
Abstract
The cannabinoid system has the ability to modulate cellular and molecular mechanisms, including excitotoxicity, oxidative stress, apoptosis, and inflammation, acting as a neuroprotective agent, by its relationship with signaling pathways associated to the control of cell proliferation, differentiation, and survival. Recent reports have raised new perspectives on the possible role of cannabinoid system in neurodegenerative diseases like Alzheimer disease’s (AD). AD is a neurodegenerative disorder characterized by the presence of amyloid plaques, neurofibrillary tangles, neuronal death, and progressive cognitive loss, which could be caused by energy metabolism impairment, changes in insulin signaling, chronic oxidative stress, neuroinflammation, Tau hyperphosphorylation, and Aβ deposition in the brain. Thus, we investigated the presumptive protective effect of the cannabinoid type 1 (CB1)-selective receptor agonist arachidonyl-2′-chloroethylamide (ACEA) against streptozotocin (STZ) exposure stimuli in an in vitro neuronal model (Neuro-2a neuroblastoma cells) and in vivo model (intracerebroventricular STZ injection), experimental models of sporadic AD. Our results demonstrated that ACEA treatment reversed cognitive impairment and increased activity of Akt and ERK triggered by STZ, and increased IR expression and increased the anti-apoptotic proteins levels, Bcl-2. In the in vitro model, ACEA was able to rescue cells from STZ-triggered death and modulated the NO release by STZ. Our study has demonstrated a participation of the cannabinoid system in cellular survival, involving the CB1 receptor, which occurs by positive regulation of the anti-apoptotic proteins, suggesting the participation of this system in neurodegenerative processes. Our data suggest that the cannabinoid system is an interesting therapeutic target for the treatment of neurodegenerative diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The cannabinoid system has been widely studied in neurodegenerative disorders due to its capacity to act as a central nervous system (CNS) modulator in different neurophysiological processes (Di Marzo et al. 2009; Di Marzo et al. 2015). Cannabinoids mediate a retrograde signaling in the brain that may inhibit the release of neurotransmitters by activating CB1 presynaptic receptors (Wilson and Nicoll, 2002; Hashimotodani et al. 2007). Heretofore, there are two well-characterized endocannabinoid receptors, type 1 (CB1) and type 2 (CB2), which along with its endogenous compounds as anandamide or exogenous cannabinoids derived from the plant Cannabis sativa, exert neuroprotective properties in different in vivo and in vitro models of neurodegeneration (Matsuda et al. 1990; Munro et al. 1993; Fernández-Ruiz et al. 2008; Sánchez and García-Merino 2012). Those studies have demonstrated the neuroprotective effect of cannabinoid compounds mediated by CB1 receptors (Abood et al. 2001; Karanian 2005; Gilbert et al. 2007), mediated by CB1 and CB2 receptors (Fernández-López et al. 2006), or non-receptor mediated (Marsicano et al. 2002), in several models of neurotoxicity (hypoxia and deprivation of glucose, 6-hydroxydopamine, ischemia, and oxidative stress).
The cytoprotective effects by cannabinoid compounds can be correlated with its homeostatic regulation and influence on cell death and survival through phosphorylation of several proteins, such as phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) and MAPK/ERK, both pro-survival signaling pathways (Derkinderen et al. 2003; Blázquez et al. 2015). The activation of the PI3K stimulates the protein kinase Akt pathway that influences several cellular signaling mechanisms (Ramalingam and Kim 2014), including regulation of glycogen metabolism, survival, and protein synthesis (Coffer et al. 1998). Some lines of evidence suggest that ERK and Akt activation can control the mechanisms of apoptosis through Bcl-2 family proteins, in maintaining the balance between pro-apoptotic and anti-apoptotic proteins (Ellert-Miklaszewska et al. 2005; Pilchova et al. 2014).
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by the presence of amyloid plaques, neurofibrillary tangles (NFTs), neuronal death, and progressive cognitive loss (Weksler et al. 2005). NFTs are formed by neuronal cytoskeleton collapse due to Tau hyperphosphorylation, a microtubule-associated protein (Kosik et al. 1987) while amyloid plaques are formed by accumulated protein fragments (amyloid-β, Aβ) resulted from amyloid precursor protein (APP) abnormal metabolism (Kang et al. 1987), especially in the hippocampus and cerebral cortex, relevant to learning and memory (De Felice and Ferreira 2002; Salkovic-Petrisic et al. 2013).
The major risk factors in AD are advanced age, genetics, and the environment (Kar et al. 2004). As advanced age is one of the major risk factors of sporadic AD (sAD), it could be associated with biochemical, molecular, and cellular abnormalities in the brain, including increased genes and cell death signaling pathway activation, energy metabolism impairment, changes in insulin signaling, mitochondrial dysfunction, chronic oxidative stress, neuroinflammation, Tau hyperphosphorylation, and Aβ deposition in the brain (De la Monte and Wands 2005; Lester-Coll et al. 2006). It is interesting to note that the key mechanism of neuronal death in sAD has been associated with an insulin-resistant brain state (IRBS), combined with cerebral glucose hypometabolism (Nitsch and Hoyer 1991; Henneberg and Hoyer 1995; Lannert and Hoyer 1998; Hoyer 2002; De Felice et al. 2014; De La Monte and Tong 2014). IRBS is a condition characterized by the reduced response of insulin receptor (IR) and its downstream effectors, PI3K and Akt pathway in the brain, which, particularly considering the neurotrophic, neuroprotective, and neuromodulatory roles of insulin in the brain, may lead to neurodegeneration and cognitive impairment as seen in AD (Salkovic-Petrisic et al. 2009). In addition to its well-known role on insulin signaling, the PI3K/Akt/glycogen synthase kinase (GSK3) cascade regulates APP metabolism and Tau phosphorylation (Ishiguro et al. 1993; Phiel et al. 2003; Salkovic-Petrisic et al. 2006).
In this context, the intracerebroventricular (icv) administration of streptozotocin (STZ) in rats is a well-established, validated, and widely accepted animal model to study sAD (Lannert and Hoyer 1998; Salkovic-Petrisic and Hoyer 2007). STZ is a glycosamine nitrosourea compound, which induces cognitive impairment and changes in the brain that resembles those found in AD patients. Previously, icv STZ injection was shown to induce oxidative stress (Zhu et al. 2003; Rai et al. 2014), neuronal cell damage (Kamat et al. 2016), and dysfunctions in learning and memory (Shoham et al. 2007; Santos et al. 2012; Crunfli et al. 2018). Accordingly, icv STZ model has been used to assess the therapeutic potential of various drugs, as well as other non-drug therapeutic strategies.
Therefore, the present study was aimed at exploring whether the presumptive protective effect of the cannabinoid type 1 (CB1)-selective receptor agonist arachidonyl-2′-chloroethylamide (ACEA) depends upon its activity on the CB1 receptor, and involving the IR/PI3K/Akt, MAPK/ERK, and pro-survival signaling pathways. To this purpose, the neuroprotective effects of ACEA was evaluated both in vivo, in a rat model of AD induced by icv STZ injection, and in vitro, in a neuronal model (Neuro-2a neuroblastoma cells) exposed to STZ.
Materials and Methods
Animals
Adult male Wistar rats (RRID: RGD_2312511; 12 weeks of age), weighing 300–350 g at the beginning of the experiments (n = 6–8/group), were obtained from the Facility for SPF rat production at Institute of Biomedical Sciences, University of São Paulo (Animal Facility Network at USP), and maintained at humidity- and temperature-controlled room (22 °C ± 2) under a 12 h light/12 h dark cycle (lights on at 06:00). A group of 3–4 animals per polyacrylic cages were kept with free access to food and water. All behavioral tests were carried out between 7:00 a.m. and 1:00 p.m. for each time point and after 30 min of habituation in the testing room.
The experimental protocol was evaluated and approved by the “Ethics Committee for Animal use” of the Institute of Biomedical Sciences, University of São Paulo (Protocol no. 33/55/02) following the Brazilian Federal Law (no. 11794; 10/08/2008). All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Intracerebroventricular Injection of STZ
Intracerebroventricular injections of STZ were conducted as previously described (Santos et al. 2012; Crunfli et al. 2018). The animals were anesthetized with 2,2,2-tribromoethanol (2.5%, 1 mL/100 g i.p., Sigma, St. Louis, MO, USA), and positioned in a stereotaxic frame (David Kopf Instruments, Tujunga, CA, USA). The injection of STZ was performed using a pressure injection system (Picospritzer) following coordinates: AP − 0.8 mm; ML ± 1.4 mm; DV − 3.4 mm relative to the bregma (Paxinos and Watson 2007). The STZ (C8H15N3O7; 3 mg/kg; Sigma) or the same volume (4 μL) of citrate buffer (0.05 mol/L; pH 4.5) was injected bilaterally into the lateral ventricles. After surgery, the animals received a polyantibiotic (0.1 mg/kg i.m.; Laboratório Bravet, Rio de Janeiro, RJ, Brazil). Clinical signs were also monitored daily after the surgery, including general body condition and dehydration.
Administration Intraperitoneal of ACEA
The animals received an intraperitoneal treatment with the selective cannabinoid type 1 (CB1) receptor agonist ACEA (3 mg/kg; Sigma) diluted in vehicle solution (saline, DMSO, Tween 8:1:1) or vehicle only. The treatments were performed during 7 days after the stereotaxic procedure. ACEA doses were based on earlier reported studies (Tsvetanova et al. 2006; Rutkowska and Gliniak 2009).
The animals then constituted the following groups: animals receiving citrate treated with vehicle solution (control = CTL); animals receiving STZ treated with vehicle solution (STZ); animals receiving citrate treated with ACEA (ACEA); animals receiving STZ treated with ACEA (STZ + ACEA).
Novel Object Recognition Behavioral Test
The object recognition test analyzes non-spatial working memory with characteristics of episodic memory that relies on intact cortical and hippocampal function (Mumby 2002; Winters et al., 2004). After surgery recovery (approximately 72 h), the animals were tested (n = 6–8 for each group) in a novel object recognition test based on the innate tendency of rodents to differentially explore novel objects over familiar ones in an open field arena. The behavioral assay consisted of three phases: training phase, testing phase 1 (short-term memory), and testing phase 2 (long-term memory) as previously described (Crunfli et al. 2018). In the training phase (third and fourth days), rats were habituated to the open field arena (Ø = 90 cm; height = 50 cm), for 10 min, three times a day with intervals of 1 h, for 2 days, in the absence of any object and then were taken back to their home cage. Each rat was tested separately and any olfactory/taste cues in the arena and objects were eliminated between each animal.
On the fifth day (testing phase 1), rats were allowed to explore two identical objects (same size, color, and material) arranged in a symmetric position from the center of the arena, for 10 min (objects A and A’). The animals returned to their home cage for a retention period. The exploratory rat behavior was measured in terms of the time spent actively exploring (sniffing or touching with the nose and/or forepaws) the objects. One hour later, the rats were placed into the arena in presence of one familiar object (object A) and a novel object (object B) of the same size, color, and material, but with a different format. The exploratory activity was measured during 5 min, in order to test the short-term memory.
Finally, 24 h after the first test (testing phase 1), the rats were placed again into the arena in the presence of one familiar object (object A) and another object of equivalent size but with a different shape and color (object C). The exploratory activity was measured during 5 min, in order to test the long-term memory (testing phase 2). The intraperitoneal treatments with ACEA or vehicle were made after completing the training phase and test phases 1 and 2. The animals were sacrificed 24 h after testing phase 2, and the hippocampi were collected for protein analysis by Immunoblotting (seventh day).
A discrimination index (DI) was obtained considering the exploration periods of each object. The DI was determined as the ratio between the time spent to explore the new object (Tnew) over the total time spent exploring both objects (Ttotal), known as discrimination index (DI = Tnew/Ttotal).
Immunoblotting
Animals tested in behavioral tasks were euthanized by decapitation and the hippocampi were rapidly dissected and homogenized in extraction buffer (Santos et al. 2012; Crunfli et al. 2018), and protein concentration of the samples was determined by Bradford method (Bio-Rad, Hercules, CA, USA). Thirty micrograms of protein were loaded on standard SDS-PAGE procedure (10% polyacrylamide minigels) followed by electrotransference to nitrocellulose membranes using a semi-dry Trans-Blot cell system (Bio-Rad, Hercules, CA, USA). The membranes were then blocked for 2 h at room temperature with phosphate-buffered saline (PBS) containing 0.05% Tween-20 (TTBS) and 5% non-fat milk and incubated overnight at 4 °C with the following specific primary antibodies: a rabbit polyclonal anti-insulin Rβ (Santa Cruz Biotechnology, cat. no. sc-711, RRID:AB_631835), a rabbit polyclonal anti-PI3K p85 (Cell Signaling Technology, cat. no. 4292, RRID:AB_329869), a rabbit polyclonal anti-Akt1/2 (Santa Cruz Technology, cat. no. sc-8312, RRID: AB_671714), a rabbit polyclonal anti-phospho Akt1/2 Ser 473 (Santa Cruz Technology, cat. no. sc-7985-R, RRID: AB_667741), a rabbit polyclonal antibody anti-phospho-p44/42 MAPK (ERK1/2) (Cell Signaling Technology, Thr202/Tyr204; cat. no. 9101; RRID: AB_331646), a mouse monoclonal antibody anti-ERK1/2 (MK1) (Santa Cruz Biotechnology, cat. no. sc-135900; RRID: AB_2141283), a mouse polyclonal anti-GSK3α/β total (Santa Cruz Biotechnology, cat. no. sc-56913, RRID:AB_783600), a rabbit polyclonal anti-phospho-GSK3α/β (Ser21/9) (Cell Signaling Technology, cat. no. 9331, RRID:AB_10171207), a rabbit polyclonal anti-BAX (Cell Signaling Technology, cat. no. 2772S, RRID: AB_10695870), a rabbit monoclonal anti- Bcl-2 (Cell Signaling Technology, cat. no. 2876, RRID:AB_2064177), all diluted 1:1000. A loading control was conducted in all experiments by using a mouse monoclonal antibody anti-GAPDH (Santa Cruz Biotechnology, 6C5; cat. no. sc-32233, RRID: AB_627679) diluted 1:5000. After incubation with the corresponding secondary antibody (anti-mouse or anti-rabbit IgG; 1:10,000; Amersham Biosciences; Little Chalfont, Buckinghamshire, UK), blots were visualized using a chemiluminescence system in a digital scanner (Li-Cor Biosciences, Lincoln, NE, USA) and densitometrically analyzed by the Image Studio Digits v.4.0 software (Li-Cor Biosciences). The corresponding bands normalized by GAPDH in each experiment were expressed as percentage of protein changes in relation to control mean value.
Cell Culture
In order to investigate the underlying molecular mechanisms of the ACEA neuroprotective effects on neuronal cells, experiments with the mouse neuroblastoma Neuro-2a cell-line (American Type Culture Collection, ATCC, Richmond, VA, USA #CCL-131, RRID: CVCL_0470) were performed. Cells were cultured as previously described (Crunfli et al. 2018; Vrechi et al. 2018) and seeded to reach 70–80% confluence by the end of the experiment in Dulbecco’s Modified Eagle Medium, DMEM (Gibco, Walthmam, MA, USA; cat. no. 12800017), at 37 °C and 5% CO2.
Cell Viability Measurements
Cell viability was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay (Mosmann 1983). Briefly, cells were seeded into 24-well plates (2.5 × 104 cells per well) and cultured for 2 days. Cells were then exposed to STZ, ACEA, and AM251 and their viability measured 24 h later with MTT solution (0.5 mg/mL; Amresco; Solon, OH, USA) in serum-free DMEM medium at 37 °C in the dark during 3 h. The absorbance was determined spectrophotometrically at 550 nm in a microtiter plate reader (BioTek, Winooski, VT, USA).
Determination of Spontaneous Nitric Oxide Donation
The total nitrite level was measured in culture medium after 24 h of the STZ and ACEA Neuro-2a cell exposition. The spectrophotometric assay was performed according to the Griess reaction principle (Green et al. 1982), which consists of incubating 50 μL of Griess’s reagent [0.1% of N-(1-naphthyl)ethylenediamine dihydrochloride, 1% sulfanilamide, 2.5% phosphoric acid] with 50 μL of the culture medium from treated cells. Incubation was performed for 10 min, followed by reading the absorbance at 548 nm in a spectrophotometer plate compared to a nitrite standard curve. The standard curve was made with known concentrations of sodium nitrite.
Statistical Analysis
Data were expressed as mean ± SEM. Statistical significance analysis was assessed by using two-way or one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test between groups. All analyses were performed using the GraphPad Prism 6.0 software (San Diego, CA, USA) and a significance level of p ≤ 0.05 was adopted.
Results
ACEA Improves Cognitive Impairment Triggered by STZ
The evaluation of short-term memory demonstrated that the selective CB1 receptor agonist ACEA was able to improve the memory deficit caused by the icv injection of STZ (STZ factor: p = 0.0158; treatment factor: p = 0.0307; and interaction: p < 0.0001). The STZ and ACEA groups showed a reduction of 34.09% (p < 0.0001) and 17.56% (p = 0.0061), respectively, in the DI of short-term memory compared to CTL group. However, the STZ + ACEA group exhibited an increase of 33.07% (p < 0.0001) in the DI of short-term memory compared to STZ group (Fig. 1a). Regarding the total time of the exploration, the two-way ANOVA showed treatment significant effects (p = 0.0236; Fig. 1b), in other words, both ACEA treatment groups (ACEA and STZ + ACEA) exhibited an increased exploration total time.

Effects of ACEA treatment after intracerebroventricular STZ injection on short- and long-term memories of rats evaluated by New Object Recognition behavioral test. The discrimination index was calculated by the time exploring the novel object/total exploring time. The ACEA treatment reverses the STZ reduction in the discrimination index of both a short- and c long-term memories. The total exploration time the objects not altered on b short- and d long-term memory tests. Data represent the mean ± SEM (n = 6–8). Statistical significance was determined by two-way ANOVA followed by Tukey’s posttest; **p < 0.01, ***p < 0.001, and ****p < 0.0001 compared to control (CTL) group; ####p < 0.0001 compared to STZ + ACEA group
We also observed that ACEA was able to improve the long-term memory deficit caused by the injection of STZ (STZ factor: p = 0.0017; treatment factor: p < 0.0001, and interaction: p = 0.0007). The evaluation of long-term memory demonstrated that STZ group exhibited a decrease of 36.82% (p = 0.0001) in the DI compared to CTL group, and ACEA treatment increased 43.21% (ACEA, p < 0.0001) and 44.94% (STZ + ACEA, p < 0.0001) when compared to STZ group (Fig. 1c). The total time of the exploration on testing phase 2 was not significantly different (Fig. 1d).
Effects of ACEA Treatments on Protein Expression Related to Insulin Signaling After Exposure to STZ
Analysis of the insulin signaling pathway showed that ACEA treatment altered the decrease of IR triggered by STZ (treatment factor: p = 0.0128; interaction: p = 0.002). The STZ + ACEA group showed an increase of 92.13% (p = 0.0012) in the IR levels when compared to the STZ group (Fig. 2a). The PI3K levels were modified only by the action of STZ (STZ factor: p < 0.01). ACEA treatment did not alter PI3K protein levels (Fig. 2b).

Effects of ACEA treatment after intracerebroventricular STZ injection on the level of IR and PI3K protein evaluated by immunoblotting, after 7 days. The ACEA treatment altered the decrease of a IR triggered by STZ and b STZ icv increased the PI3K levels. Data represent the mean ± SEM (n = 6–8). Statistical significance was determined by ANOVA two-way followed by Tukey’s posttest; #p < 0.05; ##p < 0.01 compared to STZ + vehicle group; &p < 0.05 compared to ACEA group
The PI3K activation stimulates the protein kinase Akt pathway that influences several cellular signaling mechanisms (Ramalingam and Kim 2014), including regulation of glycogen metabolism, survival, and protein synthesis (Coffer et al. 1998). We observed that ACEA reversed the increased activity of Akt triggered by STZ (interaction: p = 0.007). The STZ group showed an increase of 121.2% and 154.9% in Akt phosphorylation levels when compared to the CTL and STZ + ACEA groups, respectively (p = 0.0394; p = 0.012; Fig. 3a). The Akt kinase activity results in the phosphorylation of GSK3β in Serine 9 (Ser9) and GSK3α isoform in Ser21, which in turn, inhibits GSK3 activity (Hooper et al. 2008). There was no significant difference in GSK3 phosphorylation levels (Fig. 3b). However, we observed that STZ group showed an increase of 30.8% in total GSK3 protein levels (p = 0.0389) and there is an interaction between STZ and ACEA treatment in total GSK3 protein levels (p = 0.0162).

Effects of ACEA treatment after intracerebroventricular STZ injection on the Akt/GSK3 and ERK activities evaluated by immunoblotting, after 7 days. The ACEA treatment reversed the increase of a Akt and c ERK activity triggered by STZ, and b did not altered GSK3 levels. Data represent the mean ± SEM (n = 6–8). Statistical significance was determined by two-way ANOVA followed by Tukey’s posttest; *p < 0.05 compared to control (CTL) group; #p < 0.05; ##p < 0.01 compared to STZ + ACEA group
The CB receptor activation regulates the ERK1/2 activity protein (Laezza et al. 2010; Caffarel et al. 2012). We demonstrated that the interaction between STZ and ACEA were able to modulate ERK phosphorylation (p = 0.0004). The phosphorylation/total protein ratio of ERK was increased in 125.4% in the STZ group when compared to CTL group (p = 0.0429) and ACEA treatment reduced by 144.9% the ERK activity compared to STZ group (p = 0.0215; Fig. 3c).
ACEA Regulates Protein Expression Related to Apoptotic Pathways After Exposure to STZ
The Akt and ERK proteins could modulate anti- and pro-apoptotic Bcl-2 family members which control cell survival/proliferation processes (Granado-Serrano et al. 2006). We observed that ACEA treatment was able to increase the levels of Bcl-2, an anti-apoptotic protein (treatment factor: p < 0.0001), indicating a neuroprotective effect of this compound. The ACEA and STZ + ACEA groups showed an increase of 133.4% and 166%, respectively, when compared to the CTL group (p = 0.0016; p = 0.0002). It was also possible to observe a difference of 110.4% between the STZ + ACEA vs STZ groups (p = 0.0003, Fig. 4a). There was no significant difference in pro-apoptotic protein levels, BAX. ACEA and STZ did not alter the levels of this protein in 7 days (Fig. 4b). The balance between anti- and pro-apoptotic members of the Bcl-2 family is one of the major mechanisms that control apoptosis in cells. The analysis shown in Fig. 4c was carried out by Student’s t test comparing the percentage of Bcl-2 versus Bax of each group. Thus, we observed that ACEA and STZ + ACEA groups showed alteration in the balance of Bcl-2 and BAX proteins (p = 0.0004; p = 0.0017, respectively), in favor of cell survival (Fig. 4c).

Effects of ACEA treatment after intracerebroventricular STZ injection on the apoptosis marker evaluated by immunoblotting, after 7 days. The ACEA treatment increase the a Bcl-2 and b did not altered BAX levels. Data represent the mean ± SEM (n = 6–8). Statistical significance was determined by two-way ANOVA followed by Tukey’s posttest; **p < 0.01;****p < 0.0001 compared to control (CTL) group; ##p < 0.01 compared to STZ + ACEA group. c The ACEA treatment changed the balance between Bcl-2 and BAX proteins, in favor to cell survival. Statistical significance was determined by Student’s t test; **p < 0.01;***p < 0.001 compared the percentage of Bcl-2 vs Bax of each group
ACEA Protects Neuro-2a Cells from Neurodegeneration Conditions Triggered by STZ
The Neuro-2a cell line has already been used in several studies as an in vitro neuronal model to screen new compounds with neurotoxic and neuroprotective properties, and to evaluate the associated mechanisms (Calderón et al. 1999; Ahmad et al. 2014; Stygelbout et al. 2014; Café-Mendes et al. 2017). In addition, this lineage expresses the CB1 and TRPV1 receptors and has also been used in studies related to the cannabinoid system (Sarker and Maruyama 2003; Jordan et al. 2005).
The STZ doses were experimentally determined to set cell death in 30–40% by MTT reduction assay (Crunfli et al. 2018). The doses of cannabinoid compounds tested were based on our previous studies (Vrechi et al. 2018; Batinga et al. 2016) and described in other studies as active doses in neuronal tissue or in cell culture (Harvey et al. 2012; Caltana et al. 2015). The lower doses of cannabinoid compounds, which failed to induce cell death, were chosen for the subsequent experiments.
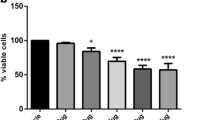
We have previously determined by the dose–response experiments, the doses of 5 mM for STZ, 1 μM for ACEA, and 3 μM for AM 251, a CB1 receptor antagonist/inverse agonist (Vrechi et al. 2018; Crunfli et al. 2018). The exposure of Neuro-2a cells to STZ (5 mM) decreased in 32.98% ± 4.07% (p = 0.0439) the cellular viability compared to control group and ACEA (1 μM) was able to rescue the cells from death, producing an improvement of 34.18% ± 10.93% (p = 0.0323) in cell viability compared to STZ group (Fig. 5a). In order to evaluate whether the ACEA protective effect against STZ was mediated by CB1 receptor, the CB1 receptor antagonist/inverse agonist AM 251 was administered in the presence and absence of ACEA. The ACEA + AM 251 treatments in 1:3 stoichiometry was able to reverse the protective effect of ACEA in Neuro-2a cells exposed to STZ decreasing in 34.03% (p = 0.0447) the cell viability compared to STZ + ACEA 1 μM treated group (Fig. 5a).

Effects of ACEA treatment and streptozotocin on Neuro-2a cell viability and NO donation after 24 h, assessed through MTT and Griess assays. a MTT reduction assay shows a decrease in Neuro-2a cell viability produced by STZ and neuroprotective effect of ACEA mediated by CB1 receptor. b Streptozotocin and ACEA donates nitric oxide once in solution. Data (expressed in percentage) represent the mean ± SEM (n = 7–9 for MTT and n = 4–5 for Griess). Statistical significance was determined by one-way ANOVA followed by Tukey’s posttest; *p < 0.05; ***p < 0.001; ****p < 0.0004 compared to control group; #p < 0.05; ####p < 0.0001 compared to STZ + ACEA group
Along with the analysis of nitrite release after treatments, it was possible to observe that the cells treated with ACEA and STZ showed a significant increase (22.58% ± 0.025, p = 0.0010; 17.15% ± 0.00327, p = 0.011, respectively) in nitrite production compared to control group. In addition, it was also observed that STZ + ACEA treatment reversed the increased release of nitrite produced by STZ, reducing in 36.03% ± 0.0173 (p < 0.0001) when compared to STZ, and 18.88% ± 0.00193 (p = 0.0052) when compared to control group, indicating that ACEA could possibly modulate NO release (Fig. 5b).
Discussion
In this study, we have shown that ACEA treatment for 7 days was able to reverse the cognitive impairment and modify some protein alterations of the IR/PI3K signaling pathway generated by STZ. In particular, the expressive increase of the Bcl-2 anti-apoptotic protein and the NO modulation revealed a possible neuroprotective mechanism of ACEA.
Endocannabinoid system has been implicated in several biological functions, and many data have suggested its relation to chronic neurodegenerative diseases, such as AD, Huntington’s disease, multiple sclerosis, and Parkinson’s disease (Pazos et al. 2004; Benito et al. 2007; Ruiz-Valdepeñas et al. 2010). Thus, the pharmacological manipulation of cannabinoid system has been proposed as possible therapeutic approach to treat symptoms and/or progression of neurodegenerative diseases. The neuroprotective role of cannabinoid system through the activation of cannabinoid CB1 receptors is supported by findings in both in vitro and in vivo models of AD (Abood et al. 2001; Karanian 2005; Gilbert et al. 2007). For example, Milton (2002) reported that anandamide and noladin ether, CB1 agonists, inhibited Aβ toxicity in a concentration-dependent manner, and the CB1 antagonist, AM251, prevented these protective effects in differentiated human teratocarcinoma cell line (Milton 2002). Moreover, Van Der Stelt et al. (2006) demonstrated that early potentiation of the endocannabinoid tone leads to neuronal loss prevention and to an anti-inflammatory response in experimental model of Aβ neurotoxicity in rats.
In AD context, low doses of STZ icv injection has been associated with morphological, molecular, and behavioral changes comparable to those observed in sporadic AD (Shoham et al. 2007; Santos et al. 2012; Crunfli et al. 2018). In particular, STZ icv causes brain energy metabolism and insulin signaling impairments that culminates in a neurodegeneration phenotype resembling sAD (De la Monte and Wands 2005; Salkovic-Petrisic and Hoyer 2007; Santos et al. 2012). In relation to AD, it was observed that chronic treatment with CB1 receptor agonists such as ACEA (Aso et al. 2012) or WIN 55,212-2 (Martín-Moreno et al. 2012) resulted in cognitive improvement in two different transgenic models of cerebral amyloidosis (AβPP/PS1 and Tg APP mice, respectively). The efficacy of cannabinoid compounds in reducing cognitive impairment was inversely proportional to disease progression stage (Aso et al. 2012). In our study, ACEA treatment was also able to reverse the cognitive impairment generated by STZ, suggesting a neuroprotective effect of the CB1 agonist. On the other hand, ACEA treatment alone caused a reduction in discrimination index on short-memory analysis. Recent researches in rodents suggested that cannabinoid receptor activation treatments could produce memory impairments (Barzegar et al. 2015; Goodman and Packard 2015). However, this view may be not oversimplified. Conflicting data on the effects of the cannabinoid system in learning and memory have been reported (Terranova et al. 1996; Puighermanal et al. 2009). The cannabinoid system is widely known as a neuromodulatory system participating in several important biological functions, like memory and learning. The CB1 receptor stimulation may promote a modulation role on CNS that cause memory beneficial or impairment effects (Puighermanal et al. 2012). In our results, ACEA treatment impaired memory acquisition but not altered the consolidation phase that was proven by ACEA effects on the long-term memory evaluation. Moreover, ACEA treatment improves the cognitive impairment triggered by STZ. Consistent with our findings, Ramírez et al. (2005) reported that the cannabinoid agonist WIN55,212-2 prevented the cognitive impairment induced by Aβ icv administration to Wistar rats. This neuroprotective effect was revealed by increased neuronal markers. In general, the modulation of the cannabinoid system may promote beneficial effects in a wide variety of pathological states, like AD. Under this conceptual paradigm, the use of agonists or by enhancing the endogenous cannabinoid tone may induce beneficial effects on the evolution of AD (Aso and Ferrer 2014; Zou and Kumar 2018).
In the last decades, the brain insulin receptor (IR) system is receiving major attention in the context of brain cognitive functions and neuronal survival regulation (Hoyer et al. 1994; De la Monte and Wands 2005). Some studies showed reduced expression of IR in the brain of patients suffering from AD (Frölich et al. 1998; Steen et al. 2005). Moreover, icv STZ injection in rats also reduced IR expression in the hippocampus at separate times, 14 days, 1 month, 3 months, and 6 months, and suggested that hippocampal IR system might be playing an important role in regulation of memory functions associated with STZ induced memory deficit (Agrawal et al. 2011; Osmanovic Barilar et al. 2015). We did not observe difference between control and STZ groups. However, the STZ group showed a tendency to a decrease in the IR expression (a difference of 40.98%). Previous laboratory data demonstrated that icv STZ injection at 3, 5, and 7 days decreased IR protein content, and we also have shown that STZ causes insulin resistance and impairment PI3K activity in the Neuro-2a cells (Crunfli et al. 2018). We observed that ACEA treatment upregulated the IR expression that could be directly related with the neuroprotection against the STZ insult conferred by the stimulation of CB1 receptors. ACEA treatment only had effect in interaction with the STZ insult. We suggested that was a neuroprotective CB1 receptors response against impaired insulin signaling triggered by STZ related to neurodegenerative processes, resulting in cognitive improvement. This result was in agreement with a previous study, revealing that metformin, an insulin sensitizer against peripheral insulin resistance and a neuroprotective drug (El-Mir et al. 2008), was also able to increase the impaired insulin-stimulated tyrosine phosphorylation of IRβ, completely restoring it to that of insulin-sensitive condition (Gupta et al. 2011). In the IR phosphorylation downstream signaling, the activities of Tau kinases, GSK3b and ERK1/2, were also restored to normal levels by metformin treatment, suggesting a neuroprotective action.
Moving downstream the IR/PIK3/Akt signaling pathway, activation of IR results in phosphorylation of insulin receptor substrate (IRS) proteins, creating binding sites for PI3K, a protein that regulates survival and growth, and influences on apoptosis and cellular metabolism pathways (Bedse et al. 2015). When PI3K is activated by phosphorylation at Tyr458 (subunit p85), it leads to activation of the downstream pathways including the Akt and MAPK cascades (Aso et al. 2012). ACEA treatment did not alter PI3K protein levels, possibly due to a temporal effect of the activation pathway. However, ACEA treatment attenuated levels of Akt and ERK, PI3K-mediated proteins, and normalized the activity of these proteins that were deregulated by STZ. ERK has been known for its effects on cell survival. However, the literature also demonstrates a critical role of ERK in neuron. ERK has been reported to be involved in the induction of cell death (Subramaniam et al. 2004); nevertheless, the mechanisms are not clear. Activated MAPK (pERK1/2) has been reported to be associated with kinase of tau and neurofibrillary tangles, hallmarks of AD-type neurodegeneration. In addition, the activity of ERK is increased in neurodegenerative diseases like AD, and its inhibitors are suggested as attractive therapeutic agents (Colucci-D’Amato et al. 2003). Our findings suggested that ACEA is a neuroprotective drug by its downregulation effect on both ERK1/2 and Akt activity, concomitant with improvement on cognitive deficits.
STZ is a glucosamine-nitrosourea compound that acts as a nitric oxide donor, wherefore the ERK activation by STZ could be a response to NO donation or to increase of APP, Aβ, and p-Tau (Ferrer et al. 2001). Ramalingam and Kim confirmed that ERK can be activated by H2O2 in glioblastoma culture and demonstrated that ERK is blocked by insulin attenuating H2O2-induced cell death through downregulation of ERK1/2 and upregulation of Akt pathways. A different neuroprotection action was observed, in which ACEA treatment downregulated and normalized both ERK1/2 and Akt activity, concomitant with improvement on cognitive deficits. Akt protein plays a critical role in cell growth, differentiation, and survival through phosphorylation of numerous cellular proteins (Ellert-Miklaszewska et al. 2005; Blázquez et al. 2015; Kokona and Thermos 2015). Akt/PKB has emerged as a central player in the signal transduction pathways to contribute to several cellular functions including metabolism, cell growth, transcriptional regulation, and cell survival. Albeit Akt is a protein related to neuronal survival (Ryu et al. 1999), some toxic substances may activate this protein, i.e., the Aβ peptide activated Akt in SH-SY5Y cells reflecting an ineffective cellular protection response of the cells to a toxic stimulus. In addition, the PI3k/Akt blocked the pathway, increased Aβ-peptide-induced cell death, suggesting that Akt basal activity levels are important to cell viability (Wei et al. 2002). In the STZ model context, we have shown previously that Akt was deregulated through S-nitrosylation (Crunfli et al. 2018) which consequently leads to the inactivation of PTEN (Numajiri et al. 2011). After 1 day of icv STZ injection, Akt activity was decreased, probably due to high NO levels donated by STZ, and after 5 and 7 days Akt activity was increased, presumably by the NO action on PTEN, through S-nitrosylation and consequent inactivation of PTEN, favoring the phosphorylation and activation of Akt and PI3K (Crunfli et al. 2018). We observed that ACEA treatment attenuated Akt levels and normalized the activity of this protein, presumably through ACEA modulation of the NO release produced by STZ.
Akt acting together with the ERK1/2 pathway influences several targets that results in attenuation of apoptosis through modulation of pro- and anti-apoptotic proteins of the Bcl-2 family (Yu et al. 2014). These proteins have emerged as key players in the regulation of apoptosis and have been proposed to integrate signals from survival-inducing and death-promoting pathways. Bad, a BH3-only pro-apoptotic protein, is a common target of Akt kinase and Erk1/2-activated ribosomal S6 kinase (p90Rsk), which is inactivated by survival-promoting factors (Downward 1999). In addition, the phosphorylation of Bad regulates effects on Bcl-xL/Bcl-2 and Bax function. Bcl-2 and Bcl-xl exert anti-apoptotic effect, at least in part, by binding to Bax and related pro-apoptotic proteins and preventing them from inducing damage to mitochondria that would result in release of cytochrome c and activation of the caspase-9/Apaf. The anti-apoptotic protein (Bcl-2) is known to regulate the activation of Bax thorough heterodimerization (Shen et al. 2010). The balance between anti- and pro-apoptotic members of Bcl-2 family is one of the major mechanisms that control apoptosis in cells.
The anti-apoptotic protein, Bcl-2, has been widely shown to have a protective effect against several insults and to be involved in neurodegenerative diseases (Paradis et al. 1996; Winter et al. 2002). Previous studies demonstrated that Bcl-2 downregulation is involved in Aβ-induced neuronal apoptosis (Ferreiro et al. 2007) and the increased Bcl-2 attenuated the processing of amyloid precursor protein and Tau, reducing the extracellular deposits of Aβ (Rohn et al. 2008). Nevertheless, neuronal-directed Bcl-2 overexpression in mice that overexpress amino-terminally truncated prion peptide reduced caspase-3 activation, glial activation, and neuronal cell death in cerebellum by improving locomotor deficits and mice life expectancy (Nicolas et al. 2007). In our study, the ACEA treatment increases levels of the anti-apoptotic Bcl-2 protein, which was an important link between the modulation of Akt and ERK activity, resulting in neuroprotection action against STZ insult. Notably, our data did not show Bcl-2 protein expression difference between control and STZ groups. It is interesting to comment, however, that our previous study showed that expression levels of Bcl-2 were decreased at 1 day and 5 days after STZ icv. Furthermore, we also showed that STZ modified the balance between Bax and Bcl-2 and its effector caspase 3, favoring the apoptosis process 5 days after STZ icv (Crunfli et al. 2018). Accordingly, it was already shown that icv injection of STZ is capable of inducing caspase-3 activation, elevating Bax levels and decreasing Bcl-2 protein expression (Song et al. 2014). The protein activation time of each signaling pathway could be different. Therefore, this could explain why STZ did not change Bcl-2 levels after 7 days of icv STZ.
The cannabinoid agonists CB1 have become potentially attractive neuroprotective agents in neurodegenerative diseases. The cannabinoid compound ACEA is the most potent and highly CB1-selective receptor agonist (Ki = 1.4 nM; Hillard et al. 1999). Its protective effects have been described against the cytotoxic effect of Aβ42 oligomers on cortical neurons in culture (Aso et al. 2012) and of LPS and tunicamycin in Neuro-2a (Vrechi et al. 2018) and against 6-OHDA damage in dopaminergic neurons (Batinga et al. 2016). The ACEA neuroprotective mechanism involves the modulation of GSK3-β (Aso et al. 2012); endoplasmic reticulum stress signaling (Elf2-α/CHOP/caspase-12) and survival pathway p44/42 MAPK, ERK1/2 (Vrechi et al. 2018); reactive oxygen species (ROS) reduction, production, and inhibition of caspase 3 protein (Batinga et al. 2016).
The present in vitro model was established to determine the potential role of ACEA against neurotoxicity damage of STZ in neuroblastoma cells. ACEA treatment conferred neuroprotection against the cytotoxic effect of STZ to neuroblastoma cells through cellular viability regulation and NO release. We also treated the cells with a selective CB1 antagonist receptor (AM251) to determine receptor-specific responses. AM251 blockaded the ACEA neuroprotective action on cellular viability, indicating the CB1 receptor participation on this mechanism. Endogenous and exogenous cannabinoids have shown to possess neuroprotective effects in different studies by binding to CB1 receptors (Esposito et al. 2006; Benito et al. 2007; Aso et al. 2012). Moreover, in addition to direct neuroprotective effect on neurons, cannabinoids have also been demonstrated to induce anti-inflammatory actions that may involve NO signaling. Importantly, growing evidence suggests that the cannabinoid system could act as a neuroprotective agent, in part, by its ability to regulate the production and release of nitric oxide, which functions as a signaling intermediate (Lipina and Hundal 2017).
However, cannabinoid compounds are able to regulate the formation and release of NO and derived ROS, in an either positive or negative depending on cell type and stimulus, by acting on distinct molecular targets (Lipina and Hundal 2017). We have observed that ACEA treatment normalized the NO release produced by STZ. On the other hand, treatment with ACEA alone increased the NO levels. Several studies suggest that cannabinoid system stimulation may act to increase NO levels under different conditions and in specific cell types. For example, stimulation of CB1 receptor by the cannabinoid agonists CP 55,940 and WIN 55,212-2 elevated NO production via nNOS in N18TG2 neuroblastoma cells (Carney et al. 2009). Further studies demonstrated that CB1 selective activation downregulated iNOS protein expression and NO release from cultured C6 rat glioma cells challenged with both LPS and HIV-1 Tat protein (Esposito et al. 2002). Therefore, in this study, the modulation of ERK activity and the upregulated Bcl-2 by ACEA could be coupled with NO release regulation and consequently the neuroprotective action against STZ neurotoxicity.
In conclusion, there is a cannabinoid system participation in cellular survival, involving the CB1 receptor, which occurs by positive regulation of the anti-apoptotic proteins, and modulation of NO release by STZ, proposing the participation of this system in neurodegenerative processes. Our data suggest that CB1 receptor is an interesting therapeutic target for the treatment of neurodegenerative diseases and reinforce the hypothesis that targeting the cannabinoid system could offer approach for the development of novel therapeutic strategies against AD.
References
Abood ME, Rizvi G, Sallapudi N, McAllister SD (2001) Activation of the CB1 cannabinoid receptor protects cultured mouse spinal neurons against excitotoxicity. Neurosci Lett 309:197–201. https://doi.org/10.1016/S0304-3940(01)02065-1
Agrawal R, Tyagi E, Shukla R, Nath C (2011) Insulin receptor signaling in rat hippocampus: a study in STZ (ICV) induced memory deficit model. Eur Neuropsychopharmacol 21:261–273. https://doi.org/10.1016/j.euroneuro.2010.11.009
Ahmad F, Nidadavolu P, Durgadoss L, Ravindranath V (2014) Critical cysteines in Akt1 regulate its activity and proteasomal degradation: implications for neurodegenerative diseases. Free Radic Biol Med 74:118–128. https://doi.org/10.1016/j.freeradbiomed.2014.06.004
Aso E, Ferrer I (2014) Cannabinoids for treatment of Alzheimer’s disease: moving toward the clinic. Front Pharmacol 5:1–11. https://doi.org/10.3389/fphar.2014.00037
Aso E, Palomer E, Juvés S, Maldonado R, Muñoz FJ, Ferrer I (2012) CB1 agonist ACEA protects neurons and reduces the cognitive impairment of AβPP/PS1 mice. J Alzheimers Dis 30:439–459. https://doi.org/10.3233/JAD-2012-111862
Barzegar S, Komaki A, Shahidi S, Sarihi A, Mirazi N, Salehi I (2015) Effects of cannabinoid and glutamate receptor antagonists and their interactions on learning and memory in male rats. Pharmacol Biochem Behav 131:87–90. https://doi.org/10.1016/j.pbb.2015.02.005
Batinga H, Zúñiga-Hertz JP, Torrão AS (2016) Cannabinoid receptor ligands prevent dopaminergic neurons death induced by neurotoxic, inflammatory and oxidative stimuli in vitro. J Biomed Sci 5:1–16
Bedse G, Di Domenico F, Serviddio G, Cassano T (2015) Aberrant insulin signaling in Alzheimer’s disease: current knowledge. Front Neurosci 9:1–13. https://doi.org/10.3389/fnins.2015.00204
Benito C, Núñez E, Pazos MR, Tolón RM, Romero J (2007) The endocannabinoid system and Alzheimer’s disease. Mol Neurobiol 36:75–81. https://doi.org/10.1007/s12035-007-8006-8
Blázquez C, Chiarlone A, Bellocchio L, Resel E, Pruunsild P, García-Rincón D, Sendtner M, Timmusk T, Lutz B, Galve-Roperh I, Guzmán M (2015) The CB1 cannabinoid receptor signals striatal neuroprotection via a PI3K/Akt/mTORC1/BDNF pathway. Cell Death Differ 22:1618–1629. https://doi.org/10.1038/cdd.2015.11
Café-Mendes CC, Ferro ES, Torrão AS, Crunfli F, Rioli V, Schmitt A, Falkai P, Britto LR, Turck CW, Martins-de-Souza D (2017) Peptidomic analysis of the anterior temporal lobe and corpus callosum from schizophrenia patients. J Proteome 151:97–105. https://doi.org/10.1016/j.jprot.2016.05.025
Caffarel MM, Andradas C, Pérez-Gómez E, Guzmán M, Sánchez C (2012) Cannabinoids: a new hope for breast cancer therapy? Cancer Treat Rev 38:911–918. https://doi.org/10.1016/j.ctrv.2012.06.005
Calderón FH, Bonnefont A, Muñoz FJ, Fernández V, Videla LA, Inestrosa NC (1999) PC12 and neuro 2ª cells have diferente susceptibilities to acetylcholinesterase-amyloid complexes, amyloid 25-35 fragment, glutamate, and hydrogen peroxide. J Neurosci Res 56:620–631. https://doi.org/10.1002/(SICI)1097-4547(19990615)56:6<620::AID-JNR8>3.0.CO;2-F
Caltana LR, Heimrich B, Brusco A (2015) Further evidence for the neuroplastic role of cannabinoids: a study in organotypic hippocampal slice cultures. J Mol Neurosci 56:773–781. https://doi.org/10.1007/s12031-015-0499-4
Carney ST, Lloyd ML, MacKinnon SE, Newton DC, Jones JD, Howlett AC, Norford DC (2009) Cannabinoid regulation of nitric oxide synthase i (nNOS) in neuronal cells. J NeuroImmune Pharmacol 4:338–349. https://doi.org/10.1007/s11481-009-9153-7
Coffer PJ, Geijsen N, M’rabet L, Schweizer RC, Maikoe T, Raaijmakers JAM, Lammers JWJ, Koenderman L (1998) Comparison of the roles of mitogen-activated protein kinase kinase and phosphatidylinositol 3-kinase signal transduction in neutrophil effector function. Biochem J 329(Pt1):121–130
Colucci-D’Amato L, Perrone-Capano C, Di Porzio U (2003) Chronic activation of ERK and neurodegenerative diseases. BioEssays 25:1085–1095. https://doi.org/10.1002/bies.10355
Crunfli F, Mazucanti CH, De Moraes RCM, Costa AP, Rodrigues AC, Scavone C, Torrão AS (2018) NO-dependent Akt inactivation by S-nitrosylation as a possible mechanism of STZ-induced neuronal insulin resistance. J Alzheimers Dis 65:1427–1443. https://doi.org/10.3233/JAD-180284
De Felice F, Ferreira S (2002) β-Amyloid production, aggergation, and clearance as targets for therapy in Alzheimer’s disease. Cell Mol Neurobiol 22:545–563
De Felice FG, Lourenco MV, Ferreira ST (2014) How does brain insulin resistance develop in Alzheimer’s disease? Alzheimers Dement 10:S26–S32. https://doi.org/10.1016/j.jalz.2013.12.004
De La Monte SM, Tong M (2014) Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochem Pharmacol 88:548–559. https://doi.org/10.1016/j.bcp.2013.12.012
De la Monte SM, Wands JR (2005) Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer’s disease. J Alzheimers Dis 7:45–61. https://doi.org/10.3233/JAD-2005-7106
Derkinderen P, Valjent E, Toutant M, Corvol JC, Enslen H, Ledent C, Trzaskos J, Caboche J, Girault JA (2003) Regulation of extracellular signal-regulated kinase by cannabinoids in hippocampus. J Neurosci 23:2371–2382 23/6/2371 [pii]
Di Marzo V, Verrijken A, Hakkarainen A, Petrosino S, Mertens I, Lundbom N, Piscitelli F, Westerbacka J, Soro-Paavonen A, Matias I, Van Gaal L, Taskinen MR (2009) Role of insulin as a negative regulator of plasma endocannabinoid levels in obese and nonobese subjects. Eur J Endocrinol 161:715–722. https://doi.org/10.1530/EJE-09-0643
Di Marzo V, Stella N, Zimmer A (2015) Endocannabinoid signalling and the deteriorating brain. Nat Rev Neurosci 16(1):30–42. https://doi.org/10.1038/nrn3876
Downward J (1999) How BAD phosphorylation is good for survival. Nat Cell Biol 1:E33–E35. https://doi.org/10.1038/10026
El-Mir MY, Detaille D, R-Villanueva G, Esteban MD, Guigas B, Attia S, Fontaine E, Almeida A, Leverve X (2008) Neuroprotective role of antidiabetic drug metformin against apoptotic cell death in primary cortical neurons. J Mol Neurosci 34:77–87. https://doi.org/10.1007/s12031-007-9002-1
Ellert-Miklaszewska A, Kaminska B, Konarska L (2005) Cannabinoids down-regulate PI3K/Akt and Erk signalling pathways and activate proapoptotic function of Bad protein. Cell Signal 17:25–37. https://doi.org/10.1016/j.cellsig.2004.05.011
Esposito G, De Filippis D, Steardo L, Scuderi C, Savani C, Cuomoa V, Iuvone T (2006) CB1 receptor selective activation inhibits β-amyloid-induced iNOS protein expression in C6 cells and subsequently blunts tau protein hyperphosphorylation in co-cultured neurons. Neurosci Lett 404:342–346. https://doi.org/10.1016/j.neulet.2006.06.012
Esposito G, Ligresti A, Izzo AA, Bisogno T, Ruvo M, Di Rosa M, Di Marzo V, Iuvone T (2002) The endocannabinoid system protects rat glioma cells against HIV-1 tat protein-induced cytotoxicity: mechanism and regulation. J Biol Chem 277:50348–50354. https://doi.org/10.1074/jbc.M207170200
Fernández-López D, Martínez-Orgado J, Nuñez E, Romero J, Lorenzo P, Moro MA, Lizasoain I (2006) Characterization of the neuroprotective effect of the cannabinoid agonist WIN-55212 in an in vitro model of hypoxic-ischemic brain damage in newborn rats. Pediatr Res 60:169–173. https://doi.org/10.1203/01.pdr.0000228839.00122.6c
Fernández-Ruiz J, Pazos MR, García-Arencibia M, Sagredo O, Ramos JA (2008) Role of CB2 receptors in neuroprotective effects of cannabinoids. Mol Cell Endocrinol 286:S91–S96. https://doi.org/10.1016/j.mce.2008.01.001
Ferreiro E, Eufrásio A, Pereira C, Oliveira CR, Rego AC (2007) Bcl-2 overexpression protects against amyloid-beta and prion toxicity in GT1-7 neural cells. J Alzheimers Dis 12:223–228. https://doi.org/10.3233/JAD-2007-12303
Ferrer I, Blanco R, Carmona M, Puig B (2001) Phosphorylated mitogen-activated protein kinase (MAPK/ERK-P), protein kinase of 38kDa (p38-P), stress-activated protein kinase (SAPK/JNK-P), and calcium/calmodulin-dependent kinase II (CaM kinase II) are differentially expressed in tau deposits in neurons. J Neural Transm 108:1397–1415. https://doi.org/10.1007/s007020100016
Frölich L, Blum-Degen D, Bernstein HG, Engelsberger S, Humrich J, Laufer S, Muschner D, Thalheimer A, Türk A, Hoyer S, Zöchling R, Boissl KW, Jellinger K, Riederer P (1998) Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J Neural Transm 105:423–438. https://doi.org/10.1007/s007020050068
Gilbert GL, Kim HJ, Waataja JJ, Thayer S (2007) Δ9-Tetrahydrocannabinol protects hippocampal neurons from excitotoxicity. Brain Res 1128:61–69. https://doi.org/10.1016/j.brainres.2006.03.011
Goodman J, Packard MG (2015) The influence of cannabinoids on learning and memory processes of the dorsal striatum. Neurobiol Learn Mem 125:1–14. https://doi.org/10.1016/j.nlm.2015.06.008
Granado-Serrano AB, Martín MA, Bravo L, Goya L, Ramos S (2006) Quercetin induces apoptosis via caspase activation, regulation of Bcl-2, and inhibition of PI-3-kinase/Akt and ERK pathways in a human hepatoma cell line (HepG2). J Nutr 136:2715–2721
Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR (1982) Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem 126:131–138. https://doi.org/10.1016/0003-2697(82)90118-X
Gupta A, Bisht B, Dey CS (2011) Peripheral insulin-sensitizer drug metformin ameliorates neuronal insulin resistance and Alzheimer’s-like changes. Neuropharmacology 60:910–920. https://doi.org/10.1016/j.neuropharm.2011.01.033
Harvey BS, Ohlsson KS, Mååg JLV, Musgrave IF, Smid SD (2012) Contrasting protective effects of cannabinoids against oxidative stress and amyloid-β evoked neurotoxicity in vitro. Neurotoxicology 33:138–146. https://doi.org/10.1016/j.neuro.2011.12.015
Hashimotodani Y, Ohno-Shosaku T, Kano M (2007) Endocannabinoids and synaptic function in the CNS. Neuroscientist 13:127–137. https://doi.org/10.1177/1073858406296716
Henneberg N, Hoyer S (1995) Desensitization of the neuronal insulin receptor: a new approach in the etiopathogenesis of late-onset sporadic dementia of the Alzheimer type (SDAT)? Arch Gerontol Geriatr 21:63–74. https://doi.org/10.1016/0167-4943(95)00646-3
Hillard CJ, Manna S, Greenberg MJ, DiCamelli R, Ross RA, Stevenson LA, Murphy V, Pertwee RG, Campbell WB (1999) Synthesis and characterization of potent and selective agonists of the neuronal cannabinoid receptor (CB1). J Pharmacol Exp Ther 289:1427–1433
Hooper C, Killick R, Lovestone S (2008) The GSK3 hypothesis of Alzheimer’s disease. J Neurochem 104:1433–1439. https://doi.org/10.1111/j.1471-4159.2007.05194.x
Hoyer S (2002) The brain insulin signal transduction system and sporadic (type II) Alzheimer disease: an update. J Neural Transm 109:341–360. https://doi.org/10.1007/s007020200028
Hoyer S, Muller D, Plaschke K (1994) Desensitization of brain insulin receptor. Effect on glucose/energy and related metabolism. J Neural Transm Suppl 44:259–268
Ishiguro K, Shiratsuchi A, Sato S, Omori A, Arioka M, Kobayashi S, Uchida T, Imahori K (1993) Glycogen synthase kinase 3 beta is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett 325:167–172
Jordan JD, He JC, Eungdamrong NJ, Gomes I, Ali W, Nguyen T, Bivona TG, Philips MR, Devi LA, Iyengar R (2005) Cannabinoid receptor-induced neurite outgrowth is mediated by Rap1 activation through Gαo/i-triggered proteasomal degradation of Rap1GAPII. J Biol Chem 280:11413–11421. https://doi.org/10.1074/jbc.M411521200
Kamat PK, Kalani A, Rai S, Tota SK, Kumar A, Ahmad AS (2016) Streptozotocin intracerebroventricular-induced neurotoxicity and brain insulin resistance: a therapeutic intervention for treatment of sporadic Alzheimer’s disease (sAD)-like pathology. Mol Neurobiol 53:4548–4562. https://doi.org/10.1007/s12035-015-9384-y
Kang J, Lemaire H-G, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Müller-Hill B (1987) The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 325:733–736. https://doi.org/10.1038/325733a0
Kar S, Slowikowski SPM, Westaway D, Mount HTJ (2004) Interactions between beta-amyloid and central cholinergic neurons: implications for Alzheimer’s disease. J Psychiatry Neurosci 29:427–441
Karanian DA (2005) Dual modulation of endocannabinoid transport and fatty acid amide hydrolase protects against excitotoxicity. J Neurosci 25:7813–7820. https://doi.org/10.1523/JNEUROSCI.2347-05.2005
Kokona D, Thermos K (2015) Synthetic and endogenous cannabinoids protect retinal neurons from AMPA excitotoxicity in vivo, via activation of CB1 receptors: involvement of PI3K/Akt and MEK/ERK signaling pathways. Exp Eye Res 136:45–58. https://doi.org/10.1016/j.exer.2015.05.007
Kosik K, Joachim C, Selkoe D (1987) Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Alzheimer Dis Assoc Disord 1:203. https://doi.org/10.1097/00002093-198701030-00022
Laezza C, Malfitano AM, Proto MC, Esposito I, Gazzerro P, Formisano P, Pisanti S, Santoro A, Caruso MG, Bifulco M (2010) Inhibition of 3-hydroxy-3-methylglutarylcoenzyme a reductase activity and of Ras farnesylation mediate antitumor effects of anandamide in human breast cancer cells. Endocr Relat Cancer 17:495–503. https://doi.org/10.1677/ERC-10-0009
Lannert H, Hoyer S (1998) Intracerebroventricular administration of streptozotocin causes long- term diminutions in learning and memory abilities and in cerebral energy metabolism in adult rats. Behav Neurosci 112:1199–1208. https://doi.org/10.1037//0735-7044.112.5.1199
Lester-Coll N, Rivera EJ, Soscia SJ, Doiron K, Wands JR, De la Monte SM (2006) Intracerebral streptozotocin model of type 3 diabetes: relevance to sporadic Alzheimer’s disease. J Alzheimers Dis 9:13–33. https://doi.org/10.3233/JAD-2006-9102
Lipina C, Hundal HS (2017) The endocannabinoid system: “NO” longer anonymous in the control of nitrergic signalling? J Mol Cell Biol 9:91–103. https://doi.org/10.1093/jmcb/mjx008
Marsicano G, Moosmann B, Hermann H, Lutz B, Behl C (2002) Neuroprotective properties of cannabinoids against oxidative stress: role of the cannabinoid receptor CB1. J Neurochem 80:448–456. https://doi.org/10.1046/j.0022-3042.2001.00716.x
Martín-Moreno AM, Brera B, Spuch C, Carro E, García-García L, Delgado M, Pozo MA, Innamorato NG, Cuadrado A, de Ceballos ML (2012) Prolonged oral cannabinoid administration prevents neuroinflammation, lowers β-amyloid levels and improves cognitive performance in Tg APP 2576 mice. J Neuroinflammation 9:8. https://doi.org/10.1186/1742-2094-9-8
Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI (1990) Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346:561–564. https://doi.org/10.1038/346561a0
Milton NGN (2002) Anandamide and noladin ether prevent neurotoxicity of the human amyloid-β peptide. Neurosci Lett 332:127–130. https://doi.org/10.1016/S0304-3940(02)00936-9
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63. https://doi.org/10.1016/0022-1759(83)90303-4
Mumby DG (2002) Hippocampal damage and exploratory preferences in rats: memory for objects, places, and contexts. Learn Mem 9:49–57. https://doi.org/10.1101/lm.41302
Munro S, Thomas KL, Abu-Shaar M (1993) Molecular characterization of a peripheral receptor for cannabinoids. Nature 365:61–65. https://doi.org/10.1038/365061a0
Nicolas O, Gavin R, Braun N, Ureña JM, Fontana X, Soriano E, Aguzzi A, del Río JA (2007) Bcl-2 overexpression delays caspase-3 activation and rescues cerebellar degeneration in prion-deficient mice that overexpress amino-terminally truncated prion. FASEB J 21:3107–3117. https://doi.org/10.1096/fj.06-7827com
Nitsch R, Hoyer S (1991) Local action of the diabetogenic drug, streptozotocin, on glucose and energy metabolism in rat brain cortex. Neurosci Lett 128:199–202. https://doi.org/10.1016/0304-3940(91)90260-Z
Numajiri N, Takasawa K, Nishiya T, Tanaka H, Ohno K, Hayakawa W, Asada M, Matsuda H, Azumi K, Kamata H, Nakamura T, Hara H, Minami M, Lipton SA, Uehara T (2011) On-off system for PI3-kinase-Akt signaling through S-nitrosylation of phosphatase with sequence homology to tensin (PTEN). Proc Natl Acad Sci U S A 108:10349–10354. https://doi.org/10.1073/pnas.1103503108
Osmanovic Barilar J, Knezovic A, Grünblatt E, Riederer P, Salkovic-Petrisic M (2015) Nine-month follow-up of the insulin receptor signalling cascade in the brain of streptozotocin rat model of sporadic Alzheimer’s disease. J Neural Transm 122:565–576. https://doi.org/10.1007/s00702-014-1323-y
Paradis E, Koutroumanis M, Goodyer C (1996) Amyloid Beta peptide of Alzheimer ’ s disease downregulates Bcl-2 and upregulates Bax expression in human neurons. J Neurosci 16:7533–7539
Paxinos G, Watson C (2007) The Rat Brain in Stereotaxic Coordinates. 6th Edition, Academic Press, San Diego
Pazos MR, Núñez E, Benito C, Tolón RM, Romero J (2004) Role of the endocannabinoid system in Alzheimer’s disease: new perspectives. Life Sci 75:1907–1915. https://doi.org/10.1016/j.lfs.2004.03.026
Phiel CJ, Wilson CA, Lee VM, Klein PS (2003) GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 423:435–439. https://doi.org/10.1038/nature01640
Pilchova I, Klacanova K, Chomova M, Tatarkova Z, Dobrota D, Racay P (2014) Possible contribution of proteins of Bcl-2 family in neuronal death following transient global brain ischemia. Cell Mol Neurobiol 35:23–31. https://doi.org/10.1007/s10571-014-0104-3
Puighermanal E, Busquets-Garcia A, Maldonado R, Ozaita A (2012) Cellular and intracellular mechanisms involved in the cognitive impairment of cannabinoids. Philos Trans R Soc B Biol Sci 367:3254–3263. https://doi.org/10.1098/rstb.2011.0384
Puighermanal E, Marsicano G, Busquets-Garcia A, Lutz B, Maldonado R, Ozaita A (2009) Cannabinoid modulation of hippocampal long-term memory is mediated by mTOR signaling. Nat Neurosci 12:1152–1158. https://doi.org/10.1038/nn.2369
Rai S, Kamat PK, Nath C, Shukla R (2014) Glial activation and post-synaptic neurotoxicity: the key events in Streptozotocin (ICV) induced memory impairment in rats. Pharmacol Biochem Behav 117:104–117. https://doi.org/10.1016/j.pbb.2013.11.035
Ramalingam M, Kim S (2014) Insulin involved Akt / ERK and Bcl-2 / Bax pathways against oxidative damages in C6 glial cells. 9893:1–7. https://doi.org/10.3109/10799893.2014.970276
Ramírez BG, Blázquez C, Gómez del Pulgar T, Guzmán M, de Ceballos ML (2005) Prevention of Alzheimer’s disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J Neurosci 25:1904–1913. https://doi.org/10.1523/JNEUROSCI.4540-04.2005
Rohn TT, Vyas V, Hernandez-Estrada T, Nichol KE, Christie LA, Head E (2008) Lack of pathology in a triple transgenic mouse model of Alzheimer’s disease after overexpression of the anti-apoptotic protein Bcl-2. J Neurosci 28:3051–3059. https://doi.org/10.1523/JNEUROSCI.5620-07.2008
Ruiz-Valdepeñas L, Benito C, Tolón RM, Martínez Orgado JA, Romero J (2010) The endocannabinoid system and amyloid-related diseases. Exp Neurol 224:66–73. https://doi.org/10.1016/j.expneurol.2010.03.024
Rutkowska M, Gliniak H (2009) The influence of ACEA: a selective cannabinoid CB1 receptor agonist on whole blood and platelet-poor plasma serotonin concentrations. Pharmazie 64:598–601
Ryu BR, Ko HW, Jou I, Noh JS, Gwag BJ (1999) Phosphatidylinositol 3-kinase-mediated regulation of neuronal apoptosis and necrosis by insulin and IGF-I. J Neurobiol 39:536–546. https://doi.org/10.1002/(SICI)1097-4695(19990615)39:4<536::AID-NEU7>3.0.CO;2-J
Salkovic-Petrisic M, Hoyer S (2007) Central insulin resistance as a trigger for sporadic Alzheimer-like pathology: an experimental approach. J Neural Transm Suppl:217–233. https://doi.org/10.1007/978-3-211-73574-9-28
Salkovic-Petrisic M, Knezovic A, Hoyer S, Riederer P (2013) What have we learned from the streptozotocin-induced animal model of sporadic Alzheimer’s disease, about the therapeutic strategies in Alzheimer’s research. J Neural Transm 120:233–252. https://doi.org/10.1007/s00702-012-0877-9
Salkovic-Petrisic M, Osmanovic J, Grünblatt E, Riederer P, Hoyer S (2009) Modeling sporadic Alzheimer’s disease: the insulin resistant brain state generates multiple long-term morphobiological abnormalities including hyperphosphorylated tau protein and amyloid-β. J Alzheimers Dis 18:729–750. https://doi.org/10.3233/JAD-2009-1184
Salkovic-Petrisic M, Tribl F, Schmidt M, Hoyer S, Riederer P (2006) Alzheimer-like changes in protein kinase B and glycogen synthase kinase-3 in rat frontal cortex and hippocampus after damage to the insulin signalling pathway. J Neurochem 96:1005–1015. https://doi.org/10.1111/j.1471-4159.2005.03637.x
Sánchez AJ, García-Merino A (2012) Neuroprotective agents: cannabinoids. Clin Immunol 142:57–67. https://doi.org/10.1016/j.clim.2011.02.010
Santos TO, Mazucanti CHY, Xavier GF, Torrão AS (2012) Early and late neurodegeneration and memory disruption after intracerebroventricular streptozotocin. Physiol Behav 107:401–413. https://doi.org/10.1016/j.physbeh.2012.06.019
Sarker KP, Maruyama I (2003) Anandamide induces cell death independently of cannabinoid receptors or vanilloid receptor 1: possible involvement of lipid rafts. Cell Mol Life Sci 60:1200–1208. https://doi.org/10.1007/s00018-003-3055-2
Shen J, Wu Y, Xu JY, Zhang J, Sinclair SH, Yanoff M, Xu G, Li W, Xu GT (2010) ERK- and Akt-dependent neuroprotection by erythropoietin (EPO) against glyoxal-AGEs via modulation of Bcl-xL, Bax, and BAD. Investig Ophthalmol Vis Sci 51:35–46. https://doi.org/10.1167/iovs.09-3544
Shoham S, Bejar C, Kovalev E, Schorer-Apelbaum D, Weinstock M (2007) Ladostigil prevents gliosis, oxidative-nitrative stress and memory deficits induced by intracerebroventricular injection of streptozotocin in rats. Neuropharmacology 52:836–843. https://doi.org/10.1016/j.neuropharm.2006.10.005
Song J, Hur BE, Bokara KK, Yang W, Cho HJ, Park KA, Lee WT, Lee KM, Lee JE (2014) Agmatine improves cognitive dysfunction and prevents cell death in a streptozotocin-induced Alzheimer rat model. Yonsei Med J 55:689–699. https://doi.org/10.3349/ymj.2014.55.3.689
Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, De la Monte SM (2005) Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease-is this type 3 diabetes? J Alzheimers Dis 7:63–80
Stygelbout V, Leroy K, Pouillon V, Ando K, D'Amico E, Jia Y, Luo HR, Duyckaerts C, Erneux C, Schurmans S, Brion JP (2014) Inositol trisphosphate 3-kinase B is increased in human Alzheimer brain and exacerbates mouse Alzheimer pathology. Brain 137:537–552. https://doi.org/10.1093/brain/awt344
Subramaniam S, Zirrgiebel U, Von Bohlen Und Halbach O, Strelau J, Laliberté C, Kaplan DR, Unsicker K (2004) ERK activation promotes neuronal degeneration predominantly through plasma membrane damage and independently of caspase-3. J Cell Biol 165:357–369. https://doi.org/10.1083/jcb.200403028
Terranova JP, Storme JJ, Lafon N, Péŕio A, Rinaldi-Carmona M, Le Fur G, Soubrié P (1996) Improvement of memory in rodents by the selective CB1 cannabinoid receptor antagonist R141716. Psychopharmacology 126(2):165–172
Tsvetanova E, Kessiova M, Alexandrova A, Petrov L, Kirkova M, Todorov S (2006) In vivo effects of CB1 receptor ligands on lipid peroxidation and antioxidant defense systems in the rat brain of healthy and ethanol-treated rats. Pharmacol Rep 58:876–883
Van Der Stelt M, Mazzola C, Esposito G, Matias I, Petrosino S, De Filippis D, Micale V, Steardo L, Drago F, Iuvone T, Di Marzo V (2006) Endocannabinoids and β-amyloid-induced neurotoxicity in vivo: effect of pharmacological elevation of endocannabinoid levels. Cell Mol Life Sci 63:1410–1424. https://doi.org/10.1007/s00018-006-6037-3
Vrechi TA, Crunfli F, Costa AP, Torrão AS (2018) Cannabinoid receptor type 1 agonist ACEA protects neurons from death and attenuates endoplasmic reticulum stress-related apoptotic pathway signaling. Neurotox Res 33. https://doi.org/10.1007/s12640-017-9839-1
Wei W, Wang X, Kusiak JW (2002) Signaling events in amyloid β-peptide-induced neuronal death and insulin-like growth factor I protection. J Biol Chem 277:17649–17656. https://doi.org/10.1074/jbc.M111704200
Weksler ME, Gouras G, Relkin NR, Szabo P (2005) The immune system, amyloid-beta peptide, and Alzheimer’s disease. Immunol Rev 205:244–256. https://doi.org/10.1111/j.0105-2896.2005.00264.x
Wilson RI, Nicoll RA (2002) Endocannabinoid signaling in the brain. Science 296:678–682. https://doi.org/10.1126/science.1063545
Winter C, Weiss C, Martin-Villalba A, Zimmermann M, Schenkel J (2002) JunB and Bcl-2 overexpression results in protection against cell death of nigral neurons following axotomy. Brain Res Mol Brain Res 104:194–202. https://doi.org/10.1016/S0169-328X(02)00378-9
Winters BD, Forwood SE, Cowell RA, Saksida LM, Bussey TJ (2004) Double dissociation between the effects of peri-postrhinal cortex and hippocampal lesions on tests of object recognition and spatial memory: heterogeneity of function within the temporal lobe. J Neurosci 24:5901–5908. https://doi.org/10.1523/JNEUROSCI.1346-04.2004
Yu HY, Kim SO, Jin CY, Kim GY, Kim WJ, Yoo YH, Choi YH (2014) β-Lapachone-induced apoptosis of human gastric carcinoma AGS cells is caspase-dependent and regulated by the PI3K/Akt pathway. Biomol Ther 22:184–192. https://doi.org/10.4062/biomolther.2014.026
Zhu X, Raina AK, Lee H-G, Chao M, Nunomura A, Tabaton M, Petersen RB, Perry G, Smith MA (2003) Oxidative stress and neuronal adaptation in Alzheimer disease: the role of SAPK pathways. Antioxid Redox Signal 5:571–576. https://doi.org/10.1089/152308603770310220
Zou S, Kumar U (2018) Cannabinoid receptors and the endocannabinoid system: signaling and function in the central nervous system. Int J Mol Sci 19. https://doi.org/10.3390/ijms19030833
Acknowledgments
Authors wish to thank the professor Dr. Julián Romero (Universidad Francisco de Vitoria, Madrid, Spain) for critical comments and his technical support during the process. We also thank to Dr. Maria Assunción de la Barreda Manso for her opinion and critical comments.
Financial Support
This work was supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo-FAPESP (grant number 2014/06372-0). T.A.V. (1279985) and F.C. (1233360) are recipients of Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) fellowships.
Author information
Authors and Affiliations
Contributions
F.C. designed the experiments, collected, and analyzed the data and wrote the paper; T.A.V. and A.P.C. collected, analyzed, and discussed the data; A.S.T. designed the experiments, discussed the data, wrote, edited, and commented on the manuscript. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
The experimental protocol was evaluated and approved by the “Ethics Committee for Animal use” of the Institute of Biomedical Sciences, University of São Paulo (Protocol no. 33/55/02) following the Brazilian Federal Law (no. 11794; 10/08/2008). All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Crunfli, F., Vrechi, T.A., Costa, A.P. et al. Cannabinoid Receptor Type 1 Agonist ACEA Improves Cognitive Deficit on STZ-Induced Neurotoxicity Through Apoptosis Pathway and NO Modulation. Neurotox Res 35, 516–529 (2019). https://doi.org/10.1007/s12640-018-9991-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-018-9991-2