronate at ca. 36–38% methyl-glucoside conversion.

MS spectrum of reaction products from methyl-glucoside (a) electro-reforming for 6 h at 0.50 V using a Pt9Bi1/C anode catalyst and MS spectrum of a fresh methyl-glucoside solution (b)
These results show that the faradaic yields to produce gluconate and methyl-glucuronate are 100%, as well as the selectivities. Under these conditions, the costs related to the energy consumption for producing 1 ton of sodium gluconate at 0.3 V and 1 ton of sodium methyl-glucuronate at 0.5 V, together with the production of ca. 9 kg and ca. 17 kg of pure hydrogen, respectively (SI 6), are ca. 11.1 € and 17.5 €, respectively, assuming a mean electricity cost of ca. 0.15 €/kWh in Europe [61]. These costs represent less than 3% of, for example, the commercial prices of industrially produced gluconic acid or sodium gluconate ranging between ca. 600 to ca. 750 US $ (ca. 550 to ca. 680 €) per ton (Dezhou Huiyang Biotechnology Co., Ltd., Wuxi Fengmin Environmental Technology Development Co., Ltd., etc.).
Moreover, since glucuronate forms are considered a door to produce glucarate or glucaric acid, key molecules for the industry of bio-sourced plastics, the green electrochemical process described in the present contribution, based on a very active and selective Pt9Bi1/C catalyst, is of paramount importance to propose alternative solution to petro-sourced resources for polymer industries.
Conclusion
Pt9Bi1/C catalyst exhibits lower onset potentials and much higher activity than Pt/C for both glucose and methyl-glucoside electrooxidation. Moreover, according to in situ infrared measurements, Pt9Bi1/C catalyst is less prone to break the C–C bond than the Pt/C one, therefore displaying also higher selectivity towards C6 and C7 oxidation compounds from glucose and methyl-glucoside, respectively. The final reaction products are in both cases carboxylate species, but these species are different for glucose and methyl-glucoside electrooxidation reactions. Chronoamperometry measurements in a real 25-cm2 electrosynthesis reactor conducted at 293 K for 6 h at the low voltages of 0.3 V and 0.5 V for glucose and methyl-glucoside electro-reforming reactions, respectively, on a Pt9Bi1/C anode allowed accumulating reaction products for further analyses by HPLC, 13C NMR, and MS. Several new important insights are pointed out:
-
Gluconate is selectively formed from glucose oxidation at potentials lower than 0.60 V vs RHE. For higher potentials, CO2 and therefore degradation products from glucose over-oxidation are formed. Reaction product analysis after chronoamperometry at 0.30 V showed 100% faradaic yield and selectivity towards gluconate.
-
Methyl-glucoside oxidation occurs without CO2 formation over the whole potential range studied, evidencing the protection of the anomeric function.
-
Methyl-glucoside oxidation leads selectively to methyl-glucuronate from 0.22 V vs RHE. Reaction product analysis after chronoamperometry at 0.50 V showed 100% faradaic yield and selectivity towards methyl-glucuronate. Pt9Bi1/C catalyst shows remarkably high activity and selectivity for the oxidation of primary alcohol groups, reaction considered one of the important problems in organic chemistry.
Therefore, Pt9Bi1/C material corresponds to the outstanding electrocatalyst for glucose and methyl-glucoside oxidation since onset potentials lower than 0.05 V and close to 0.22 V vs RHE for the selective oxidations of glucose and methyl-glucoside, respectively, are much lower than those obtained for Pt- [62], Pd- [63], and Au-based [39, 40, 53, 64] catalysts.
At last, the low energetic cost and the high yield towards methyl-glucuronate of the methyl-glucoside electrocatalytic process based on a very active and selective Pt9Bi1/C catalyst open the door to green alternative solutions to fossil resources for bio-sourced polymer industries.
References
M.A. Andrews, S.A. Laeren, G. L. Gould, in Carbohydrates as organic raw materials II, ed. G. Descotes (VCH, Weinheim, 1993), pp. 3–27
C. Chatterjee, F. Pong, A. Sen, Chemical conversion pathways for carbohydrates. Green Chem. 17, 40 (2015)
I. M. O’Hara, Z. Zhang, W.O. Doherty, C.M. Fellows, in Green chemistry for environmental remediation, ed. R. Sanghi, V. Singh (John Wiley & Sons, Inc., Salem, 2011), pp. 506–560
M. Benoit, A. Rodrigues, Q. Zhang, E. Fourré, K. De Oliveira Vigier, J.M. Tatibouët, F. Jérôme, Depolymerization of cellulose assisted by a nonthermal atmospheric plasma, Angewandte Chemie Int. Ed. 50, 8964 (2011)
M. Benoit, A. Rodrigues, K. De Oliveira Vigier, E. Fourré, J. Barrault, J.M. Tatibouët, F. Jérôme, Combination of ball-milling and non-thermal atmospheric plasma as physical treatments for the saccharification of microcrystalline cellulose. Green Chem. 14, 2212 (2012)
Q. Zhang, M. Benoit, K. De Oliveira Vigier, J. Barrault, F. Jérôme, Pretreatment of microcrystalline cellulose by ultrasounds: effect of particle size in the heterogeneously-catalyzed hydrolysis of cellulose to glucose. Green Chem. 15, 963 (2013)
S. Carrettin, P. Mc Morn, P. Johnston, K. Griffin, C.J. Kiely, G.A. Attard, G.J. Hutchings, Oxidation of glycerol using supported gold catalysts. Top. Catal. 27, 131 (2004)
R.A. Sheldon, J. Dakka, Heterogeneous catalytic oxidations in the manufacture of fine chemicals. Catal. Today 19, 215 (1994)
R.A. Sheldon, The E factor 25 years on: the rise of green chemistry and sustainability. Green Chem. 19, 18 (2017)
P. Gallezot, Conversion of biomass to selected chemical products. Chem. Soc. Rev. 41, 1538 (2012)
M.J. Climent, A. Corma, S. Iborra, Converting carbohydrates to bulk chemicals and fine chemicals over heterogeneous catalysts. Green Chem. 13, 520 (2011)
A. Tathod, T. Kane, E.S. Sanil, P.L. Dhepe, Solid base supported metal catalysts for the oxidation and hydrogenation of sugars. J. Mol. Catal. A Chem. 388-389, 90 (2014)
N.J. Davis, S.L. Flitsch, Selective oxidation of monosaccharide derivatives to uronic acids. Tetrahedron Lett. 34, 1181 (1993)
E.J. Arjan, D. Nooy, A.C. Besemer, H. van Bekkum, Selective oxidation of primary alcohols mediated by nitroxyl radical in aqueous solution. Kinetics and Mechanism. Tetrahedron 51(8023) (1995)
Y. Hua, L. Huaqing, D. Zhiping, W. Yuanxin, Synthesis of glucuronic acid from methyl glucoside by heterogeneous selective catalytic oxidation. Chem. Eng. J. 207-208, 72 (2012)
P.J. Chen, C.H. Chiu, J.K. Tseng, K.T. Yang, Y.C. Chen, Ameliorative effects of D-glucuronolactone on oxidative stress and inflammatory/fibrogenic responses in livers of thioacetamide-treated rats. J. Funct. Foods 14, 154162 (2015)
R. Beerthuis, G. Rothenberg, N.R., Shiju, Catalytic routes towards acrylic acid, adipic acid and ε-caprolactam starting from biorenewables, Green Chem. 17, 1341 (2015)
P.N. Amaniampong, Q.T. Trinh, K. De Oliveira Vigier, D.Q. Dao, N.H. Tran, Y. Wang, M.P. Sherburne, F. Jérôme, Synergistic effect of high-frequency ultrasound with cupric oxide catalyst resulting in a selectivity switch in glucose oxidation under argon. J. Am. Chem. Soc. 141, 14772 (2019)
Y. Hua, D. Zhiping, Y. Zhiguo, W. Yuanxin, Selective oxidation of primary alcohol from methyl glucoside over Pd/La0.5Pb0.5Mn0.9Sn0.1O3 catalyst. Acta Chim. Sin. 67, 345 (2009)
M. Omri, M. Becuwe, C. Davoisne, G. Pourceau, A. Wadouachi, Nitroxide supported on nanometric metal oxides as new hybrid catalysts for selective sugar oxidation. J. Colloid Interface Sci. 536, 526 (2019)
M.M. Zhao, J. Li, E. Mano, Z.J. Song, D.M. Tschaen, Oxidation of primary alcohols to carboxylic acids with sodium chlorite catalyzed by TEMPO and bleach: 4-methoxyphenylacetic acid. Org. Synth. 81, 195 (2005)
D. S. P. Cardoso, B. Šljukic, D. M. Santos ́and C. A. C. Sequeira, Organic electrosynthesis: from laboratorial practice to industrial applications, Org. Process. Res. Dev. 21, 1213 (2017)
M. Simões, S. Baranton, C. Coutanceau, Electrochemical valorisation of glycerol. ChemSusChem 5, 2106 (2012)
A. Zalineeva, M. Padilla, U. Martinez, A. Serov, K. Artyushkova, S. Baranton, C. Coutanceau, P.B. Atanassov, Self-supported PdxBi catalysts for the electrooxidation of glycerol in alkaline media. J. Am. Chem. Soc. 136(3937), 3937–3945 (2014)
T. Rafaïdeen, S. Baranton, C. Coutanceau, Highly efficient and selective electrooxidation of glucose and xylose in alkaline medium at carbon supported alloyed PdAu nanocatalysts. Appl. Catal. B Environ. 243(641) (2019)
P. Gallezot, Selective oxidation with air on metal catalysts. Catal. Today 37, 405 (1997)
J. Cobos-Gonzalez, S. Baranton, C. Coutanceau, A systematic in situ infrared study of the electrooxidation of C3 alcohols on carbon-supported Pt and Pt−Bi catalysts. J. Phys. Chem. C 120, 7155 (2016)
J. Cobos-Gonzalez, S. Baranton, C. Coutanceau, Development of bismuth-modified PtPd nanocatalysts for the electrochemical reforming of polyols into hydrogen and value-added chemicals. Chem. Electro. Chem 3, 1694 (2016)
B.S.R. Kouamé, S. Baranton, P. Brault, C. Canaff, W. Chamorro-Coral, A. Caillard, K. De Oliveira Vigier, C. Coutanceau, Insights on the unique electro-catalytic behavior of PtBi/C materials. Electrochim. Acta 329, 135161 (2019)
S. Lankiang, M. Chiwata, S. Baranton, H. Uchida, C. Coutanceau, Oxygen reduction reaction at binary and ternary nanocatalysts based on Pt, Pd and Au. Electrochim. Acta 182, 131 (2015)
B. Beden, C. Lamy, in Spectroelectrochemistry: theory and practice, ed. R. J. Gale (Plenum Press, New York, 1988), pp 189–2.61
A. Kabbabi, R. Faure, R. Durand, B. Beden, F. Hahn, J.M. Léger, C. Lamy, In situ FTIRS study of the electrocatalytic oxidation of carbon monoxide and methanol at platinum–ruthenium bulk alloy electrodes. J. Electroanal. Chem. 444, 41 (1998)
B.E. Warren, X-ray diffraction (Dover Publications, Inc., New York, 1990) pp. 251-254
M. Simoes, S. Baranton, C. Coutanceau, Enhancement of catalytic properties for glycerol electrooxidation on Pt and Pd nanoparticles induced by Bi surface modification. Appl. Catal. B Environ. 110, 40 (2011)
M.M. Tusi, N.S.O. Polanco, S.G. Da Silva, E.V. Spnacé, A.O. Nato, The high activity of PtBi/C electrocatalysts for ethanol electro-oxidation in alkaline medium. Electrochem. Commun. 13, 143 (2011)
J. Clavilier, J.M. Feliu, A. Aldaz, An irreversible structure sensitive adsorption step in bismuth underpotential deposition at platinum electrodes. J. Electroanal. Chem. 243, 419 (1988)
W. Zuo, W. Zhu, D. Zhao, Y. Sun, Y. Li, J. Liu, X. W. Lou, Bismuth oxide: a versatile high-capacity electrode material for rechargeable aqueous metal-ion batteries, energy environ. Sci. 9, 2881 (2016
M. Simões, S. Baranton, C. Coutanceau, Influence of bismuth on the structure and activity of Pt and Pd nanocatalysts for the direct electrooxidation of NaBH4. Electrochim. Acta 56, 580 (2010)
S. Ghosh, Y. Holade, H. Remita, K. Servat, P. Beaunier, A. Hagège, K.B. Kokoh, T.W. Napporn, One-pot synthesis of reduced graphene oxide supported gold-based nanomaterials as robust nanocatalysts for glucose electrooxidation. Electrochim. Acta 212(864) (2016)
L. Yan, A. Brouzgou, Y. Meng, M. Xiao, P. Tsiakaras, S. Song, Efficient and poison-tolerant PdxAuy/C binary electrocatalysts for glucose electrooxidation in alkaline medium. Appl. Catal. B Environ. 150-151, 268 (2014)
L. Demarconnay, S. Brimaud, C. Coutanceau, J.M. Léger, Ethylene glycol electrooxidation in alkaline medium at multi-metallic Pt based catalysts. J. Electroanal. Chem. 601, 169 (2007)
B. Beden, F. Largeaud, K.B. Kokoh, C. Lamy, Fourier transform infrared reflectance spectroscopic investigation of the electrocatalytic oxidation of d-glucose: identification of reactive intermediates and reaction products. Electrochim. Acta 41, 701 (1996)
C.J. Pouchert, The Aldrich Library of Infrared Spectra, 3rd edn. (Aldrich Chemical Company, Inc., Milwaukee, 1981)
Spectral Database for Organic Compounds (SDBS), National Institute of Advanced Industrial Science and Technology (AIST-Japan); http://sdbs.db.aist.go.jp/sdbs/cgi-bin/cre_index.cgi. (Accessed September 2019)
A. Couto, A. Rincón, M.C. Pérez, C. Gutiérrez, Adsorption and electrooxidation of carbon monoxide on polycrystalline platinum at pH 0.3–13. Electrochim. Acta 46, 1285 (2001)
Y.-X. Jiang, S.-G. Sun, N. Ding, Novel phenomenon of enhancement of IR absorption of CO adsorbed on nanoparticles of Pd confined in supercages of Y-zeolite. Chem. Phys. Lett. 344, 463 (2001)
A. Dailey, J. Shin, C. Korzeniewski, Ethylene glycol electrochemical oxidation at platinum probed by ion chromatography and infrared spectroscopy. Electrochim. Acta 44, 1147 (1998)
L. Dubau, F. Hahn, C. Coutanceau, J.M. Léger, C. Lamy, On the structure effects of bimetallic PtRu electrocatalysts towards methanol oxidation. J. Electroanal. Chem. 554-555, 407 (2003)
N.W. Smirnova, O.A. Petrii, A. Grzejdziak, Effect of ad-atoms on the electro-oxidation of ethylene glycol and oxalic acid on platinized platinum. J. Electroanal. Chem. 251, 73 (1988)
V. Climent, N. García-Aríá and J.-M. Feliu, in Fuel cell catalyst, a surface science approach, ed. M. T. M. Koper, (Wiley, Hoboken, 2009), pp. 209–244
H.W. Lei, B. Wu, C.S. Cha, H. Kita, Electro-oxidation of glucose on platinum in alkaline solution and selective oxidation in the presence of additives. J. Electroanal. Chem. 384, 103 (1995)
M. Pasta, F. La Mantia, Y. Cui, Mechanism of glucose electrochemical oxidation on gold surface. Electrochim. Acta 55, 5561 (2010)
Y. Holade, K. Servat, T.W. Napporn, C. Morais, J.-M. Berjeaud, K.B. Kokoh, Highly selective oxidation of carbohydrates in an efficient electrochemical energy converter: cogenerating organic electrosynthesis. ChemSusChem 9, 252 (2016)
S. Ernst, J. Heitbaum, C.H. Hamann, The electrooxidation of glucose in phosphate buffer solutions: Part I. Reactivity and kinetics below 350 mV/RHE. J. Electroanal. Chem. Interfacial Electrochem. 100, 173 (1979)
S. Blais, G. Jerkiewicz, E. Herrero, J.M. Feliu, New insight into the electro-oxidation of the irreversibly chemisorbed bismuth on Pt (111) through temperature-dependent research. J. Electroanal. Chem. 519, 111 (2002)
M.B.C. de Souza, R.A. Vicente, V.Y. Yukuhiro, C.T.G.V.M.T. Pires, W. Cheuquepan, J.L. Bott-Neto, J. Solla-Gullon, P.S. Fernandez, Bi-modified Pt electrodes toward glycerol electrooxidation in alkaline solution: effects on activity and selectivity. ACS Catal. 9, 5104 (2019)
M. Watanabe, S. Motoo, Electrocatalysis by ad-atoms: Part II. Enhancement of the oxidation of methanol on platinum by ruthenium ad-atoms. J. Electroanal. Chem. 60, 267 (1975)
Y.Y. Tong, H.S. Kim, P.K. Babu, P. Waszczuk, A. Wieckowski, E. Oldfield, An NMR investigation of CO tolerance in a Pt/Ru fuel cell catalyst. J. Am. Chem. Soc. 124, 468 (2002)
Z. Zhang, P. Gibson, S. Clark, G. Tian, P. Zanonato, L. Rao, Lactonization and protonation of gluconic acid: a thermodynamic and kinetic study by potentiometry, NMR and ESI-MS. J. Solut. Chem. 36, 1187 (2007)
C. Coutanceau, S. Baranton, Electrochemical conversion of alcohols for hydrogen production: a short overview, WIRE Energy and Environment, 388 (2016)
Energy prices and costs in Europe - 2019 – Enerdata. Report from the commission to the European parliament, the Council, the European Economic and Social Committee and the Committee of the regions: energy prices and costs in Europe. https://www.enerdata.net › about-us › company-news (Accessed October 2019)
A.T. Governo, L. Proença, P. Parpot, M.I.S. Lopes, I.T.E. Fonseca, Electro-oxidation of D-xylose on platinum and gold electrodes in alkaline medium. Electrochim. Acta 49, 1535 (2004)
S. Song, K. Wang, L. Yan, A. Brouzgou, Y. Zhang, Y. Wang, P. Tsiakaras, Ceria promoted Pd/C catalysts for glucose electrooxidation in alkaline media. Appl. Catal. B Environ. 176-177, 233 (2015)
M. Tominaga, M. Nagashima, K. Nishiyama, I. Taniguchi, Surface poisoning during electrocatalytic monosaccharide oxidation reactions at gold electrodes in alkaline medium. Electrochem. Commun. 9, 1892 (2007)
Funding
The authors received funding from the INCREASE Federation (FR CNRS 3707), the technogreen chair, the European communities (FEDER) through the ECONAT project, the “Région Nouvelle Aquitaine,” and the PEPS CNRS Energy.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(DOCX 2788 kb)
Rights and permissions
About this article

Cite this article
Neha, N., Kouamé, B.S.R., Rafaïdeen, T. et al. Remarkably Efficient Carbon-Supported Nanostructured Platinum-Bismuth Catalysts for the Selective Electrooxidation of Glucose and Methyl-Glucoside. Electrocatalysis 12, 1–14 (2021). https://doi.org/10.1007/s12678-020-00586-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12678-020-00586-y