
Abstract
An inherent challenge that mitochondria face is the continuous exposure to diverse stresses which increase their likelihood of dysregulation. In response, human cells have evolved sophisticated quality control mechanisms to identify and eliminate abnormal dysfunctional mitochondria. One pivotal mitochondrial quality control pathway is PINK1/Parkin-dependent mitophagy which mediates the selective removal of the dysfunctional mitochondria from the cell by autophagy. PTEN-induced putative kinase 1 (PINK1) is a mitochondrial Ser/Thr kinase that was originally identified as a gene responsible for autosomal recessive early-onset Parkinson’s disease (PD). Notably, upon failure of mitochondrial import, Parkin, another autosomal-recessive PD gene, is recruited to mitochondria and mediates the autophagic clearance of deregulated mitochondria. Importantly, recruitment of Parkin to damaged mitochondria hinges on the accumulation of PINK1 on the outer mitochondrial membrane (OMM). Normally, PINK1 is imported from the cytosol through the translocase of the outer membrane (TOM) complex, a large multimeric channel responsible for the import of most mitochondrial proteins. After import, PINK1 is rapidly degraded. Thus, at steady-state, PINK1 levels are kept low. However, upon mitochondrial import failure, PINK1 accumulates and forms a high-molecular weight > 700 kDa complex with TOM on the OMM. Thus, PINK1 functions as sensor, tagging dysfunctional mitochondria for Parkin-mediated mitophagy. Although much has been learned about the function of PINK1 in mitophagy, the biochemical and structural basis of negative regulation of PINK1 operation and functions is far from clear. Recent work unveiled new players as PTEN-l as negative regulator of PINK1 function. Herein, we review key aspects of mitophagy and PINK1/Parkin-mediated mitophagy with highlighting the role of negative regulation of PINK1 function and presenting some of the key future directions in PD cell biology.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cellular homeostasis is accomplished through a sustained balance between biogenesis and turnover. Defects in damaged organelles and protein aggregate removal can cause cellular stress and eventually cell death. Lysosomal-mediated degradation and intracellular component recycling are under a tight control of regulated, highly conserved process termed autophagy (Mizushima et al. 2008; Mizushima 2018). There are three forms of intracellular autophagy in mammalian cells, including macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). Autophagy could be either a non-selective randomly uptake process (bulk autophagy) or a special process to remove or degrade specific organelles, aggregated proteins, DNA, and pathogens (selective autophagy). The classification of these forms depends on the size of the substrates to be removed, their lysosomal degradation scale, and the delivery mechanism of the substrates to the lysosome. Macroautophagy is considered to be the major route for cytoplasmic proteins and organelles degradation; it is orchestrated by a group of proteins encoded by autophagy-related genes (ATGs) (Nakatogawa et al. 2009). Subcellular components in macroautophagy are engulfed by double-membrane vesicles termed autophagosomes and be vulnerable to lysosomal enzymes. The second form of autophagy (microautophagy) arises through the entrance of cellular constituents (including whole organelles) to lysosome via the direct invaginations of lysosomal membranes (Li et al. 2012). The latter form of autophagy is chaperone-mediated autophagy, in which soluble cytosolic proteins containing a specific targeting motif are delivered by the cytosolic chaperone heat shock cognate 70 (HSC70) to the lysosomal surface (Kaushik et al. 2012). Several types of selective autophagy, specific for the substrate, have been found; for instance, mitochondria (mitophagy), lipids (lipophagy), pathogens (xenophagy), peroxisomes (pexophagy), ribosomes (ribophagy), and endoplasmic reticulum (reticulophagy or ERphagy) (Tasdemir et al. 2007; Weidberg et al. 2009; Bauckman et al. 2015; Eldeeb et al. 2021a).
Mitochondria are not only the powerhouse of the cell that provide energy for a variety of different processes, but also are key triggers for programmed cell death, regulators of calcium homeostasis, and providers of diverse cellular metabolic and chemicals for the cell (Lill 2009; Schmidt et al. 2010; Harbauer et al. 2014; Hou et al. 2017). To fulfill such multitasking, mitochondrial quality control must be tightly controlled to achieve normal cellular activities. One major aspect of evolutionarily conserved macroautophagy is mitophagy, which involves monitoring quality control of mitochondria, either by regulating their number or, specifically, by removing those that are damaged (De Duve et al. 1996; Lemasters 2005; Eldeeb et al. 2018; Eldeeb et al. 2020a, b, c; Eldeeb and Ragheb 2020). Owing to its pivotal function in sustaining mitochondrial homeostasis and strong association with multiple human diseases, such as Parkinson’s disease (PD) and Alzheimer’s disease (AD), the mitophagy machinery has gained considerable attention throughout the last two decades. Cells possess numerous non‐redundant mechanisms of mitophagy which imply that different stimuli can trigger mitophagy via various signaling cascades (Fig. 1) (Palikaras et al. 2017). For instance, the PINK1/Parkin-dependent mitophagy is the main modulator of depolarized mitochondria turnover. Additionally, several mitochondrial proteins, such as BNIP3, NIX, and FUNDC1, could function as mitophagy receptors, and they are constitutively localized at the outer membrane of mitochondria (OMM) and interact directly with autophagosomal membrane protein light chain 3 (LC3) to stimulate mitophagy. There are also lipid-mediated mitophagy and ubiquitin-mediated mitophagy (Chu et al. 2013; Strappazzon et al. 2015; Villa et al. 2018). Collectively, these pathways are deregulated in human diseases, including cancer, neurodegenerative disorders, metabolic disorders, and aging revealing the significance of mitophagy as a cellular housekeeping function (Valente et al. 2004; Narendra et al. 2008; Chourasia et al. 2015; Springer et al. 2016; Pickles et al. 2018a). In the current review, we provide an overview of the key pathways involved in mitophagy regulation, and we discuss the potential role of the newly identified PTEN isoforms, PTEN-Short, and PTEN-Long, in the fine-tuning of mitophagy.

The major pathways of mitophagy, including ubiquitin-mediated mitophagy (pathway 1, PINK1-Parkin-dependent mitophagy; and pathway 2, Parkin-independent mitophagy), receptor-mediated mitophagy, and lipid-mediated mitophagy. In PINK1-Parkin-dependent mitophagy, the stabilized PINK1 on the OMM of the damaged mitochondria facilitates the recruitment of Parkin from the cytosol to the OMM, resulting in the phospho-ubiquitination of proteins on the OMM via PINK1 and Parkin activities and finally the formation of mitophagosome. In ubiquitin-mediated Parkin-independent mitophagy, MUL1 (E3 ubiquitin ligase) located at damaged mitochondria can bind directly to GABAA receptor-associated protein (GABARAP), resulting in recruitment of phagophore to engulf damaged mitochondria. In receptor-mediated mitophagy, OMM receptors, such as BNIP3, NIX/BNIP3L, FUNDC1, and FKBP8, bind directly to LC3s allowing finally the formation of mitophagosome. In lipid-mediated mitophagy, cardiolipin is translocated from IMM to OMM, and binds directly to LC3s, resulting in mitophagy initiation
Molecular Pathways of Mitophagy
PINK1-Parkin-Mediated Ubiquitin-Driven Mitophagy
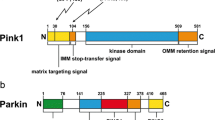
Ubiquitin (ub) is a small protein that plays crucial role in multi-cellular processes including protein degradation and immune system signaling. Among the ubiquitin enzymes, ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin ligase (E3), E3 Ub ligases are the most abundant which designates that E3 ligases are the principal factors affecting the substrate specificity essential to the ubiquitin pathway (Scheffner et al. 1995). Parkin (encoded by the PARK2 gene) is an E3 Ub ligase, which was discovered in 1998, and it contains five domains: an N-terminal Ub-like domain (UBL), a RING1 domain, an IBR domain, a RING2 domain, and a RING0 domain which is a Parkin unique domain (Hristova et al. 2009; Trempe et al. 2013; Walden et al. 2017) and has important roles in the pathogenesis of autosomal recessive Parkinson’s disease (ARPD) (Kitada et al. 1998; Lucking et al. 1998; Abbas et al. 1999). Another ARPD-associated gene, PINK1 (PTEN-induced putative kinase 1) which encoded by PARK6 gene and was discovered in 2001 (Unoki et al. 2001), encodes a mitochondrial serine/threonine kinase that regulates Parkin activity via phosphorylation cascades. PINK1 comprises of different domains, including an N-terminal mitochondrial targeting sequence (MTS) and a transmembrane domain (TMD) followed by a serine/threonine kinase domain and a regulatory domain at the C-terminal (Okatsu et al. 2015). Considering mitochondrial quality maintenance, the PINK1/Parkin-pathway is considered to play a key role in the removal of dysfunctional mitochondria and to constitute a mitochondrial quality-control system via PINK1-Parkin-mediated mitophagy initiation (Harper et al. 2018; Pickles et al. 2018a; Wang et al. 2020). The discovery of PINK1-Parkin-mediated pathway has been persuasive due to its contributions in understanding the key molecular mechanisms of mitophagy (Narendra et al. 2008, 2010; Vives-Bauza et al. 2010).
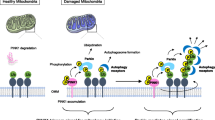
In normal healthy condition, Parkin is located in the cytosol and is in an autoinhibited state. Also, PINK1 is regularly maintained at a low level owing to mitochondrial import, protease cleavage, and proteasomal degradation, where PINK1 gets imported by the translocase of the outer membrane (TOM) complex into the inter membrane space (IMS) and the mitochondrial inner membrane (MIM), and then degraded by matrix processing peptidase (MPP), presenilin-associated rhomboid like (PARL), and the proteasome at the N-terminal part between Ala103 and Phe104 (Jin et al. 2010; Deas et al. 2011; Lazarou et al. 2012). The resulting N-terminal destabilizing amino acid is constitutively recognized by N-end rule E3 ubiquitin ligases (UBR1, UBR2, and UBR4) for protein degradation (Eldeeb and Ragheb 2018; Yamano et al. 2013). However, reduced potential of mitochondrial membrane results in accumulation of PINK1 on the outer mitochondrial membrane (OMM), and the accumulated PINK1 then undergoes dimerization and autophosphorylation at Ser228 and Ser402 thus resulting in its activation (Okatsu et al. 2012, 2013; Aerts et al. 2015; Rasool et al. 2018). Therefore, PINK1 functions as a mitochondrial damage sensor resulting in mitophagy propagation. Once activated, PINK1 leads to downstream phosphorylation events including Ser65 in the UBL domain of Parkin. Unfortunately, this alone is not enough to fully induce mitophagy, so PINK1 phosphorylate free ubiquitin as well as (poly-ubiquitin) chains at Ser65, which are already present on OMM proteins. Phosphoubiquitin (pSer65-Ub) then serves as a key receptor to initiate Parkin recruitment from cytosol to mitochondria (Okatsu et al. 2013; Kane et al. 2014; Kazlauskaite et al. 2014; Koyano et al. 2014; Shiba-Fukushima et al. 2014). Subsequently, activated Parkin linked more Ub onto OMM proteins for PINK1 phosphorylation that modulates more rounds of Parkin translocation to mitochondria, thereby forming a positive feedforward loop of PINK1, pSer65-Ub, and Parkin to trigger mitophagy. Intriguingly, PINK1-mediated pSer65-Ub is not the only identified phosphorylation; multiple other PINK1-independent phosphorylation Ub sites, including pSer20-Ub, pThr7-Ub, and pSer57-Ub, have been recognized (Wauer et al. 2015). Between them, it has been reported that pSer57-Ub hyperactivate Parkin (George et al. 2017).
After fully activation, Parkin polyubiquitylates different proteins on OMM such as Mfn1/2, TOM20/40/70, and VDAC 1 (Geisler et al. 2010; Sarraf et al. 2013). The bulk ubiquitylation of OMM proteins facilitates two main downstream events: recruitment of receptor proteins and activation of the ubiquitin–proteasome system. Receptor proteins, such as p62, interact on one side with the polyubiquitin chains directly and on the other side with LC3s or GABARAPs (Stolz et al. 2014). Initially, p62 was identified as the main adapter for Pink1/Parkin-mediated mitophagy (Geisler et al. 2010). Recently, comprehensive study reported the importance of five well-known receptors: TAX1BP1, NDP52, NBR1, p62, and OPTN. Among them, NDP52 and OPTN were found to be the most important receptors for PINK1/Parkin-dependent mitophagy (Lazarou et al. 2015). The recruitment of autophagy receptors such as NDP52 and OPTN to damaged mitochondria is TANK-binding kinase 1 (TBK1)-dependent process (Heo et al. 2015; Lazarou et al. 2015; Richter et al. 2016). TBK1 is a serine/threonine kinase that enhances the binding ability of autophagy receptors to various Ub chains through their phosphorylation (Heo et al. 2015; Richter et al. 2016). In the presence of PINK1 and Parkin, TBK1 activation also requires OPTN binding to Ub chains (Heo et al. 2015; Richter et al. 2016). In the current mitophagy model, OPTN and NDP52 recruit phagophore onto mitochondria by directly binding to LC3 through their LC3-interacting region (LIR) domain after binding to polyubiquitin chains (Gatica et al. 2018; Palikaras et al. 2018). A very recent study has highlighted the role of NDP52 in the recruitment of ULK1 complex to damaged mitochondria (Vargas et al. 2019). NDP52 directly interacts with FIP200 in a TBK1-dependent manner to recruit ULK1 complex, leading to autophagosome biogenesis on damaged mitochondria and initiation of autophagy machinery. Therefore, receptor proteins ensure the removal of mitochondria by autophagosomes.
Receptor-Mediated Mitophagy
BNIP3/NIX-Mediated Mitophagy
Several mitophagy receptors, such as ATG32 in yeast (Okamoto et al. 2009) as well as BNIP3 (BCL2 and adenovirus E1B 19-kDa-interacting protein 3) (Hanna et al. 2012), NIX (also known as BNIP3L) (Chen et al. 2010), and FUNDC1 in mammalian cells, have recently been identified. One major characteristic of mitophagy receptors is that they contain LIR that interacts with LC3, thereby enhancing the mitochondrial sequestration into phagophore (Wei et al. 2015; Bhujabal et al. 2017). The mechanism of BNIP3‐ and NIX‐mediated mitophagy is distinguished from that of the Parkin/PINK1 pathway in that these proteins act as direct adaptors targeting mitochondria to the autophagosome. BNIP3 (a member of pro-death BCL2 family proteins) (Boyd et al. 1994) and NIX (a homolog of BNIP3 with ~ 56% sequence similarity) (Matsushima et al. 1998) have BH3 domain and C-terminal transmembrane domain (TMD), which is crucial for their proapoptotic functions and mitochondrial localization (Yasuda et al. 1998; Imazu et al. 1999). Furthermore, BNIP3 and NIX have an identical N-terminus LIR domain exposed to the cytosol that facilitate interacting with LC3s (microtubule-associated protein 1A/1B light chain) for both receptors, or to GABARAP (gamma aminobutyric acid receptor-associated protein) for NIX, leading to recruitment of autophagosomes to induce mitophagy (Novak et al. 2010; Hanna et al. 2012; Birgisdottir et al. 2013). In these stress response pathways, the expression of BNIP3 is transcriptionally regulated by HIF‐1, PPARγ, Rb/E2F, FoxO3, activated Ras, and p53, whereas NIX is regulated by HIF‐1 and p53 (Sowter et al. 2001; Mammucari et al. 2007; Zhang et al. 2008). Although BNIP3 and NIX are predominantly under transcriptional control, they are post-translationally modified for their mitophagic activity. Evidently, it has been shown that serine phosphorylation at positions 17 and 24 adjacent to the LIR of BNIP3 and at positions 34 and 35 in the LIR domain of NIX enhances the interaction of these receptors with LC3 augmenting mitophagy (Rogov et al. 2017). LIR motif mutation prevents the BNIP3/NIX interaction with LC3 and thus mitigates the mitochondrial removal (Novak et al. 2010; Hanna et al. 2012; Zhu et al. 2013), while LIR motif phosphorylation promotes the interaction with LC3 and enhances mitophagy (Zhu et al. 2013; Rogov et al. 2017). NIX is implicated in the clearance of mitochondria from reticulocytes which is a crucial step for the red blood cell maturation, and it was confirmed as mitochondria were not cleared in reticulocytes when NIX is deficient (Diwan et al. 2007; Schweers et al. 2007; Zhang et al. 2008).
Furthermore, recent studies emphasized that both BNIP3 and NIX have a significant role in the progression of cancer and metastasis (Chourasia et al. 2015). In addition, it is believed that BNIP3-mediated mitophagy delays the metastatic disease progression. It is accepted that mitophagy, in general, is a tumor suppression mechanism (Bernardini et al. 2017; Roperto et al. 2019).
BNIP3 and NIX are implicated in hypoxia-induced tumor cell death. BNIP3 was identified in a subtractive hybridization screen in Chinese hamster ovary-K1 cells exposed to hypoxia, and hypoxia strongly induced BNIP3 mRNA (Bruick 2000). Furthermore, BNIP3 protein was induced by hypoxia in these cells, and the kinetics of induction correlated with cell death. The BNIP3 promoter has two HIF-1α-binding sites, and the site at − 234 relative to the translational start codon is required for transactivation by hypoxia and HIF-1α. In another study, hypoxia induced BNIP3 expression in tumor cell lines, and BNIP3 was expressed in the perinecrotic areas of several epithelial cell carcinomas (Sowter et al. 2001). In this study, BNIP3 was suppressed by Von Hippel-Lindau protein in a renal cell carcinoma cell line, consistent with its regulation through the HIF-1α pathway. Hypoxia in tumors is a negative prognostic indicator; accordingly, deregulation of BNIP3 expression is associated with aggressive disease (reviewed by Burton and Gibson (Burton et al. 2009).
Under hypoxic conditions, NIX level is fine-tuned by various post-transcriptional and post-translational mechanisms (Bruick 2000; Sowter et al. 2001; Fei et al. 2004). For instance, in U2OS osteosarcoma cells, NIX abundance appear to be regulated transcriptionally and post-transcriptionally by two factors including hypoxia and p53 (Fei et al. 2004). Although the transcriptional mechanism by which NIX levels is regulated appears to involve HIF-1α-dependent recruitment of CBP to the Nix gene, followed by recruitment of p53, the post-transcriptional mechanism remains yet to be fully elucidated. In line with this, it was found that repressing NIX level experimentally augments the growth of these cells in a tumor transplant model, underscoring a potential role for NIX in restraining tumor growth upon hypoxic circumstances. Tellingly, in studies of human cancer, hypermethylation of the BNIP3 promoter was found in pancreatic cancer (Okami et al. 2004), and the Nix gene was found to be mutated in a panel of primary breast and ovarian tumors (Lai et al. 2003). Thus, BNIP3 and NIX are regulated by hypoxia in tumor cells, and their expression is associated with tumor cell death.
In heart muscle, the BNIP3 and NIX have been shown to play a regulatory role in pathological cell death and this has been demonstrated in rat cardiomyocytes (Guo et al. 2001; Kubasiak et al. 2002; Regula et al. 2002).
Numerous researches suggest a possible crosstalk between BNIP3/NIX receptor-mediated pathway and PINK1-Parkin-mediated axis (Ding et al. 2010; Lee et al. 2011); NIX was connected to Pink1/Parkin-mediated mitophagy as a substrate of Parkin that recruits NBR1 to the mitochondria (Gao et al. 2015). Additionally, BNIP3-induced mitophagy is reduced in Parkin-deficient cells (Lee et al. 2011) and BNIP3 can stabilize PINK1 on OMM and inhibit PINK1 proteolytic degradation (Zhang et al. 2016). These results indicate that these pathways could cooperate with each other to ensure effective mitophagy.
FUNDC1-Mediated Mitophagy
FUN14 domain containing 1 (FUNDC1), an integral mitochondrial outer-membrane protein, is another important receptor for hypoxia-mediated mitophagy. FUNDC1 composed of three TMD and an LIR domain in its N-terminus exposed to the cytosol which interacts with LC3 for autophagosome recruitment (Liu et al. 2012). Like other key regulators of mitophagy, the activity of FUNDC1 is also fine-tuned by phosphorylation and dephosphorylation. The phosphorylation states of the three key residues, Ser13, Ser17, and Tyr18, in the outer membrane region of FUNDC1 have been reported to play essential roles in impacting the binding affinity for LC3 and controlling mitophagy (Chen et al. 2014; Wu et al. 2016). Under normal conditions, the LIR motif of FUNDC1 is phosphorylated at Ser13 by CSNK2/CK2 kinase and at Tyr18 by SRC kinase, which leads to inhibition of its interaction with LC3 and prevents mitophagy. Conversely, hypoxia elicits dephosphorylation of FUNDC1, which can then bind to LC3 and provoke mitophagy (Chen et al. 2014; Lv et al. 2017).
Another study showed that hypoxia leads to upregulation of ULK1 and initiates its translocation to damaged mitochondria; ULK1 directly phosphorylates FUNDC1 at serine‐17, which is required for FUNDC1 and LC3 binding leading to mitophagy (Chen et al. 2014). Furthermore, Chen et al. confirmed that phosphoglycerate mutase family member 5 (PGAM5) dephosphorylates Ser13 upon induction of hypoxia in mitochondria, which results in the enhanced interaction of FUNDC1 with LC3, and eventually selective removal of dysfunctional mitochondria, while (casein kinase 2) CK2 phosphorylates the Ser13 of FUNDC1 in normal cells to reverse the effect of PGAM5 in mitophagy activation (Chen et al. 2014).
Many studies have demonstrated that contact between the mitochondria and the ER plays a crucial role in mitochondrial fission (Friedman et al. 2011; Murley et al. 2013; Naon et al. 2014). During physiological mitochondrial fission, several mitochondrial receptors, including MFF, MID49/51, and FIS1, have been reported to recruit DRP1, a highly conserved dynamin‐related GTPase which is essential for the mitochondrial fission process (Smirnova et al. 2001). In contrast, mitochondrial fission under hypoxic conditions is still elusive, and thus, further studies are warranted to enhance our understanding of the molecular mechanisms of mitochondrial fission upon hypoxia (Kim et al. 2011).
Interestingly, it was reported that FUNDC1 integrates mitophagy and mitochondrial fission at the interface of the ER–mitochondrial contact site (MAM) through the association with ER‐membrane protein calnexin to recruit DRP‐1 (Wu et al. 2016). Another interactor of FUNDC1 is the mitochondrial E3 ligase MARCH5 (known as MITOL), which is a mitochondrially localized RING‐finger E3 ligase that is involved in mitochondrial dynamics by ubiquitylating Fis1 (Yonashiro et al. 2006), Mfn1 (Park et al. 2014), and Mfn2 (Sugiura et al. 2013). Recently, MARCH5 was found to play a role in ubiquitin‐mediated degradation of MiD49 and recruitment of Drp1 (Xu et al. 2016). The MARCH5/FUNDC1 interaction mediate FUNDC1 ubiquitylation at lysine 119 for subsequent degradation hence reducing mitophagy activity. Therefore, the regulation of MARCH5/FUNDC1 axis desensitizes mitochondrial degradation and prevents improper clearness of undamaged mitochondria.
PTEN-Short as a Negative Regulator of Mitophagy
PTEN (phosphatase and tensin homolog deleted on chromosome ten) was shown to be instrumental for several signal transduction networks. PTEN contains 403 amino acids with a N-terminal phosphatidylinositol (4,5)-bisphosphate [PI(4,5)P2]-binding domain (PBD), a catalytic phosphatase domain, a C2 domain, a C-tail domain, and a PDZ-binding motif (Lee et al. 1999). PTEN is a potent tumor suppressor with both lipid phosphatase and protein phosphatase activity, which was identified in 1997 (Li et al. 1997; Li et al. 1997; Steck et al. 1997). In addition, it is the second most common tumor suppressor, after P53, and is closely associated to tumorigenesis (Nakamura et al. 2000). Furthermore, PTEN is also widely expressed in the central nervous system (Song et al. 2012), and plays an important role in the development of the nervous system and maintenance of its normal functions. Concomitantly, its deregulation has been implicated in neurological disorders, such as Alzheimer’s disease (Sonoda et al. 2010). Previous studies demonstrated that autophagy signaling depends upon the activity of the tumor suppressor PTEN. Crucially, the role of PTEN in controlling autophagy is dependent upon its lipid phosphatase activity, which downregulates the inhibitory effect of the PI3-K/AKT pathway on the autophagic pathway by dephosphorylating phosphatidylinositol 3,4,5-trisphosphate (PIP3) to phosphatidylinositol-4,5-bisphosphate (PIP2) (Cantley et al. 1999; Arico et al. 2001; Ueno et al. 2008; Rodon et al. 2013).
Previous studies identified Ser72 in RAB7A, a RAB linked with mitophagy pathway, as influential target of TBK1 during mitochondrial depolarization. RAB7A could be a direct target for phosphorylation by TBK1 at Ser72 and provoke PINK1-Parkin-mediated mitophagy, but non-phosphorylated RAB7A failed to initiate mitophagy (Heo et al. 2018). PTEN has been suggested to regulate RAB7A dephosphorylation in the context of epidermal growth factor receptor (EGFR) signaling through the endosome (Shinde et al. 2016). Based on the findings of previous studies (Erland et al. 2018), mitophagy can be activated by Mitofusin‐2 (Mfn2) and helps injured mitochondria fuse with the lysosome (Chandra et al. 2018). Furthermore, other studies have also indicated that Mfn2 is primarily activated by the AMPK pathway (Daniel et al. 2018), which increases the phosphorylation of CREB, a transcriptional promoter (Edwards et al. 2018). Phosphorylated CREB translocates into the nucleus where it interacts with and activates the promoter of Mfn2, leading to the upregulation of Mfn2 expression and mitophagy activity (Fernández Vázquez et al. 2018). Inhibition of PTEN could promote endothelial survival via activating the AMPK–CREB–Mfn2‐mitophagy signaling pathway providing a beneficial influence on mitochondrial homeostasis, cellular survival, and endothelial migration (Li et al. 2020). A recent study by W Tang et al. concluded that inhibition of PTEN function induced by bv (phen)-suppressed PINK1/Parkin-mediated mitophagy, which resulted in an increased apoptosis and release of mitochondrial Cytochrome C in H/R-injured H9c2 cells (Tang et al. 2019). Another study has also suggested that inflammation-induced PTEN downregulation resulted in TLR4-JNK-Bnip3-mitophagy pathway activation, which eventually amplified the cellular death signals in nasal epithelial cells(Li et al. 2018).
PTEN-Long as a Pivotal Regulator of PINK1–Parkin-Mediated Mitophagy
PINK1-mediated phosphorylation and Parkin-mediated ubiquitination are the two key molecular events positively regulating mitophagy. The PINK1/Parkin pathway of mitophagy is subject to intricate regulation, primarily via the action of a number of deubiquitinating enzymes (DUBs), including USP15, USP30, USP35, and PTEN-L (PTENα) (Bingol et al. 2014; Cornelissen et al. 2014; Wang et al. 2015; Wang et al. 2018a, b). Unlike PTEN which is typically initiated at AUG codons (Kozak 1999), PTEN-L translation initiation occurs at non-AUG codons, which enhances genome coding capacity and protein diversity (Hann et al. 1988; Németh et al. 2007; Gerashchenko et al. 2010). Besides the same five functional domains with the canonical PTEN, PTEN-L contains an alternatively translated region (ATR) adding 173 amino acids at the N-terminus that encode a secretion signal sequence that allows this enzyme to be secreted into the extracellular environment (Fig. 2) (Hopkins et al. 2013). The extended ATR of PTEN-L consists of a secreted polyalanine signal sequence (Poly-A), a cell permeable polyarginine motif (Poly-R), a nuclear localization sequence (NLS), and a membrane-binding α-helix (MBH) (Hopkins et al. 2013; Malaney et al. 2013; Masson et al. 2016; Shen et al. 2019). In addition, PTEN-L may modify distinct substrates compared with PTEN as most parts of the ATR contain various post-translational modification sites and protein-binding motifs (Malaney et al. 2013; Masson et al. 2016). Significant proportion of PTEN-L is present in the mitochondrial fraction which enhances the possible regulatory role of PTEN-L in mitophagy (Wang et al. 2018a, b). PTEN-L is a canonical PTEN isoform located at outer mitochondrial membrane (OMM) and dephosphorylates Ub, hence may act to oppose PINK1/Parkin-mediated mitophagy. PTEN-L serves as the phosphatase to dephosphorylate pSer65-Ub mediated by PINK1, which is the key step for subsequent events including Parkin translocation, phosphorylation, conformational changes, and E3 ligase activation and ultimately mitophagy. Thus, PTEN-L-mediated dephosphorylation of pSer65-Ub eventually disrupts the feedforward loop and suppresses mitophagy (Fig. 3). In vitro analysis showed the role of PTEN-L on pSer65-Ub chains, a key element in the feedforward mechanism in mitophagy, and PTEN-L dephosphorylates pSer65-mono-Ub, pSer65-tetra-Ub, and pSer65-poly-Ub chains (Wang et al. 2018a, b). PTEN-L was found to be involved in many cell functions; for instance, PTEN-L was found to be regulating mitochondrial energy metabolism. Concomitantly, somatic deletion of PTEN-L impairs mitochondrial respiratory chain function, as it is involved in the electron transfer reaction and ATP production, likely through regulation of COX activity, the rate-limiting enzyme in the respiratory chain. Multiple mechanisms may be involved in PTENα regulation of COX activity (Liang et al. 2014). Based on previous studies, PTEN-L is proposed to be a membrane-permeable lipid phosphatase that is released from cells and then taken up into other cells. PTEN-L antagonized PI3K signaling and induced tumor cell death in vitro and in vivo. Recently, studies report that PTEN-L is a required component of MFN1-Bak signaling for apoptosis which resulted in mitochondrial fragmentation. This study, in conjunction to the Pink1-Parkin mitophagy-associated functions of PTEN-L, further solidifies a role for PTEN-L in regulating the targeted elimination of dysfunctional mitochondria (Sivakumar et al. 2020). Thus, understanding this novel function of PTEN-L provides a key missing piece in the molecular mitophagy pathway, a critical process in several human diseases.

Protein domain structure and isoforms of phosphatase and tensin homolog (PTEN). (A) PTEN-short (canonical PTEN) consists of five functional domains: a PIP2-binding domain (PBD), catalytic phosphatase domain, C2 lipid/membrane-binding domain, C-tail domain, and PDZ-binding motif. Canonical PTEN is translated from AUG start codon. (B) PTEN-long (PTEN-L) composed of the same five functional domains of the canonical PTEN and an alternatively translated region (ATR) which adds 173 amino acids to the N-terminus. PTEN-L is translated from a CUG start codon upstream from the classic AUG start codon. (C) ATR region structure of PTEN-L contains a polyalanine signal sequence (Poly-A), a cell permeable polyarginine stretch (Poly-R), a nuclear localization sequence (NLS), and a membrane-binding a-helix (MBH)

Key effectors involved in mitophagy machinery during healthy and damaged phases of mitochondria. Under basal mitochondrial healthy condition, PINK1 is imported into the mitochondria, cleaved by proteases, and degraded by proteasome, while Parkin keeps in an inactive conformation in the cytosol. Upon mitochondrial deregulation, PINK1 is stabilized and activated at the outer mitochondrial membrane (OMM), which leads to the phosphorylation of its downstream targets, such as ubiquitin (Ub). Parkin has a high affinity to phosphorylated Ub (pSer65-Ub), which recruits Parkin from cytosol to mitochondria. Several other factors, such as mitofusin 2 (MFN2), Miro, Rab7A, and BCL2/adenovirus E1B 19 kDa protein-interacting proteins 3 (BNIP3), are also involved in Parkin mitochondrial recruitment. Binding to pSer65-Ub releases the Ub-like (UBL) domain of Parkin from RING1 domain, partially activating Parkin. Then, PINK1 phosphorylates the UBL domain at Ser65, which drives the phospho-UBL to rebind fully activation of Parkin. On the other hand, PTEN-L located at OMM dephosphorylates Ub to suppress mitophagy
Intriguingly, recognition of PTENα helps understand the complexity of PTEN function. Tellingly, previous studies revealed that PTENα and PTEN-L have distinct functions in response to stress and might be involved in different molecular mechanisms of neuroprotection (Jochner et al. 2019). PTEN family proteins are not only involved in the regulation of PINK1-Parkin-mediated Ub-driven but also implicated in BNIP3-mediated mitophagy. Additional studies to further understand the key tenets of PTEN family proteins are thus needed. These future studies would evaluate the potential role of targeting PTEN-L as potential molecular therapeutic targets in the regulation of mitophagy to benefit mitophagy-related human diseases.
Concluding Remarks and Future Perspectives
Optimal mitochondrial functioning is critical for cellular homeostasis, and abrogation of mitochondrial operation have long been widely linked to the pathogenesis of neurodegenerative diseases such as AD, PD, and ALS. Nevertheless, the detailed molecular mechanisms by which mitochondrial integrity is compromised in neurodegeneration are still far from clear.
To combat mitochondrial damage and maintain healthy mitochondrial operation, mammalian cells have evolved sophisticated mitochondrial quality control mechanisms. In neuronal cells, mitophagy represents a major quality control strategy for the clearance of aged and deficient mitochondria through lysosomal proteolysis. While the molecular mechanisms governing mitophagy have been extensively studied in the past decade, ablation in mitophagy progression has emerged recently as a pivotal hallmark in aging-linked neurodegeneration. Importantly, approaches to enhance protection of mitochondrial function have been recently recognized as a potential practical strategy to promote neuroprotection and halt disease pathology (Eldeeb et al. 2021b). For instance, mitochondrially targeted antioxidants have been proposed to exert protective effect against neurodegeneration in mice models. Remarkably, the antioxidant MitoQ, a redox active ubiquinone targeted to mitochondria, has been demonstrated to exhibit protective role in several of aging and neurodegenerative disorders (Kelso et al. 2001; McManus et al. 2011; Miquel et al. 2014; Ng et al. 2014).
Importantly, given the findings that PTEN-L is negative regulator of mitophagy, targeting PTEN-L could be another promising target for future drug discovery investigations. Lastly, further detailed molecular studies to elucidate mitophagy in physiological-relevant cellular models not only advance our understanding of molecular basis of diseases, but also unearth novel strategies to circumvent neurodegeneration (Cai et al. 2020).
References
Abbas N, Lucking CB, Ricard S, Durr A, Bonifati V, De Michele G, Al E (1999) A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson’s disease genetics study group and the european consortium on genetic susceptibility in Parkinson’s Disease. Hum Mol Genet 8:567–574
Aerts L, Craessaerts K, De Strooper B, Morais VA (2015) PINK1 kinase catalytic activity is regulated by phosphorylation on serines 228 and 402. J Biol Chem 290:2798–2811
Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, Ogier-Denis E (2001) The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem 276:35243–35246
Bauckman KA, Owusu-Boaitey N, Mysorekar IU (2015) Selective autophagy: xenophagy. Methods 75:120–127
Bernardini J, Lazarou M, Dewson G (2017) Parkin and mitophagy in cancer. Oncogene 36:1315–1327
Bhujabal Z, Birgisdottir ÅB, Sjøttem E, Brenne HB, Øvervatn A, Habisov S, Kirkin V, Lamark T, Johansen T (2017) FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep 18:947–961
Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M (2014) The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510:370–375
Birgisdottir AB, Lamark T, Johansen T (2013) The LIR motif - crucial for selective autophagy. J Cell Sci 126:3237–3247
Boyd JM, Malstrom S, Subramanian T, Venkatesh LK, Schaeper U, Elangovan B, Al E (1994) Adenovirus E1B 19 kDa and Bcl-2 proteins interact with a common set of cellular proteins. Cell 79:341–351
Bruick RK (2000) Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci U S A 97:9082–9087
Burton TR, Gibson SB (2009) The role of Bcl-2 family member BNIP3 in cell death and disease: NIPping at the heels of cell death. Cell Death Differ 16:515–523
Cai Q, Jeong YY (2020) Mitophagy in Alzheimer’s disease and other age-related neurodegenerative diseases. Cells 9:150
Cantley LC, Neel BG (1999) New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A 96:4240–4245
Chandra M, Escalante-Alcalde D, Bhuiyan MS, Orr AW, Kevil C, Morris AJ, Nam H, Dominic P, Mccarthy KJ, Miriyala S (2018) Cardiac-specific inactivation of LPP3 in mice leads to myocardial dysfunction and heart failure. Redox Biol 14:261–271
Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H, Huang L, Zhou C, Cai X, Fu C (2014) A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell 54:362–377
Chen Y, Lewis W, Diwan A, Cheng EH-Y, Matkovich SJ, Dorn GW (2010) Dual autonomous mitochondrial cell death pathways are activated by Nix/BNip3L and induce cardiomyopathy. Proc Natl Acad Sci U S A 107:9035–9042
Chourasia AH, Tracy K, Frankenberger C, Boland ML, Sharifi MN, Drake LE, Sachleben JR, Asara JM, Locasale JW, Karczmar GS (2015) Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep 16:1145–1163
Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, Tyurin VA, Yanamala N, Shrivastava IH, Mohammadyani D (2013) Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol 15:1197–1205
Cornelissen T, Haddad D, Wauters F, Van Humbeeck C, Mandemakers W, Koentjoro B, Sue C, Gevaert K, De Strooper B, Verstreken P (2014) The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum Mol Genet 23:5227–5242
Daniel E, Azizoglu DB, Ryan AR, Walji TA, Chaney CP, Sutton GI, Carroll TJ, Marciano DK, Cleaver O (2018) Spatiotemporal heterogeneity and patterning of developing renal blood vessels. Angiogenesis 21:617–634
De Duve C, Wattiaux R (1996) Functions of lysosomes. Annu Rev Physiol 28:435–492
Deas E, Plun-Favreau H, Gandhi S, Desmond H, Kjaer S, Loh SH, Al E (2011) PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet 20:867–879
Ding W-X, Ni H-M, Li M, Liao Y, Chen X, Stolz DB, Al E (2010) Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem 285:27879–27890
Diwan A, Koesters AG, Odley AM, Pushkaran S, Baines CP, Spikes BT, Al E (2007) Unrestrained erythroblast development in Nix-/- mice reveals a mechanism for apoptotic modulation of erythropoiesis. Proc Natl Acad Sci USA 104:6794–6799
Edwards KS, Ashraf S, Lomax TM, Wiseman JM, Hall ME, Gava FN, Hall JE, Hosler JP, Harmancey R (2018) Uncoupling protein 3 deficiency impairs myocardial fatty acid oxidation and contractile recovery following ischemia/reperfusion. Basic Res Cardiol 113:47
Eldeeb MA, Bayne AN, Trempe J-F, Fon EA (2020a) Fine-tuning TOM-mitochondrial import via ubiquitin. Trends Cell Biol 30:425–427
Eldeeb MA, Fahlman RP (2018) Does too much MAGIC lead to mitophagy? Trends Biochem. Sci 43:485–487
Eldeeb MA, Fahlman RP, Michalak M (2020b) ER-associated protein degradation at atomic resolution. Trends Biochem Sci 45:723–725
Eldeeb MA, Ragheb MA (2018) Post-translational N-terminal arginylation of protein fragments: a pivotal portal to proteolysis. Curr Protein Pept Sci 19:1214–1223
Eldeeb MA, Ragheb MA (2020) N-degron-mediated degradation and regulation of mitochondrial PINK1 kinase. Curr Genet 66:693–701
Eldeeb MA, Ragheb MA, Esmaili M (2020c) How does protein degradation regulate TOM machinery-dependent mitochondrial import? Curr Genet 66:501–505
Eldeeb MA, Thomas RA, Ragheb MA, Fallahi A, Fon EA (2021b) Mitochondrial quality control in health and in Parkinson's disease. Physiol Rev. https://doi.org/10.1152/physrev.00041.2021
Eldeeb MA, Zorca CE, Ragheb MA et al (2021a) Fine-tuning ER-phagy by post-translational modifications. BioEssays 43:2000212. https://doi.org/10.1002/bies.202000212
Erland LA, Shukla MR, Singh AS, Murch SJ, Saxena PK (2018) Melatonin and serotonin: mediators in the symphony of plant morphogenesis. J Pineal Res 64:e12452
Fei P, Wang W, Kim S-H, Wang S, Burns TF, Sax JK, Buzzai M, Dicker DT, Mckenna WG, Bernhard EJ, El-Deiry WS (2004) Bnip3L is induced by p53 under hypoxia, and its knockdown promotes tumor growth. Cancer Cell 6:597–609
Fernández Vázquez G, Reiter RJ, Agil A (2018) Melatonin increases brown adipose tissue mass and function in Zücker diabetic fatty rats: implications for obesity control. J Pineal Res 64:e12472
Friedman JR, Lackner LL, West M, Dibenedetto JR, Nunnari J, Voeltz GK (2011) ER tubules mark sites of mitochondrial division. Science 334:358–362
Gao F, Chen D, Si J, Hu Q, Qin Z, Fang M, Al E (2015) The mitochondrial protein BNIP3L is the substrate of PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum Mol Genet 24:2528–2538
Gatica D, Lahiri V, Klionsky DJ (2018) Cargo recognition and degradation by selective autophagy. Nat Cell Biol 20:233–242
Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12:119–131
George S, Wang SM, Bi Y, Treidlinger M, Barber KR, Shaw GS (2017) Ubiquitin phosphorylated at Ser57 hyper-activates parkin. Biochim Biophys Acta Gen Subj 1861:3038–3046
Gerashchenko MV, Su D, Gladyshev VN (2010) CUG start codon generates thioredoxin/glutathione reductase isoforms in mouse testes. J Biol Chem 285:4595–4602
Guo K, Searfoss G, Krolikowski D, Pagnoni M, Franks C, Clark K, Yu KT, Jaye M, Ivashchenko Y (2001) Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ 8:367–376
Hann SR, King MW, Bentley DL, Anderson CW, Eisenman RN (1988) A non-AUG translational initiation in c-myc exon 1 generates an N-terminally distinct protein whose synthesis is disrupted in Burkitt’s lymphomas. Cell 52:185–195
Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson ÅB (2012) Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem 287:19094–19104
Harbauer AB, Zahedi RP, Sickmann A, Pfanner N, Meisinger C (2014) The protein import machinery of mitochondria—a regulatory hub in metabolism, stress, and disease. Cell Metab 19:357–372
Harper JW, Ordureau A, Heo JM (2018) Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Biol 19:93–108
Heo J-M, Ordureau A, Swarup S, Paulo J, Shen K, Sabatini D, Harper J (2018) RAB7A phosphorylation by TBK1 promotes mitophagy via the PINK-PARKIN pathway. Sci Adv 4:eaav0443
Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW (2015) The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell 60:7–20
Hopkins BD, Fine B, Steinbach N, Dendy M, Rapp Z, Shaw J, Pappas K, Jennifer SY, Hodakoski C, Mense S (2013) A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science 341:399–402
Hou J, Eldeeb M, Wang X (2017) In: Sun H, Wang X (eds) Mitochondrial DNA and Diseases. Springer, Singapore, pp 133–148
Hristova VA, Beasley SA, Rylett RJ, Shaw GS (2009) Identification of a novel Zn2+-binding domain in the autosomal recessive juvenile Parkinson-related E3 ligase parkin. J Biol Chem 284:14978–14986
Imazu T, Shimizu S, Tagami S, Matsushima M, Nakamura Y, Miki T, Al E (1999) Bcl-2/E1B 19 kDa-interacting protein 3-like protein (Bnip3L) interacts with bcl-2/Bcl-xL and induces apoptosis by altering mitochondrial membrane permeability. Oncogene 18:4523–4529
Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 191:933–942
Jochner MC, An J, Lättig-Tünnemann G, Kirchner M, Dagane A, Dittmar G, Dirnagl U, Eickholt BJ, Harms C (2019) Unique properties of PTEN-L contribute to neuroprotection in response to ischemic-like stress. Sci Rep 9:1–14
Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205:143–153
Kaushik S, Cuervo AM (2012) Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol 22:407–417
Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Al E (2014) Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J 460:127–139
Kelso GF, Porteous CM, Coulter CV, Hughes G, Porteous WK, Ledgerwood EC, Smith RA, Murphy MP (2001) Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J Biol Chem 276:4588–4596
Kim H, Scimia MC, Wilkinson D, Trelles RD, Wood MR, Bowtell D, Dillin A, Mercola M, Ze’ev AR, (2011) Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol Cell 44:532–544
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Al E (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608
Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Al E (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510:162–166
Kozak M (1999) Initiation of translation in prokaryotes and eukaryotes. Gene 234:187–208
Kubasiak LA, Hernandez OM, Bishopric NH, Webster KA (2002) Hypoxia and acidosis activate cardiac myocyte death through the Bcl-2 family protein BNIP3. Proc Natl Acad Sci U S A 99:12825–12830
Lai J, Flanagan J, Phillips WA, Chenevix-Trench G, Arnold J (2003) Analysis of the candidate 8p21 tumour suppressor, BNIP3L, in breast and ovarian cancer. Br J Cancer 88:270–276
Lazarou M, Jin SM, Kane LA, Youle R (2012) Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell 22:320–333
Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524:309–314
Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, Al E (1999) Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell 99:323–334
Lee Y, Lee H-Y, Hanna RA, Gustafsson ÅB (2011) Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol 301:H1924–H1931
Lemasters JJ (2005) Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 8:3–5
Li J, Yen C, Liaw D, PK, Bose S, Wang SI & Al E, (1997) PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 267:1943–1947
Li M, Yang X, Wang S (2018) PTEN enhances nasal epithelial cell resistance to TNFα-induced inflammatory injury by limiting mitophagy via repression of the TLR4-JNK-Bnip3 pathway. Mol Med Rep 18:2973–2986
Li P, Wang J, Zhao X, Ru J, Tian T, An Y, Tang L, Bai Y (2020) PTEN inhibition attenuates endothelial cell apoptosis in coronary heart disease via modulating the AMPK–CREB–Mfn2-mitophagy signaling pathway. J Cell Physiol 235:4878–4889
Li W-W, Li J, Bao J-K (2012) Microautophagy: lesser-known self-eating. Cell Mol Life Sci 69:1125–1136
Liang H, He S, Yang J, Jia X, Wang P, Chen X, Zhang Z, Zou X, Mcnutt MA, Shen WH (2014) PTENα, a PTEN isoform translated through alternative initiation, regulates mitochondrial function and energy metabolism. Cell Metab 19:836–848
Lill R (2009) Function and biogenesis of iron–sulphur proteins. Nature 460:831–838
Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Al E (2012) Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 14:177–185
Lucking CB, Abbas N, Durr A, Bonifati V, Bonnet AM, De Broucker T, Al E (1998) Homozygous deletions in parkin gene in European and North African families with autosomal recessive juvenile parkinsonism. The European Consortium on Genetic Susceptibility in Parkinson’s Disease and the French Parkinson’s Disease Genetics Study Group. Lancet 352:1355–1356
Lv M, Wang C, Li F, Peng J, Wen B, Gong Q, Shi Y, Tang Y (2017) Structural insights into the recognition of phosphorylated FUNDC1 by LC3B in mitophagy. Protein Cell 8:25–38
Malaney P, Uversky VN, Davé V (2013) The PTEN Long N-tail is intrinsically disordered: increased viability for PTEN therapy. Mol Biosyst 9:2877–2888
Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Al E (2007) FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 6:458–471
Masson GR, Perisic O, Burke JE, Williams RL (2016) The intrinsically disordered tails of PTEN and PTEN-L have distinct roles in regulating substrate specificity and membrane activity. Biochem J 473:135–144
Matsushima M, Fujiwara T, Takahashi E, Minaguchi T, Eguchi Y, Tsujimoto Y, Al E (1998) Isolation, mapping, and functional analysis of a novel human cDNA (BNIP3L) encoding a protein homologous to human NIP3. Genes Chromosomes Cancer 21:230–235
Mcmanus MJ, Murphy MP, Franklin JL (2011) The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J Neurosci 31:15703–15715
Miquel E, Cassina A, Martínez-Palma L, Souza JM, Bolatto C, Rodríguez-Bottero S, Logan A, RaJ S, Murphy MP, Barbeito L, Radi R, Cassina P (2014) Neuroprotective effects of the mitochondria-targeted antioxidant MitoQ in a model of inherited amyotrophic lateral sclerosis. Free Radic Biol Med 70:204–213
Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self-digestion. Nature 451:1069–1075
Mizushima N (2018) A brief history of autophagy from cell biology to physiology and disease. Nat Cell Biol 20:521–527
Murley A, Lackner LL, Osman C, West M, Voeltz GK, Walter P, Nunnari J (2013) ER-associated mitochondrial division links the distribution of mitochondria and mitochondrial DNA in yeast. eLife 2:e00422
Nakamura N, Ramaswamy S, Vazquez F, Signoretti S, Loda M, Sellers WR (2000) Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol Cell Biol 20:8969–8982
Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y (2009) Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol 10:458–467
Naon D, Scorrano L (2014) At the right distance: ER-mitochondria juxtaposition in cell life and death. Biochim Biophys Acta Mol Cell Res 1843:2184–2194
Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183:795–803
Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8:e1000298
Németh AL, Medveczky P, Tóth J, Siklódi E, Schlett K, Patthy A, Palkovits M, Ovádi J, Tõkési N, Németh P (2007) Unconventional translation initiation of human trypsinogen 4 at a CUG codon with an N-terminal leucine: a possible means to regulate gene expression. FEBS J 274:1610–1620
Ng LF, Gruber J, Cheah IK, Goo CK, Cheong WF, Shui G, Sit KP, Wenk MR, Halliwell B (2014) The mitochondria-targeted antioxidant MitoQ extends lifespan and improves healthspan of a transgenic Caenorhabditis elegans model of Alzheimer disease. Free Radic Biol Med 71:390–401
Novak I, Kirkin V, Mcewan DG, Zhang JW, Wild P, Rozenknop A, Al E (2010) Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 11:45–51
Okami J, Simeone DM, Logsdon CD (2004) Silencing of the hypoxia-inducible cell death protein BNIP3 in pancreatic cancer. Cancer Res 64:5338–5346
Okamoto K, Kondo-Okamoto N, Ohsumi Y (2009) Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell 17:87–97
Okatsu K, Oka T, Iguchi M, Imamura K, Kosako H, Tani N, Al E (2012) PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat Commun 3:1016
Okatsu K, Uno M, Koyano F, Go E, Kimura M, Oka T, Al E (2013) A dimeric PINK1-containing complex on depolarized mitochondria stimulates Parkin recruitment. J Biol Chem 288:36372–36384
Okatsu K, Kimura M, Oka T, Tanaka K, Matsuda N (2015) Unconventional PINK1 localization to the outer membrane of depolarized mitochondria drives Parkin recruitment. J Cell Sci 128:964–978
Palikaras K, Daskalaki I, Markaki M, Tavernarakis N (2017) Mitophagy and age-related pathologies: development of new therapeutics by targeting mitochondrial turnover. Pharmacol Ther 178:157–174
Palikaras K, Lionaki E, Tavernarakis N (2018) Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol 20:1013–1022
Park Y, Nguyen O, Kang H, Cho H (2014) MARCH5-mediated quality control on acetylated Mfn1 facilitates mitochondrial homeostasis and cell survival. Cell Death Dis 5:e1172–e1172
Pickles S, Vigie P, Youle RJ (2018a) Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol 28:R170–R185
Rasool S, Soya N, Truong L, Croteau N, Lukacs GL, Trempe JF (2018) PINK1 autophosphorylation is required for ubiquitin recognition. EMBO Rep 19:e44981
Regula KM, Ens K, Kirshenbaum LA (2002) Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ Res 91:226–231
Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, Al E (2016) Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci USA 113:4039–4044
Rodon J, Dienstmann R, Serra V, Tabernero J (2013) Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol 10:143–153
Rogov VV, Suzuki H, Marinković M, Lang V, Kato R, Kawasaki M, Buljubašić M, Šprung M, Rogova N, Wakatsuki S, Hamacher-Brady A, Dötsch V, Dikic I, Brady NR, Novak I (2017) Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci Rep 7:1131
Roperto S, De Falco F, Perillo A, Catoi C, Roperto F (2019) Mitophagy mediated by BNIP3 and BNIP3L/NIX in urothelial cells of the urinary bladder of cattle harbouring bovine papillomavirus infection. Vet Microbiol 236:108396
Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW (2013) Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496:372–376
Scheffner M, Nuber U, Huibregtse JM (1995) Protein ubiquitination involving an E1–E2–E3 enzyme ubiquitin thioester cascade. Nature 373:81–83
Schmidt O, Pfanner N, Meisinger C (2010) Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol 11:655–667
Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Al E (2007) NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA 104:19500–19505
Shen S-M, Zhang C, Ge M-K, Dong S-S, Xia L, He P, Zhang N, Ji Y, Yang S, Yu Y (2019) PTENα and PTENβ promote carcinogenesis through WDR5 and H3K4 trimethylation. Nat Cell Biol 21:1436–1448
Shiba-Fukushima K, Arano T, Matsumoto G, Inoshita T, Yoshida S, Ishihama Y, Al E (2014) Phosphorylation of mitochondrial polyubiquitin by PINK1 promotes Parkin mitochondrial tethering. PLoS Genet 10:e1004861
Shinde SR, Maddika S (2016) PTEN modulates EGFR late endocytic trafficking and degradation by dephosphorylating Rab7. Nat Commun 7:1–11
Sivakumar A, Shanmugarajan S, Subbiah R, Balakrishnan R (2020) Cardiac Mitochondrial PTEN-L determines cell fate between apoptosis and survival during chronic alcohol consumption. Apoptosis 1–15
Smirnova E, Griparic L, Shurland D-L, Van Der Bliek AM (2001) Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 12:2245–2256
Song MS, Salmena L, Pandolfi PP (2012) The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol 13:283–296
Sonoda Y, Mukai H, Matsuo K, Takahashi M, Ono Y, Maeda K, Akiyama H, Kawamata T (2010) Accumulation of tumor-suppressor PTEN in Alzheimer neurofibrillary tangles. Neurosci Lett 471:20–24
Sowter HM, Ratcliffe PJ, Watson P, Greenberg AH, Harris AL (2001) HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res 61:6669–6673
Springer MZ, Macleod KF (2016) In brief: mitophagy: mechanisms and role in human disease. J Pathol 240:253–255
Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Al E (1997) Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet 15:356–362
Stolz A, Ernst A, Dikic I (2014) Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 16:495–501
Strappazzon F, Nazio F, Corrado M, Cianfanelli V, Romagnoli A, Fimia GM, Campello S, Nardacci R, Piacentini M, Campanella M (2015) AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ 22:419–432
Sugiura A, Nagashima S, Tokuyama T, Amo T, Matsuki Y, Ishido S, Kudo Y, Mcbride HM, Fukuda T, Matsushita N (2013) MITOL regulates endoplasmic reticulum-mitochondria contacts via Mitofusin2. Mol Cell 51:20–34
Tang W, Lin D, Chen M, Li Z, Zhang W, Hu W, Li F (2019) PTEN-mediated mitophagy and APE1 overexpression protects against cardiac hypoxia/reoxygenation injury. In Vitro Cell Dev Biol Anim 55:741–748
Tasdemir E, Maiuri MC, Tajeddine N, Vitale I, Criollo A, Vicencio JM, Hickman JA, Geneste O, Kroemer G (2007) Cell cycle-dependent induction of autophagy, mitophagy and reticulophagy. Cell Cycle 6:2263–2267
Trempe J-F, Sauvé V, Grenier K, Seirafi M, Tang MY, Ménade M, Al E (2013) Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 340:1451–1455
Ueno T, Sato W, Horie Y, Komatsu M, Tanida I, Yoshida M, Ohshima S, Mak TW, Watanabe S, Kominami E (2008) Loss of Pten, a tumor suppressor, causes the strong inhibition of autophagy without affecting LC3 lipidation. Autophagy 4:692–700
Unoki M, Nakamura Y (2001) Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene 20:4457–4465
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Al E (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304:1158–1160
Vargas JNS, Wang C, Bunker E, Hao L, Maric D, Schiavo G, Al E (2019) Spatiotemporal control of ULK1 activation by NDP52 and TBK1 during selective autophagy. Mol Cell 74:347–362. e346
Villa E, Marchetti S, Ricci J-E (2018) No Parkin zone: mitophagy without Parkin. Trends Cell Biol 28:882–895
Vives-Bauza C, Zhou C, Huang Y, Cui M, De Vries RL, Kim J, Al E (2010) PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA 107:378–383
Walden H, Muqit MMK (2017) Ubiquitin and Parkinson’s disease through the looking glass of genetics. Biochem J 474:1439–1451
Wang L, Cho Y-L, Tang Y, Wang J, Park J-E, Wu Y, Wang C, Tong Y, Chawla R, Zhang J (2018a) PTEN-L is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1–Parkin-mediated mitophagy. Cell Res 28:787–802
Wang L, Wang J, Tang Y, Shen H-M (2018b) PTEN-L puts a brake on mitophagy. Autophagy 14:2023–2025
Wang L, Qi H, Tang Y, Shen H-M (2020) Post-translational modifications of key machinery in the control of mitophagy. Trends Biochem Sci 45:58–75
Wang Y, Serricchio M, Jauregui M, Shanbhag R, Stoltz T, Di Paolo CT, Kim PK, Mcquibban GA (2015) Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy 11:595–606
Wauer T, Swate KN, Wagstaff JL, Gladkova C, Pruneda JN, Michel MA (2015) Ubiquitin Ser65 phosphorylation affects ubiquitin structure, chain assembly and hydrolysis. EMBO J 34:307–325
Wei H, Liu L, Chen Q (2015) Selective removal of mitochondria via mitophagy: distinct pathways for different mitochondrial stresses. Biochim Biophys Acta Mol Cell Res 1853:2784–2790
Weidberg H, Shvets E, Elazar Z (2009) Lipophagy: selective catabolism designed for lipids. Dev Cell 16:628–630
Wu W, Lin C, Wu K, Jiang L, Wang X, Li W, Al E (2016) FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J 35:1368–1384
Xu S, Cherok E, Das S, Li S, Roelofs BA, Ge SX, Polster BM, Boyman L, Lederer WJ, Wang C (2016) Mitochondrial E3 ubiquitin ligase MARCH5 controls mitochondrial fission and cell sensitivity to stress-induced apoptosis through regulation of MiD49 protein. Mol Biol Cell 27:349–359
Yamano K, Youle RJ (2013) PINK1 is degraded through the N-end rule pathway. Autophagy 9:1758–1769
Yasuda M, Theodorakis P, Subramanian T, Chinnadurai G (1998) Adenovirus E1B–19K/BCL-2 interacting protein BNIP3 contains a BH3 domain and a mitochondrial targeting sequence. J Biol Chem 273:12415–12421
Yonashiro R, Ishido S, Kyo S, Fukuda T, Goto E, Matsuki Y, Ohmura-Hoshino M, Sada K, Hotta H, Yamamura H (2006) A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J 25:3618–3626
Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Al E (2008) Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 283:10892–10903
Zhang T, Xue L, Li L, Tang C, Wan Z, Wang R, Al E (2016) BNIP3 Protein suppresses PINK1 kinase proteolytic cleavage to promote mitophagy. J Biol Chem 291:21616–21629
Zhu Y, Massen S, Terenzio M, Lang V, Chen-Lindner S, Eils R, Al E (2013) Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J Biol Chem 288:1099–1113
Author information
Authors and Affiliations
Contributions
MAE conceived the idea of the article. MAE, ME, MH, and MAR wrote the article. ME and MAE crafted the figures. All authors edited the article.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Eldeeb, M.A., Esmaili, M., Hassan, M. et al. The Role of PTEN-L in Modulating PINK1-Parkin-Mediated Mitophagy. Neurotox Res 40, 1103–1114 (2022). https://doi.org/10.1007/s12640-022-00475-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-022-00475-w