
Abstract
The electrooxidation of glucose on gold (Au) and platinum (Pt) nanoparticles (NPs) is investigated in alkaline medium by cyclic voltammetry after chronoamperometry at different potentials (+0.100 V, +0.200 V, and +0.400 V vs the reversible hydrogen electrode, RHE), in situ Fourier transform infrared spectroscopy, and differential electrochemical mass spectrometry measurements. We show that glucose can adsorb on both metallic Au and Pt surfaces at low potentials, but that the adsorbed species are different: hydrogen atoms, carbon monoxide (CO), lactones and gluconate species on Pt-NPs, and only hydrogen atoms and gluconate species on Au-NPs. On Pt-NPs, the first oxidation peak between +0.050 V vs RHE and +0.250 V vs RHE is due to glucose adsorption and hydrogen atoms oxidation into protons (H+), whereas the second electrochemical feature between +0.250 V vs RHE and +0.800 V vs RHE is due to the oxidation of glucose into lactone and gluconate and of adsorbed CO into carbon dioxide (CO2). For Au-NPs, adsorbed hydrogen atoms are not oxidized into H+ but transformed into molecular hydrogen H2, and glucose is adsorbed as gluconate species that are desorbed into gluconates for potentials higher than +0.300 V vs RHE.
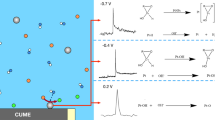
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The increasing demand for energy and raw materials and the depletion of the fossil fuel resources [1] have made the use of substantial alternative resources mandatory. Non-edible lignocellulosic biomass issued from agriculture residues, grass, and forests represents annually more than 12 billion tons [2]. Therefore, it is then considered a rich renewable hydrogen and carbon reservoir to harvest both energy and chemicals [3]. Non-edible lignocellulosic biomass is composed of around 20% lignin, 30–45% cellulose (linear crystalline polymers of glucose units [4]), and 20–50% hemicellulose (branched polymers of mainly xylose and glucose [5]), depending on the feedstock [4]. Therefore, hydrolysates after lignocellulosic pretreatments to separate lignin from cellulose and hemicellulose [6] contain more than 50 mol. % of glucose; glucose accounts then for more than 40% of the whole lignocellulosic biomass (lignin accounting for ca. 20%), which represents a very huge quantity of raw materials for production of chemicals. Noteworthy, electroreforming of glucose in an electrolysis cell can produce high value-added chemicals at the anode together with pure hydrogen at the cathode [7, 8]. In this context, unveiling the oxidation mechanism of glucose (considered as a model molecule) is of paramount importance and will further allow developing active and selective catalysts in view of the production of value-added compounds, such as gluconic acid, at high conversion rate and selectivity.
The electrooxidation of glucose in alkaline media has been extensively studied on bulk platinum (Pt) and gold (Au) [9,10,11,12,13,14], because of their high activity. The mechanism generally accepted on both metal surfaces involves the adsorption of glucose on a metallic Pt or Au site (M) through the anomeric function according to the following Reaction (1):

followed by an attack of the C-M bond by a hydroxyl anion in solution to produce weakly bonded gluconate species that later desorb in the electrolyte according to reaction Scheme 1.

The attack of the C-M bond by a hydroxyl anion in solution to produce weakly bonded gluconate species that later desorb in the electrolyte
A second mechanism was proposed for potential higher than +0.600 V vs RHE on Pt [9] and as an alternative path on Au [13]. It consists of the oxidation of the adsorbed dehydrogenated glucose species into \(\delta\)-gluconolactone. After desorption, \(\delta\)-gluconolactone is hydrolyzed in the presence of a hydroxyl anion in the electrolyte into gluconate, according to reaction Scheme 2.

The hydrolyzation of \(\delta\)-gluconolactone in the presence of a hydroxyl anion in the electrolyte into gluconate
However, considering that Pt proved to be extremely non-selective compared to Au at low electrode potentials and that the cyclic voltammograms recorded on both materials differ dramatically in terms of shapes and oxidation waves/peaks potentials, one may find very surprising that similar mechanisms are proposed (and accepted) for the glucose oxidation reaction (GOR) on both Pt and Au [9, 13, 14]. In our view, these experimental features rather suggest the existence of electrocatalyst-dependent glucose oxidation mechanisms.
The present contribution aims at going beyond these uncertainties by providing new insights into the adsorption modes of glucose, the intermediates, and the final products formed both as a function Pt or Au surfaces and of the electrode potentials to propose mechanisms of the GOR on Pt and Au nanoparticles (Pt-NPs and Au-NPs). The knowledge of the reaction mechanisms is paramount important because it is mandatory to be able to design very active catalysts with great selectivity towards the desired product. To our knowledge, it is the first time that the herein-employed complementary analyses are performed to obtain important information allowing proposing different GOR processes according to the catalytic material and the electrode potential. These analyses consist of (i) cyclic voltammograms recorded after chronoamperometry at different potentials to verify the poisoning of the surfaces, (ii) spectra from in situ Fourier transform infrared spectroscopy (in situ FTIRS) to determine intermediates/products formed at the electrode/electrolyte interface, and (iii) differential electrochemical mass spectrometry (DEMS) measurements to determine volatile compounds formed.
Experimental
Materials
Hexachloroplatinic acid hexahydrate (H2PtCl6 · 6 H2O) and tetrachloroauric acid trihydrate (HAuCl4 · 3 H2O), both 99.99% in purity, were purchased from Alfa Aesar, polyethylene glycol dodecyl ether (Brij® L4), acetone (Chromasolv®, 99.8% in purity), sodium borohydride (NaBH4, Reagent Plus, 99% in purity), NaOH (semiconductor grade, 99.99% in purity), glucose (99% in purity) and Nafion 5 wt.% in aliphatic alcohols from Sigma-Aldrich, and n-heptane (99% in purity) from Acros Organics. A Milli-Q-Millipore system was used to provide ultra-high purity water (18.2 MΩ cm, < 2 ppb total organic carbon).
All chemicals were used as received, except the carbon powder (Vulcan XC72 from Cabot Corp., further called C), which was heat-treated at 673 K under N2 atmosphere (U-quality from Air Liquide) for 4 h to remove adsorbed impurities.
Synthesis and Characterization of Carbon-supported Catalysts
The protocol of the water-in-oil route for the synthesis of Pt-NPs/C and Au-NPs/C catalysts was described elsewhere, as well as the characterization methods to determine the metal loading, the particle, and the crystallite sizes [7, 10, 15]. The results are given in Table 1.
Electrochemical Measurements
Chronoamperometry (CA) and cyclic voltammograms (CV) were recorded at 293 K in a three-electrode cell using a Voltalab PGZ402 potentiostat (Radiometer Analytical). The reference electrode was a home-made reversible hydrogen electrode (RHE), the counter electrode was a 3 cm2 geometric surface area glassy carbon plate, and the working electrode consisted of a 100 μgmetal cm−2 catalytic layer deposited on a 0.071 cm2 geometric surface area glassy carbon disc. The electrolyte consisted of a N2-purged 0.10 mol L−1 NaOH aqueous solution for catalyst characterization, to which 0.10 mol L−1 of glucose was added for electrooxidation measurements. More experimental details are given elsewhere [7, 10, 15].
In Situ FTIR Spectroscopy
FTIRS measurements were performed using a Bruker IFS 66 FTIR spectrometer modified for beam reflection on the electrode surface. The incident angle and the spectral resolution were 65° and 4 cm−1, respectively. Detailed experimental conditions can be found elsewhere [16, 17]. Five hundred twelve interferograms recorded for 35 s during the linear voltammetry at 0.001 V s−1 carried out in a 0.10 mol L−1 NaOH + 0.10 mol L−1 glucose electrolyte were averaged every 0.050 V from +0.050 to +1.200 V vs RHE. Spectra were normalized considering the averaged one recorded at +0.050 V vs RHE as the reference. By this way, negative absorption bands correspond to the formation of species and positive absorption bands to the consumption of species at the electrode surface [7, 10].
Differential Electrochemical Mass Spectrometry Measurements (DEMS)
A Pfeiffer Vacuum QMG 220 M1 quadrupole mass spectrometer was used for on-line differential electrochemical mass spectrometry measurements. The description of the method can be found elsewhere [18,19,20]. The current vs potential curves were recorded simultaneously with the mass intensity vs potential curves, for selected values of m/z (mass/charge) ionic signals (here m/z = 2 for dihydrogen detection and m/z = 44 for carbon dioxide detection have been particularly surveyed). The working electrodes were prepared in the form a Pt or Au nanoporous layer (0.28 cm2 area, 50-nm +/− 10-nm average thickness), obtained by sputtering onto a porous polytetrafluoroethylene (PTFE) membrane (pore size 0.02 µm, thickness 20 µm, Cobetter Filtration®), which serves as interface between the electrochemical cell and the high vacuum chamber of the mass spectrometer. The Pt/PTFE or Au/PTFE working electrode (WE) was connected to a VMP2Z potentiostat (Biologic®) via a ring-shaped Au (for an Au WE) or Pt (for a Pt WE) current collector. The counter electrode (CE, Pt grid) was located near the surface of the electrolyte, as far as possible from the working electrode to avoid any interference with the H2 bubbles that can be formed at the CE when the glucose oxidation reaction proceeds on the WE. The electrochemical measurements were first performed in 0.01 L of 0.10 mol L−1 NaOH supporting electrolyte to determine the m/z = 2 and m/z = 44 signal baselines. A 171-µL aliquot of 0.595 mol L−1 glucose in 0.10 mol L−1 NaOH solution was added at open circuit potential (ocp) conditions to reach 0.010 mol L−1 glucose, the changes of potential and m/z signals being measured concomitantly. The amounts of evolved H2 and CO2 were measured during a CV (0.010 V s−1) of glucose oxidation reaction from the ocp to +0.050 V vs RHE as lower potential limit and +1.500 V vs RHE as upper potential limit.
Results
The mechanism generally proposed for the electrooxidation of glucose at Pt and Au surfaces involves the adsorption of glucose as the first step. To test this hypothesis, chronoamperometry (CA) was performed at different electrode potentials until a stable current density was reached. The CAs were followed by CVs recorded in the negative scan direction from the CA potential to +0.050 V vs RHE and then between +0.050 V vs RHE and +1.200 V vs RHE (Figs. 1 and 2 for Pt-NPs/C and Au-NPs/C, respectively). For clarity reasons, only the first and second CVs are represented in Figs. 1 and 2, because additional CVs perfectly superimposed with the second one.

CV of the Pt-NPs/C electrode in 0.10 mol L−1 NaOH electrolyte (plain black line); CA performed at a +0.100 V vs RHE, b +0.200 V vs RHE, and c +0.400 V vs RHE followed by a first CV (dotted black line), and a second CV (plain gray line) recorded in N2-purged 0.10 mol L−1 NaOH electrolyte in the presence of 0.10 mol L−1 glucose at a Pt-NPs/C electrode. Scan rate = 0.005 V s−1, T = 293 K

Cyclic voltammograms recorded in N2-purged 0.10 mol L−1 NaOH electrolyte at an Au-NPs/C electrode in the presence of 0.10 mol L−1 glucose (black line for the first cycle and gray line for the second one), after chronoamperometry (black dotted line) at a +0.100 V vs RHE, b +0.200 V vs RHE, and c +0.400 V vs RHE; scan rate = 0.005 V s−1, T = 293 K
On a Pt-NPs/C electrode (Fig. 1), two oxidation waves, the first one between +0.050 V and +0.200 V vs RHE and the second one between +0.200 V and +0.500 V vs RHE, and an oxidation peak at ca. +0.600 V vs RHE can be seen in the potential range from +0.050 to +0.800 V vs RHE. These electrochemical features were already observed by Beden et al. [9] on a bulk Pt surface, although with different relative intensities compared to those reported for Pt-NPs/C. After the CA at +0.100 V vs RHE, the first CV is slightly shifted towards higher potentials compared to the second one, but all features are still visible between +0.050 and +0.800 V vs RHE, indicating that the surface is only slightly covered by adsorbed species. When the CA potential is increased, the oxidation wave of the first CV is delayed towards more positive potentials compared with the second one, which seems to indicate either that there are more adsorbed species on the Pt surface or that they are more strongly adsorbed, or even that the adsorbed species are not the same according to the CA potential.
In the case of an Au-NPs/C electrode (Fig. 2), the first CVs recorded after CA at different potentials display an onset potential at ca. +0.300 V vs RHE, a peak current at ca. 0.600 V vs RHE, and a diffusion plateau from +0.800 V vs RHE. Such behavior was already observed for bulk CO oxidation on Pt-based catalyst and is typical of strong poisoning of the surface by adsorbed species at low potentials [21] and limitation by diffusion in the plateau region. The adsorption of glucose on metallic gold has already been described in the literature [9, 13]. The peak current density and the current densities in the plateau region between +0.800 and +1.200 V vs RHE are higher than those for the second stable cycle. The higher peak current density of the first cycle suggests that more species are adsorbed on Au surface during all CAs at constant potential than during linear potential variation for the second cycle. However, conversely to the case of the Pt-NPs/C electrode, where the great dependence of the shape of the first CV on the CA potential was explained in terms of changes in amount of adsorbed species on the Pt surface or in their adsorption strength, or in their nature, the similarity in the case of the Au-NPs/C electrode of CV shapes and achieved current densities whatever the CA potential speaks rather towards invariance of the nature of the adsorbates and of their coverage on the Au-NPs/C electrode. Taken together, the results also point towards different initial steps of glucose oxidation at Au-NPs and Pt-NPs surface.
Fourier transform infrared (FTIR) spectra were recorded at Pt-NPs/C and Au-NPs/C electrodes during a linear scan voltammetry (LSV) at 0.001 V s−1 in 0.10 mol L−1 NaOH +0.10 mol L−1 glucose aqueous electrolyte (Fig. 3).

Fourier transform infrared spectra recorded every 0.050 V during 0.10 mol L−1 glucose electrooxidation in 0.10 mol L−1 NaOH electrolyte from +0.100 V to +1.200 V vs RHE on a Pt-NPs/C and b Au-NPs/C catalysts. Scan rate: 0.001 V s−1, resolution 4 cm−1, T = 293 K. Vertical scale: ΔR/R
Figure 3a displays the spectra recorded from +0.100 V to +1.200 V vs RHE on a Pt-NPs/C electrode. A negative IR band located 1581 cm−1 is visible starting from +0.150 V vs RHE. Simultaneously, IR bands appear in the 1340–1420 cm−1 wavelength region. The negative bands are located at 1346 cm−1 and 1415 cm−1, and a positive one at 1388 cm−1. The spectrum of glucose recorded in alkaline solution (supporting information, SI1) shows a weak band at ca. 1363 cm−1, which could translate into a positive band during glucose consumption. However, looking closely at the positive band height, it always reaches the same value as that of the baseline at ca. 1500 cm−1. This is even more obvious when looking at the in situ IR spectra recorded on Pt9Bi1/C catalyst during glucose electrooxidation [10], where both negative bands display high intensities. Moreover, the in situ infrared spectra of methylglucoside (supporting information, SI 2) display two negative bands corresponding to the formation of carboxylate species in the same wavenumber region as for glucose oxidation (1340–1420 cm−1), without the positive one between those two negative bands. At last, the bands at 1415 cm−1 (band b) and at 1581 cm−1 (band c) were assigned to the formation of gluconate (\({\nu }_{\mathrm{sym}.}\) and \({\nu }_{\mathrm{asym}.}\) O-C-O, respectively) and the one at 1346 cm−1 (band a) attributed to the \(\delta\)(CH2) mode of gluconate or gluconolactone by Beden et al. [9]. But it is possible that a small positive band related to glucose consumption exists in the spectra, together with the two negative bands already attributed by Beden et al. [9]. The intensities of these three negative bands increase with an increase in the electrode potential. Gluconate is then formed at low electrode potential at a Pt-NPs/C electrode. The absorption band in the 2000 to 2100 cm−1 wavenumber range (band f) appearing from +0.150 V vs RHE is assigned to the formation of linearly adsorbed CO (COads) [22, 23]. This band appears positive from +0.200 V vs RHE, shifts by 51.5 cm−1 V−1 (not linearly) towards lower wavenumbers between +0.200 V and +0.750 V vs RHE, and stabilizes at 2052 cm−1 at higher potentials. This result suggests that COads species are formed at potentials lower than +0.200 V vs RHE and progressively depleted from the surface as the potential is increased. The red shift of the signal may correspond to a decrease of the CO surface coverage upon increasing potentials, sign of progressive consumption of the COads species formed at low potential. The small band g (pointed by an arrow) and the band h, located at 1304 cm−1 and 2343 cm−1, respectively, are compatible with the formation of hydrogenocarbonate ions (HCO3−) [24] from ca. +0.250 V vs RHE and CO2 [25] for potentials higher than +0.500 V vs RHE on Pt-NPs/C electrode. Another interesting band (band e) at 1785 cm−1, in the carbonyl region, can be attributed to the formation of \(\gamma\)-gluconolactone (\({\nu }_{\mathrm{C}=\mathrm{O}}\)) [9] and is detected negative from +0.150 V vs RHE. A shoulder (band d) around 1720 cm−1 that could be attributed to \({\nu }_{\mathrm{C}=\mathrm{O}}\) of \(\delta\)-gluconolactone [9] is detected at high potentials (but the band exists already form ca. +0.150/+0.200 V vs RHE). Chemical structures of \(\delta\)- and \(\gamma\)-gluconolactones are given in Scheme 3.

Chemical structures of \(\delta\)-gluconolactone and \(\gamma\)-gluconolactones
Importantly, the band characteristics of such species do not appear for the oxidation of methylglucoside on a Pt-NPs/C electrode, i.e., when the anomeric function of glucose is protected by a methyl group, and when the C6 primary alcohol is oxidized (supporting information, SI2) [10]. Their presence in Fig. 3a is strong evidence that the anomeric function was oxidized and that gluconolactones did form from very low electrode potentials at the Pt-NPs/C electrode.
IR spectra recorded at an Au-NPs/C electrode from +0.100 V to +1.200 V vs RHE (Fig. 3b) display the same a, b, and c absorption bands as those observed at Pt-NPs/C in the wavenumber range from 1000 to 1650 cm−1, but their intensity increases for potentials higher than +0.350 V vs RHE, suggesting that gluconate is continuously formed on Au/C electrode for potential higher than +0.300 V vs RHE. Now, comparing the IR spectra recorded on Au-NPs/C and on Pt-NPs/C, three bands are missing for the Au surfaces: the shoulder at ca. 1720 cm−1 and the band at 1785 cm−1 related to the formation of gluconolactone, and the band at ca. 2050 cm−1 related to the formation of COads. This confirms the hypothesis made from the results of Fig. 1, Pt and Au surfaces are likely not covered by same species for the early stage of glucose oxidation, and Au-NPs/C catalyst does not produce lactone conversely to Pt-NPs/C catalyst. The pseudo-band j observed at ca. 1680 cm−1 cannot be attributed to any carbonyl function and is likely due to the growth of the positive interfacial water band at ca. 1640 cm−1 [26].
DEMS measurements were performed at m/z = 2 and m/z = 44 to detect the formation of dihydrogen, H2, and/or of carbon dioxide, CO2, as a function of the electrode potential (Fig. 4). To avoid any possible signal perturbation due to carbon support oxidation into CO2 at potentials higher than +0.207 V vs the normal hydrogen electrode (NHE) [27], DEMS experiments were carried out on unsupported sputtered Pt-NPs and Au-NPs (i.e., in the absence of any carbon support that would render uncertain the CO2 origin from the GOR or from the oxidation of the carbon support). In the case of 0.010 mol L−1 glucose electrooxidation on Pt-NPs, little H2 is detected, but only for potentials lower than +0.100 V vs RHE (Fig. 4B, gray line hidden by the blank experiment), between +0.050 V and +0.075 V vs RHE. The formation of H2 at so low potentials is more likely due to the hydrogen evolution reaction (HER) from the water electrochemical reduction than to glucose adsorption and this hypothesis was confirmed by a blank experiment in 0.10 mol L−1 NaOH (black line in Fig. 4b). Note that similar results were observed during borohydride oxidation [18]. CO2 is detected over the potential range from ca. +0.450 V vs RHE to +0.900 V vs RHE (Fig. 4c), in the potential region that corresponds to the third oxidation process in CVs of Figs. 1 and 4a, and where IR spectra (Fig. 3a) indicate a strong shift of the COads band towards lower wavenumber values due to lowering of the COads surface coverage. In the case of 0.010 mol L−1 glucose electrooxidation on Au-NPs (Fig. 4d), H2 is detected as soon as glucose is added in the electrolyte at the ocp (supporting information, SI2), and then a large peak with high intensity is recorded in the potential range from +0.300 V to +0.900 V vs RHE (Fig. 4e) that corresponds to the first oxidation peak in CVs of Figs. 2 and 4d. This H2 production is also witnessed in the backward cycle (although in smaller extent), which demonstrates that it originates from the glucose oxidation mechanism and not from pre-adsorbed species formed at low potentials. For higher potentials, only CO2 is detected in the course of the glucose electrooxidation reaction (Fig. 4f).

CVs of 0.10 mol L−1 glucose oxidation in 0.10 mol L−1 NaOH electrolyte on a sputtered Pt-NPs and d sputtered Au-NPs; DEMS measurement performed at m/z = 2 for the electrooxidation of 0.010 mol L−1 glucose oxidation in 0.10 mol L−1 NaOH electrolyte on b sputtered Pt-NPs and e sputtered Au-NPs; DEMS measurement performed at m/z = 44 for the electrooxidation of 0.010 mol L−1 glucose oxidation in 0.10 mol L−1 NaOH electrolyte on d sputtered Pt-NPs and f sputtered Au-NPs. Scan rate: 0.010 V s−1, T = 293 K
Discussion
The sequential CAs and CVs displayed in Fig. 1 provide clear evidence that COads is formed at potentials lower than +0.200 V vs RHE on Pt-NPs/C. This hypothesis is further supported by CO stripping measurements (Fig. 5) showing that the charge involved in the oxidation of the COads layer (i.e., the coverage with adsorbed CO at the Pt-NPs/C surface) decreases with an increase of the CO adsorption potential, from ca. 0.14 to ca. 0.13 mC cm−2 and to ca. 0.06 mC cm−2 for CO adsorption at +0.100 V, +0.200 V, and +0.400 V vs RHE, respectively (Fig. 5d). Additionally, the results indicate that adsorbed CO molecules are partly oxidized at E > +0.200 V vs RHE.

CO stripping measurements recorded in N2-purged 0.10 mol L−1 NaOH electrolyte at a Pt-NPs/C electrode after 20-min chronoamperometry at a +0.100 V vs RHE, b +0.200 V vs RHE, and c +0.400 V vs RHE (scan rate = 0.005 V s−1, T = 293 K). d Charge density under the CO stripping peak as a function of the CO adsorption potential
Nevertheless, the lower COads coverage achieved upon glucose dissociation and dehydrogenation at +0.400 V vs RHE is unlikely to account for the positive shifts of both the onset potential and the oxidation wave on Pt-NPs/C. Rather, we postulate that other species are co-adsorbed on the Pt surface, and the IR spectroscopy measurements of Fig. 3a suggest \(\delta\)- and \(\gamma\)-gluconolactones as possible candidates.
Also, as no H2 formation was detected by DEMS measurements on Pt-NPs (while current is detected in this potential region by voltammetry), one can infer that adsorbed hydrogen is oxidized into protons on the Pt surface. Based on these results, the following mechanism is proposed for the adsorption of glucose on Pt-NPs/C at E ≤ +0.200 V vs RHE, corresponding to the first voltammetric feature of the second cycles in Fig. 1:
This mechanism is in line with that proposed by Beden et al. [9] on bulk Pt. Interestingly, it may be noticed that the formation of lactones (IR measurements, Fig. 3a) coincides with the onset of the second electrochemical feature of the second cycle in Fig. 1, at ca. +0.150/+0.200 V vs RHE. According to Scheme 2, the formation of lactones (from the first aldose adsorbed specie) involves the attack by a hydroxyl anion (OH−). Strmcnik et al. [28] reported that OH species could adsorb on low-coordinated Pt atoms in alkaline media, which can help for the formation of lactone on Pt-NPs. But the formation of lactone on Pt-NPs could also proceed via an Eley–Rideal mechanism, similar to that for the oxidation of adsorbed COads [29]. Moreover, as the position of the IR absorption bands assigned to lactone does not change with potential, lactone species are likely in solution or very weakly adsorbed to the Pt surface, and therefore can desorb to be hydrolyzed into gluconate in the electrolyte (Scheme 2). As no COads and no lactones are produced during the electrooxidation of methylglucoside on Pt-NPs/C (supporting information, SI2) [10], we propose that COads species form, at least partially, from the carbonyl function of lactones.
For potentials higher than +0.450 V vs RHE, Pt adsorbs OH− species (OHads) in alkaline electrolyte [30]; hence, a Langmuir–Hinshelwood mechanism likely accelerates the oxidation of adsorbed lactones into gluconate and adsorbed CO into CO2. As the three IR adsorption bands located at 1346 cm−1 (band a), 1415 cm−1 (band b), and 1581 cm−1 (band c) in Fig. 3a assigned to the formation of gluconate appear at E > + 0.150 V vs RHE, and because their position is independent on the electrode potential, the parallel formation of weakly adsorbed gluconate-like species (Scheme 1) to produce gluconate by the mechanisms proposed above cannot be discarded.
The CVs recorded after CAs at different potentials (+0.100 V, +0.200 V, and +0.400 V vs RHE) on Au-NPs/C catalyst indicate that some species are adsorbed at the metallic surface, but in contrast to Pt-NPs/C where COads and lactones are concomitantly formed, none of these species is detected during the glucose oxidation on Au-NPs/C (Fig. 3b). Indeed, Au does not adsorb OH species at E < +0.300/+0.400 V vs RHE [31]. In the low potential region from +0.050 V to +0.300 V vs RHE, the same first step as on Pt-NPs/C (Eqs. (1) and (2)) can occur on metallic Au-NPs/C (Eqs. (4) and (5)). For potentials comprised between +0.300 V and +0.900 V vs RHE corresponding to the first oxidation peaks in CVs, the DEMS measurements clearly indicate that H2 is formed on Au-NPs, not on Pt-NPs. As atomic H can adsorb on metallic Au surface in the presence of organic species [32], and as DFT calculations have shown that Hads at metallic Au surface are less stable than free H2 [33], we postulate that adsorbed H atoms diffuse on the metallic Au surface and combine to produce hydrogen molecules (Eq. (6)) [34], similar to what happens during the electrooxidation of borohydride at Au electrode [20]. For potential lower than +0.300 V vs RHE, the DEMS measurements indicate that glucose adsorbs on and blocks the metallic Au surface, producing too few H2 to be detected during the CV (the whole baseline of the m/z = 2 is raised in the presence of glucose; the transient signal of H2 production was however detected during the glucose addition steps at ocp (supplementary information, SI 3)). For potentials comprised between +0.300 V vs RHE and +0.900 V vs RHE, the competition between glucose and OH− adsorption likely leads to the coexistence of metallic Au and Au-OH patches at the surface of NPs (this hypothesis is supported by the IR baseline perturbation that starts around +0.350 V– +0.400 V vs RHE and the formation of the positive band at ca. 1640 cm−1 in Fig. 3b). Therefore, it may be postulated that glucose adsorbs on the metallic sites of the Au surface while the Au-OH surface species oxidize adsorbed species into gluconate and water via a Langmuir–Hinshelwood mechanism. The following mechanism is then proposed for Au-NPs:
-
For +0.050 V < E < +0.900 V vs RHE
-
from +0.300 V vs RHE to +0.900 V vs RHE
The first step of this mechanism (Eq. (4)) agrees with former propositions of Beden et al. [9], Pasta et al. [13, 35], and Adzic et al. [36]. This mechanism explains why an oxidation current peak/wave is observed at low potentials on Pt-NPs (Fig. 1) and not on Au-NPs (Fig. 2). As no gluconolactone was detected by IR measurements (Fig. 3b) in the 1640 to 1800 cm−1 wavenumber range, the reaction process described in reaction Scheme 1 seems thus to be favored on Au-NPs/C compared to Pt-NPs/C. In addition, our results suggest that adsorbed lactone is not formed on Au-NPs/C, conversely to what was proposed by Pasta et al. [13] and Adzic et al. [36]. For potentials higher than +0.900 V vs RHE, CO2 is detected both by in situ FTIRS and DEMS on Au-NPs, indicating that the mechanism is more complex, owing to the ability of Au to break the C–C bond and to produce C1 to C6 oxidized species. At such high overpotentials, the glucose oxidation mechanism is also unclear on Pt-NPs.
Conclusion
In conclusion, by combining cyclic voltammetry, in situ FTIRS, and DEMS results, we have brought new insights into the glucose oxidation mechanisms on Pt and Au nanoparticles. We provided spectrometric evidence that different species are adsorbed on Pt-NPs and Au-NPs; hence, different mechanisms are involved. In particular, lactones seem to be (at least some of) the adsorbed intermediates during the oxidation of glucose into gluconate on Pt-NPs, whereas only adsorbed gluconate species are formed on Au-NPs. The proposed glucose oxidation mechanisms account for the difference in electrocatalytic behavior between Pt-NPs and Au-NPs, and particularly for the lower onset potential of glucose electrooxidation observed on Pt-NPs than on Au-NPs. Because the knowledge of the reaction mechanisms is a prerequisite for the development of more selective and more active electrocatalysts, the findings described in the present contribution are of paramount importance concerning the selective electrooxidation of glucose into value-added compounds, and that of aldoses in general.
Pt-NPs led to poor selectivity whatever the electrode potential, whereas Au-NPs have shown high selectivity towards gluconate in the +0.300 V < E < +0.800 V vs RHE potential range. Moreover, -Au NPs are less prone to poisoning by strong adsorbed species such as COads. In addition, DEMS evidenced hydrogen evolution on gold in the same electrode potential range, with the maximum of production coinciding with the first current peak of glucose oxidation into gluconate around +0.500 – +0.600 V vs RHE. The electrocatalytic behavior of gold could then help to integrate electrochemical methods in biorefinery by demonstrating high activity and selectivity, and lower energy cost. Indeed, electrochemical reactors often consist of divided cells separated by an ionic conductive separator allowing biomass oxidation reaction into value-added compounds at the anode and either hydrogen production [7, 10] or reduction reaction of biomass molecules into value-added compounds (paired electrochemical processes) [37, 38] at the cathode. The production of valuable compounds at both electrodes allows decreasing the total energy cost [39]. Using gold at the anode of a glucose electroreforming reactor [40], as an example, will lead to the simultaneous production of two valuable compounds, hydrogen and gluconate, at the anode and of one valuable compound, hydrogen, at the cathode of the reactor, i.e., twice more hydrogen per glucose molecule oxidized into gluconate than in conventional systems. Of course, gold is an expensive strategical metal, but its amount in catalysts can certainly be lowered keeping its properties by material engineering.
References
X. Pang, C. Jia, K. Zhang, M. Li, Y. Wang, J. Peng, B. Li, J. Chen, The depth limit for the formation and occurrence of fossil fuel resources. Earth Syst. Sci. Data (2020). https://doi.org/10.5194/essd-2019-72
N. Dahmen, I. Lewandowski, S. Zibek, A. Weidtmann, GCB Bioenerg. (2019). https://doi.org/10.1111/gcbb.12586
C. Dollé, N. Neha, C. Coutanceau, Curr. Op. Electrochem. (2022). https://doi.org/10.1016/j.coelec.2021.100841
E. Taarning, C.M. Osmundsen, X. Yang, B. Voss, S.I. Andersen, C.H. Christensen, Energy Environ. Sci. (2011). https://doi.org/10.1039/c004518g
V.B. Agbor, N. Cicek, R. Sparling, A. Berlin, D.B. Levin, Biotechnol. Adv. (2011). https://doi.org/10.1016/j.biotechadv.2011.05.005
T. Rafaïdeen, S. Baranton, C. Coutanceau, ChemElectroChem (2022). https://doi.org/10.1002/celc.202101575
T. Rafaïdeen, S. Baranton, C. Coutanceau, Appl. Catal. B: Env. (2019). https://doi.org/10.1016/j.apcatb.2018.11.006
W.J. Liu, Z. Xu, D. Zhao, X.-Q. Pan, H.-C. Li, X. Hu, Z.-Y. Fan, W.-K. Wang, G.-H. Zhao, S. Jin, G.W. Huber, H.-Q. Yu, Nat. Comm. (2020). https://doi.org/10.1038/s41467-019-14157-3
B. Beden, F. Largeaud, K.B. Kokoh, C. Lamy, Electrochim. Acta. (1996). https://doi.org/10.1016/0013-4686(95)00359-2
N. Neha, S.R.B. Kouamé, T. Rafaïdeen, S. Baranton, C. Coutanceau, Electrocatalysis (2021). https://doi.org/10.1007/s12678-020-00586-y
G. Moggia, T. Kenis, N. Daems, T. Breugelmans, ChemElectroChem (2020). https://doi.org/10.1002/celc.201901960
N. Schlegel, G.K. Wiberg, M. Arenz, On the electrooxidation of glucose on gold: Towards an electrochemical glucaric acid production as value-added chemical. Electrochim. Acta (2022). https://doi.org/10.1016/j.electacta.2022.140023
M. Pasta, F. La Mantia, Y. Cui, Electrochim. Acta (2010). https://doi.org/10.1016/j.electacta.2010.04.069
Y.B. Vassilyev, O.A. Khazova, N.N. Nikolaeva, J. Electroanal. Chem. Interfac. Electrochem. (1985). https://doi.org/10.1016/0022-0728(85)85085-3
B. S. R. Kouamé, S. Baranton, P. Brault, C. Canaff, W. Chamorro-Coral, A. Caillard, K. De Oliveira Vigier, C. Coutanceau, Electrochim. Acta (2020). https://doi.org/10.1016/j.electacta.2019.135161
B. Beden, C. Lamy, in Spectroelectrochemistry: theory and practice, ed. By R. J. Gale (Plenum Press, New York, 1988) pp 189–261. https://doi.org/10.1007/978-1-4613-0985-7_5
A. Kabbabi, R. Faure, R. Durand, B. Beden, F. Hahn, J.M. Léger, C. Lamy, J. Electroanal. Chem. (1998). https://doi.org/10.1016/S0022-0728(97)00558-5
G. Braesch, A. Bonnefont, V. Martin, E.R. Savinova, M. Chatenet, Electrochim. Acta (2018). https://doi.org/10.1016/j.electacta.2018.04.068
R. Ianniello, V.M. Schmidt, Ber. Bunsenges. Phys. Chem. Chem. Phys. (1995). https://doi.org/10.1002/bbpc.19950990114
M. B. Molina Concha, M. Chatenet, F. H. B. Lima, E. A. Ticianelli, Electrochim. Acta (2013). https://doi.org/10.1016/j.electacta.2012.11.027
W.T. Napporn, J.-M. Léger, C. Lamy, J. Electroanal. Chem. (1996). https://doi.org/10.1016/0022-0728(96)04543-3
A. Couto, A. Rincon, M.C. Perez, C. Gutierrez, Electrochim. Acta (2001). https://doi.org/10.1016/S0013-4686(00)00714-3
T. Elgayyar, R. Atwi, A. Tuel, F.C. Meunier, Catal. Today (2021). https://doi.org/10.1016/j.cattod.2021.01.009
C. Lafforgue, F. Maillard, V. Martin, L. Dubau, M. Chatenet, ACS Catal. (2019). https://doi.org/10.1021/acscatal.9b00439
L. Dubau, F. Hahn, C. Coutanceau, J.-M. Léger, C. Lamy, J. Electroanal. Chem. (2003). https://doi.org/10.1016/S0022-0728(03)00308-5
A. Rodes, E. Pastor, T. Iwasita, J. Electroanal. Chem. (1994). https://doi.org/10.1016/0022-0728(94)03585-7
L. Castanheira, W.O. Silva, F.H.B. Lima, A. Crisci, L. Dubau, F. Maillard, ACS Catal. (2015). https://doi.org/10.1021/cs501973j
D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, D. van der Vliet, A.P. Paulikas, V.R. Stamenkovic, N.M. Markovic, Nat Chem. (2013). https://doi.org/10.1038/nchem.1574
P. Urchaga, S. Baranton, C. Coutanceau, G. Jerkiewicz, Langmuir (2012). https://doi.org/10.1021/la302388p
S. Kusano, D. Matsumura, K. Ishii, H. Tanaka, J.I. Mizuki, Nanomaterials (2019). https://doi.org/10.3390/nano9040642
B. Beden, I. Cetin, A. Kahyaoglu, D. Takky, C. Lamy, J. Catal. (1987). https://doi.org/10.1016/0021-9517(87)90334-4
N. Arjona, G. Trejo, J. Ledesma-García, L.G. Arriaga, M. Guerra-Balcázar, RSC Adv. (2016). https://doi.org/10.1039/C5RA23780G
S.A.C. Carabineiro, B.E. Nieuwenhuys, Gold Bull. (2009). https://doi.org/10.1007/BF03214951
W. Watkins, Y. Borensztein, Phys. Chem. Chem. Phy. (2017). https://doi.org/10.1039/C7CP04843B
M. Pasta, R. Ruffo, E. Falletta, C. M. Mari, C. Della Pina, Gold Bull. (2010). https://doi.org/10.1007/BF03214967
R.R. Adzic, M.W. Hsiao, E.B. Yeager, J. Electroanal. Chem. Inter. Electrochem. (1989). https://doi.org/10.1016/0022-0728(89)87164-5
K. Park, P. N. Pintauro, M. M. Baizer, K. Nobe. J. Electrochem. Soc. 132, (1985). https://doi.org/10.1149/1.2114229
Y. Cao, J. Knijff, A. Delparish, M. F. N. D’Angelo, T. Noёl. ChemSusChem 14, (2021). https://doi.org/10.1002/cssc.202002833
Y. Kwon, K. J. P. Schouten, J. C. van der Waal, E. de Jong, M. T. M. Koper. ACS Catal. 6, (2016). https://doi.org/10.1021/acscatal.6b01861
C. Lamy, C. Coutanceau, S. Baranton. in Hydrogen and Fuel cells primers, ed. by B. Pollet (Elsevier, Amsterdam, 2020) pp 82–106. https://doi.org/10.1016/B978-0-12-821500-5.00001-1
Funding
This work was supported by a grant overseen by the French National Research Agency (ANR) through the GLUCONIC Project (ANR-20-CE43-0005). Part of this work also pertains to the French government program “Investissements d’Avenir” (EUR INTREE, reference ANR-18-EURE-0010). Some of us (NN, TR, CC) acknowledge the INCREASE Federation (FR CNRS 3707), the Technogreen chair, the European communities (FEDER) through the ECONAT project, and the “Région Nouvelle Aquitaine” for their support. TF, FM, and MC thank Vincent Martin for his assistance in the DEMS measurements and Bruno Gilles for his support in making the sputtered deposits. Some of this work (in LEPMI) has been performed within the framework of the Centre of Excellence of Multifunctional Architectured Materials “CEMAM,” Grenoble France n° ANR-10-LABX-44–01.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Neha, N., Rafaïdeen, T., Faverge, T. et al. Revisited Mechanisms for Glucose Electrooxidation at Platinum and Gold Nanoparticles. Electrocatalysis 14, 121–130 (2023). https://doi.org/10.1007/s12678-022-00774-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12678-022-00774-y