
Abstract
Mature double-stranded DNA bacteriophages have capsids with symmetrical shells that typically resist disruption, as they must to survive in the wild. However, flexibility and associated dynamism assist function. We describe biochemistry-oriented procedures used to find previously obscure flexibility for capsids of the related phages, T3 and T7. The primary procedures are hydration-based buoyant density ultracentrifugation and purified particle-based cryo-electron microscopy (cryo-EM). We review the buoyant density centrifugation in detail. The mature, stable T3/T7 capsid is a shell flexibility-derived conversion product of an initially assembled procapsid (capsid I). During DNA packaging, capsid I expands and loses a scaffolding protein to form capsid II. The following are observations made with capsid II. (1) The in vivo DNA packaging of wild type T3 generates capsid II that has a slight (1.4%), cryo-EM-detected hyper-expansion relative to the mature phage capsid. (2) DNA packaging in some altered conditions generates more extensive hyper-expansion of capsid II, initially detected by hydration-based preparative buoyant density centrifugation in Nycodenz density gradients. (3) Capsid contraction sometimes occurs, e.g., during quantized leakage of DNA from mature T3 capsids without a tail.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Bacteriophages (abbreviated, phages) are viruses that infect bacteria. Phages are used to investigate basic aspects of all biology. Double-stranded DNA phages, such as T3 and T7, consist of a protein capsid and a genome of double-stranded DNA. The genome is packaged in an internal cavity of the capsid (Harvey et al. 2009; Black and Thomas 2012; Serwer and Jiang 2012; Fokine and Rossmann 2014; Veesler and Johnson 2012).
Capsids must be stable in natural environments, which include gastrointestinal tracts in the case of coliphages, i.e., phages that infect Escherichia coli. For example, the related (Studier 1979) coliphages, T3 and T7, are not inactivated by the proteases, trypsin and subtilisin, although their outer shells are made of protein (Serwer et al. 1982). Here, we will focus on these two phages (Fig. 1d). To discuss a specific protein, the protein will be identified by “gp” (gene product), followed by the number of the encoding gene. Homologous T3 and T7 genes have been given the same gene numbers (Pajunen et al. 2002). The capsid of a mature phage consists of (1) an icosahedral (T = 7) shell of gp10, (2) a small, external tail, and (3) a co-axial internal stack of protein rings with an axial hole (gp8 connector and gp14/15/16 core stack; Fig. 1e; Guo et al. 2013; Serwer and Jiang 2012).

T3 and T7 phages and related particles. a Procapsid (capsid I). b Capsid II packaging a DNA molecule. c Nycodenz low-density (NLD) capsid II detached from the DNA molecule that it was packaging. d A mature capsid with a packaged genome and no tail (head). e The mature, complete phage
Phages T3 and T7 are chosen for analysis because of the relative simplicity of isolating intermediates in their assembly. This simplicity arises from the following. (1) The growth period is relatively short, 13–15 min at 37 °C, which is useful for experimentation with both wild type phages and especially mutants/conditions with prolonged growth periods. (2) Host DNA is degraded (Center et al. 1970; Sadowski and Kerr 1970) and host protein synthesis is stopped (Schweiger et al. 1972) during infection, which reduces host interference with detecting, isolating, and characterizing assembly intermediates (Serwer 1980). (3) The DNA genome is short enough so that viscosity of lysates is not problematic, even given that T3/T7 DNA packaging occurs with DNA genomes that are joined end-to-end to form a concatemer (Serwer and Watson 1981); concatemers are not further discussed here. These advantages do not co-exist with any other studied phage to our knowledge.
Two activities of phages T3 and T7 classify as the activities of bio-nanomachines. The first is the injection of DNA that initiates an infection. The second is the packaging of DNA that occurs during the assembly of an infective phage particle. Although phages provide relatively tractable model systems, the mechanisms of phage bio-nanomachine activities are not known. In determining these mechanisms, we adhere to the principle that one has to have a complete picture of the relevant biochemistry. We have been pursuing this biochemistry via the isolation of the relevant bio-nanomachines and related particles in intermediate states. With this strategy, isolation technology and the concepts derived from it are so tightly linked that we discuss them together here. We focus on gp10 shell-related details potentially useful to understanding both DNA injection and DNA packaging. We begin with a more detailed description of phage DNA injection and DNA packaging.
DNA injection
Infection of a host bacterial cell begins with DNA injection from a phage particle. After interaction with membrane proteins (González-García et al. 2015), the DNA molecule is injected through the outer membrane, murein layer, periplasmic space, and inner membrane of the Gram-negative host cell. To understand T3/T7 injection, attention has been focused on the gp14/15/16 core stack (Fig. 2a for T3/T7). The reasons are that (1) the T7 core stack proteins are seen by electron microscopy (EM) to externalize while forming a needle with a central channel (Serwer 1978; Serwer et al. 2008; Hu et al. 2013), as illustrated in Fig. 2b, and (2) the T7 core stack proteins subsequently become associated with host outer and inner membranes (for reviews, see Molineux 2001; Leptihn et al. 2016). Thus, the assumption is that T7 DNA is injected through the core stack-derived externalized needle, which we will call a DNA injection needle.

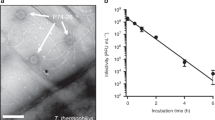
Phage T3 has the capacity to form a similar needle, as shown by the following. In the case of some T3 mutants, tails have a reduced frequency of assembly on capsids that had previously packaged a complete DNA genome (heads; Fig. 1d; Serwer et al. 2014a). During storage, T3 heads sometimes spontaneously extend a T7-like apparent DNA injection needle (Fig. 3). This needle is longer than the phage tail. A tail is seen on an occasional contaminating phage particle (arrow in Fig. 3b). Thus, we will assume here that the genomically homologous phages, T3 and T7, have homologous DNA injection mechanisms.

An apparent injection needle of phage T3. Phage T3 heads were obtained from lysates by a procedure that ends with buoyant density centrifugation in a cesium chloride density gradient and is described by Serwer et al. (2014a). The heads were dialyzed against 0.2 M NaCl, 0.01 M Tris-Cl, pH 7.4, 0.001 M MgCl2, and stored at 4 °C. We performed electron microscopy (EM) by negative staining with 1% sodium phosphotungstate, pH 7.6, as previously described (Serwer and Wright 2017). a–f Several different fields are shown, each with at least one particle that has an extension that appears to be a DNA injection needle
The assumption is almost always made that the gp10 shell of T3 and T7 is inert during DNA injection (inert shell model; for reviews see Molineux 2001; Leptihn et al. 2016). The likely reason for this assumption is environmental stability of the phage shell. In the Data and means for obtaining it section, we will further discuss DNA injection.
DNA packaging: changes of the shell
During in vivo production of mature, infective T3/T7 phage particles, progeny capsids are assembled in a state (procapsid state) that differs from the mature capsid state. Capsids in the procapsid state (capsid I; Fig. 1a) bind a DNA genome and package it (for reviews, see Fujisawa and Morita 1997; Serwer and Jiang 2012). This sequence of events has been found to be universal among double-stranded DNA phages (for reviews, see Harvey et al. 2009; Serwer and Jiang 2012; Veesler and Johnson 2012; Rixon and Schmid 2014; Rao and Feiss 2015). This sequence has also been found for some eukaryotic viruses, including herpesviruses (for reviews, see Veesler and Johnson 2012; Rixon and Schmid 2014). Early in packaging, the procapsid converts to a more mature capsid (capsid II for T3/T7) (Fig. 1b, c). In the case of T3/T7, the details of this conversion include (1) expansion observed by low-angle X-ray scattering (Stroud et al. 1981) and cryo-EM (Guo et al. 2014), (2) loss of a scaffolding protein, gp9 (Fig. 1), (3) change in the conformation of the gp10 major shell protein observed by cryo-EM (Guo et al. 2014), and (4) conversion from a round to angular outline observed by negative-staining EM (Stroud et al. 1981) and cryo-EM (Agirrezabala et al. 2005; Guo et al. 2014; illustrated in Fig. 1).
Intermediates in the gp10 shell dynamics of the T3/T7 capsid I-to-capsid II conversion are not, at this point, published. Some phage HK97 intermediates are accessible after procapsid assembly from recombinant proteins. Covalent inter-subunit cross-linking stabilizes them (Veesler et al. 2012). The cross-linking occurs spontaneously during an HK97 infection (Popa et al. 1991). In an ongoing study (P. Serwer and W. Jiang, unpublished), we have used a hybrid strategy (biochemical isolation/cryo-EM) to obtain and determine the structure of T3 capsids in multiple intermediate states (five major states were found) between the capsid I and capsid II states.
DNA packaging: additional shell dynamics
An inert shell model is also used, usually tacitly, for DNA packaging events that occur after the procapsid-to-mature capsid conversion. For example, nanometry of phage phi29 and lambda DNA packaging sometimes reveals a sudden decrease to near zero of the force opposing packaging (for a review, see Chemla and Smith 2012). The assumption is made that this decrease is the result of rupture of the capsid. The word “trepanation” is sometimes used to describe the presumed hole (Liu et al. 2014). A trepanation is a hole drilled in a skull, as done by Louis Pasteur to produce rabies vaccines (Smith 2012). A skull is a rigid object. Thus, the word “trepanation” implies capsid shell rigidity. However, one can also explain the force drop by assuming hyper-expansion of the capsid’s shell, i.e., shell flexibility.
Furthermore, the gp10 shell of T3 capsid II has recently been found to exhibit DNA packaging-associated flexibility beyond what is seen in the major wild type capsid I-to-capsid II transition. The flexibility is observed in capsids that are classified as capsid II, based on protein composition. The evidence begins with an unusually low capsid density during hydration-based buoyant density centrifugation in Nycodenz density gradients (Serwer and Wright 2016, 2017). Nycodenz is a tri-iodinated benzene ring-based compound developed for buoyant density centrifugation (Rickwood et al. 1982). The unusually low density is caused by unusually high hydration, caused, in turn, by impermeability of this capsid II to Nycodenz. Impermeability-derived hydration causes the density to progressively decrease as the gp10 shell radius increases (Serwer and Wright 2016). We will review details of this technique, given its development via the investigation of capsid shell flexibility.
Data and means for obtaining it
Spontaneous DNA leakage from phage T3
When T3 heads are stored, they undergo partial DNA leakage (illustrated in Fig. 4, left side; Serwer et al. 2014a) in quantized amounts. Analysis of leakage begins with preparative rate zonal centrifugation in a sucrose gradient. Sedimentation velocity decreases as leaked DNA length increases. Leakage quantization is detected via agarose gel electrophoresis of the un-leaked DNA in each fraction of the sucrose gradient. For this analysis, the leaked DNA end is identified by digestion of the un-leaked DNA with a one-site restriction endonuclease, Bgl II (digestion site 44% of the genome length from the left genomic end), before electrophoresis. One obtains the un-leaked DNA for this analysis by (1) DNase I-digestion of leaked DNA segments and, then, (2) inhibition of the DNase I and expulsion of un-leaked (DNase-resistant) DNA segments. The electrophoretic DNA profile versus sedimentation rate (Fig. 4, right side; Serwer et al. 2014a) unambiguously shows the formation by the un-leaked DNA of bands: five major bands and one minor band. The bands imply quantization of DNA leakage.

Quantized leakage of DNA from T3 heads. Phage T3 heads, obtained and stored as described in the caption to Fig. 3, were analyzed by a procedure that started with rate zonal centrifugation in a sucrose gradient and is described in detail by Serwer et al. (2014a). The left side of the figure outlines the procedure used for analysis. The right side has the gel electrophoresis of un-leaked DNA that had been digested with a one-hit restriction endonuclease, Bgl II. The fractions with the minimum leaked DNA and the maximum leaked DNA are indicated by Min.-leaked and Max.-leaked, respectively. Lanes marked M have the following length standards: (1) monomeric, mature phage T3 DNA, two lanes on the left and (2) an Xba I digest of phage T7 DNA on the right. [Figure reproduced from Serwer et al. (2014a) with permission]
The leakage potentially occurs at only one of the two DNA ends. The left end of the packaged genome is (1) the last end packaged when the T7 packaging substrate is a concatemer in vivo (Serwer et al. 1992) and (2) the first end injected into a host cell (Pao and Speyer 1973; Zavriev and Shemyakin 1982).
The un-leaked DNA is found to have the right end (RE in Fig. 4). This is concluded because the RE restriction fragment is not progressively shortened as the sedimentation velocity decreases in the DNA profile of Fig. 4. The left end (LE) restriction fragment is progressively shortened. Therefore, the end that leaks from the capsid is the left end. The spontaneous leaking of DNA thus has the same polarity as DNA injection during infection.
Quantization of DNA leakage is a relatively new phenomenon observed, thus far, only with T3. Indeed, very unusual is the finding of any phage capsid with incompletely packaged DNA (ipDNA). Usually, DNA leakage is an all-or-none phenomenon. To our knowledge, an isolated non-T3 capsid with stable ipDNA has previously been observed only once (Sternberg and Weisberg 1975) and was not characterized.
The following are points to consider for determining how quantization occurs. In addition to DNA, T3 heads have only a gp10 shell, gp8 connector, and gp14/15/16 core stack (Fig. 1d). The connector/core stack is in contact with only a small percentage of the packaged DNA. Thus, the connector/core stack cannot directly sense how much DNA is packaged. However, the connector/core stack might cause quantization via one of the following two possibilities: (1) measuring the amount of DNA that has leaked through it or (2) indirectly sensing the amount of packaged DNA via DNA conformation.
A signal to stop DNA leakage would most likely have no way to be quantized via DNA conformation, i.e., possibility (2), above. The reason is that DNA being packaged is disordered by thermal motion (Petrov and Harvey 2007; Forrey and Muthukumar 2006; Spakowitz and Wang 2005). Thus, thermal motion is likely to be a major factor in disordering DNA during DNA expulsion also.
Measuring of the DNA leakage amount [possibility (1), above] is also unlikely. The reason is that the longer quantized DNAs of Fig. 4 are separated in length by at least 10× the length of the core stack. Details of how the core stack could be the source of quantization are, therefore, difficult to even conceive. Thus, the connector/core stack is not likely to be the source of quantization.
An alternative hypothesis is that the T3 gp10 shell is the source of quantization. The details of this hypothesis are the following. (1) The gp10 shell has several quasi-stable states of different radius. After contraction from one state to a state of the next smaller capsid radius, the contraction stalls. (3) A stall triggers the gp8 connector (Fig. 1) to clamp the DNA molecule, making the capsid stably less full by a quantized amount.
Shell-derived quantization is the most plausible for the following reasons. (1) The number of major intermediate states during expansion of the gp10 shell (5; above) equals the number of major, leakage-generated ipDNA bands in Fig. 4 and Serwer et al. (2014a). (2) In Serwer et al. (2014a), native two-dimensional agarose gel electrophoresis (2d-AGE) demonstrates DNA leakage-associated contraction of the T3 gp10 shell. The source of this conclusion is differential sieving by the gel; sieving increases as particle radius increases. The resolution of the 2d-AGE is, however, not sufficient to resolve the quantization. The procedure of 2d-AGE has previously been reviewed (Serwer et al. 1995). We note that the total number of quantized states might be higher than 5. We have possibly detected only the major states here.
To explain why quantization of DNA leakage occurs, we will make the assumption that this quantization is an evolutionarily selected, not an accidental, property. We think that this assumption is covered by the following general principle. Order in biological nanomachine-associated events is likely to be evolutionarily selected. The reason is that, otherwise, thermal motion would have obliterated the order.
Then, the “why” of leakage quantization is possibly the following. (1) Quantization of biological DNA injection is the original source of leakage quantization. (2) DNA leakage is accompanied by activation of the process of quantization that occurs during biological injection. By this hypothesis, quantization of biological DNA injection is a product of evolutionary selection for periodic stalling of biological DNA injection. The selective advantage of the stalling is avoidance of malfunctions caused by DNA injection too rapid for phage-induced modifications of the host cell. This hypothesis includes the proposals that (1) each stall is accompanied by clamping of DNA by the connector/core stack and (2) a stall continues until the capsid receives feedback that needed modifications have been made in the host cell.
DNA packaging-associated shell flexibility and dynamism
Isolation/characterization studies reveal flexibility of the shell of T3 capsid II during in vivo DNA packaging. Some flexibility is found via the isolation of hyper-expanded capsid II (Serwer and Wright 2016), as reviewed below. Additional flexibility, accompanied by dynamism, is observed when some ipDNA-containing, hyper-expanded capsid II is contracted in vitro by incubation with magnesium ATP at physiological ATP concentration (Serwer and Wright 2017). The contraction does not occur with ADP at the same concentration. Supra-physiological ATP concentration also has an effect, but a destructive one; the capsids disintegrate (Serwer and Wright 2017). Investigations of these phenomena are part of the analysis of DNA packaging. Further tests of models are ongoing. Here, we insert two subsections different from the above sections in that we present some details of the means by which novel, radius-altered capsids can be isolated. The fractionation procedure is buoyant density ultracentrifugation (BDU). This procedure has the advantage of concentrating particles during fractionation.
BDU: typical use
BDU typically fractionates by particle density, not by particle size. The density of a particle is determined primarily by its composition, including co-sedimenting water of hydration. However, DNA water of hydration decreases as water activity decreases. Within a single density gradient, water activity is a decreasing function of density (Costello and Baldwin 1972; Hearst and Vinograd 1961a, b). If this function is relatively weak, DNA can have an equilibrium hydration of as much as 3.1 g water/g sodium DNA (Serwer 1975). DNA has an equilibrium density of ~ 1.7 g/mL during buoyant density centrifugation in a cesium chloride density gradient, which has a relatively strong water activity versus density relationship (Hearst and Vinograd 1961a). However, when the density gradient-forming compound is made either more massive or more dense (or both; water activity higher at any given density), the equilibrium DNA density can be as low as ~ 1.14 g/mL because of higher hydration (Serwer 1975).
If the water activity is high enough, one can fractionate by steric hydration restriction (Costello and Baldwin 1972; Serwer 1975). For DNA packaged in a phage particle, an example of steric hydration restriction is the absence of room for all water that unpackaged DNA binds. Steric hydration restriction has been used during BDU to fractionate infective phages lambda and T7 by the amount of DNA packaged. Steric hydration restriction is typically not observed in cesium chloride density gradients because of low DNA hydration in all packaged states (Serwer 1975).
The larger compounds used for density gradients include (1) lithium silicotungstate (Hearst and Vinograd 1961a), (2) sodium iothalamate (Serwer 1975), (3) Metrizamide (Rickwood and Birnie 1975), and (4) the Metrizamide- and iothalamate-related, radio-opaque compound, Nycodenz (Rickwood et al. 1982). The latter three all have tri-iodinated benzene rings with solubilizing groups added. Even larger, dimerized versions exist, but are not further discussed here. The original work with sodium iothalamate, Metrizamide, and Nycodenz was done by preparative centrifugation. One way to obtain fractions of these gradients is by dripping through the bottom of a centrifuge tube. A device used is sketched in Fig. 5. Details for its use are in the caption to Fig. 5.

Centrifuge tube puncturing and density gradient collecting device. The centrifuge tube is placed in the glass sleeve (truncated glass storage tube) that has a rubber cork at the top. The blunt end of the syringe needle (pink) protrudes from this rubber cork. To close the sleeve, a second rubber cork is snugly inserted in the sleeve’s open end and pressed against the centrifuge tube. This bottom cork has an 18-gauge needle inserted through its middle; the tip of the needle has been cut away. The centrifuge tube is sealed against the bottom cork by placing grease, typically silicone grease, around the needle in the bottom cork. To puncture the tube bottom, a 28-gauge needle is inserted through the 18-gauge needle in the bottom cork. The 28-gauge needle is pushed against the centrifuge tube bottom until puncture occurs. The atmospheric pressure inside of the sleeve is controlled with a finger placed on the blunt end of the top needle. At the time of puncture, the pressure is raised to prevent an air bubble from rising in the gradient and causing mixing. To collect and fractionate the gradient, the pressure is lowered to the point that drops fall from the bottom needle at the desired rate
Restriction of the dripping rate to < 1 drop per 1.5 s is needed to avoid viscosity-driven asymmetric band spreading with some density gradients (Serwer 1975). Included are density gradients of Nycodenz, Metrizamide, and iothalamate salts. The refractive index is sometimes used to determine the densities of fractions. If used with density gradients formed by the latter three compounds, then the refractive index should be measured within 30 min of fractionation. This strategy avoids effects of evaporation. Viscosity-driven band spreading is not a problem with cesium chloride density gradients. Also, evaporation is slower with cesium chloride density gradients because of lower water activity.
BDU: fractionation by capsid permeability
If a BDU-fractionated capsid is impermeable to the density gradient-forming compound, then the capsid’s internal cavity has no density gradient-forming compound. Thus, the capsid behaves as though more hydrated and less dense than it would have been if not impermeable. Also, the capsid’s outer shell cannot have a hole larger than the density gradient-forming compound. Thus, capsid density (θ) becomes a criterion for intactness of the capsid’s shell.
In addition, the density of such an impermeable capsid decreases as the capsid radius (and hydrated volume, Vh) increases. The reason for this is the increase in effective hydration (Γ) as the radius of the internal cavity increases. This relationship exists even if anhydrous capsid mass (M) remains constant. Equations 1 and 2 quantify these relationships for a capsid of invariant M and anhydrous specific volume, v (Costello and Baldwin 1972; Serwer 1975):
For any given particle being fractionated by BDU, the probability of impermeability increases as the density gradient-forming compound’s molecular weight increases. The molecular weights of Metrizamide and Nycodenz are 789 and 821, respectively. One detects impermeability in one or more of the following ways. (1) Γ (Eq. 2) is higher than expected from bound water of hydration. (2) The density reversibly reverts to an expected, higher density when slightly smaller compounds are used to form the density gradient (Serwer 1975, 1980). (3) Assays for binding to the particle interior reveal permeability restriction (Khan et al. 1992).
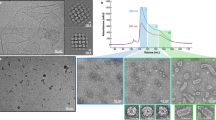
The above points are illustrated by analytical BDU in Nycodenz density gradients. Recently, fluorescence detection has made this possible. Fluorescence detection is needed because Nycodenz density gradients are too ultraviolet light adsorptive and too refractive (light is refracted out of the range of the detector) to use other optics. Figure 6 shows Nycodenz analytical BDU, with fluorescence detection, of (1) a Nycodenz-impermeable (by all three criteria above; Serwer 1980; Khan et al. 1992), Nycodenz low-density (NLD) wild type T3 capsid II, (2) T3 DNA (D), (3) a Nycodenz-permeable, Nycodenz high-density (NHD) T3 capsid II, and (4) a complex of T3 DNA and a Nycodenz-permeable T3 capsid (D-C). These four samples are in three separate cells in the same rotor, all with the same starting density. Note that the DNA–capsid complex is closer to the DNA than it is to the NHD capsid, even though this complex has roughly equal amounts of anhydrous protein and DNA. The primary reason is that the DNA has more bound water than the capsid. Parenthetically, the largest hole in NLD capsid II is at the connector, based on assays for the binding of dyes to the particle interior (Khan et al. (1992).

Analytical Nycodenz buoyant density ultracentrifugation (BDU). Four 0.25-mL solutions of Nycodenz (initial density = 1.313 g/mL) were centrifuged to form an equilibrium Nycodenz density gradient at 50,000 rpm, 20 °C in Epon–charcoal two-channel centerpieces (Beckman-Coulter) with a bottom position at 7.2 cm, meniscus position at 6.5 cm. Particles were stained with either SYBR Green (1/10,000 dilution) or Alexa 488 obtained from Life Technologies (product #A20000; Grand Island, NY). The SYBR Green was added to the Nycodenz solution used to form the density gradient and revealed nucleic acids via non-covalent binding and fluorescence enhancement. The Alexa 488 was covalently bound to proteins; unbound Alexa-488 was removed by dialysis before centrifugation. The sample was detected with confocal microscope optics and a scanning laser that had a 488-nm output wavelength and that excited green fluorescence in GelStar and Alexa 488. We used an AVIV fluorescence detection system (AVIV Biomedical, Inc., Lakewood, NJ). The fluorescence profiles of the following three samples are shown: (1) Alexa 488-stained, dialyzed T7 Nycodenz low-density (NLD) capsid II (black plot with peak labeled NLD), (2) Alexa 488-stained, dialyzed T7 Nycodenz high-density (NHD) capsid II (red plot with peak labeled NHD) (3) DNA expelled from T7 phage at 51 °C and stained with SYBR Green, undialyzed (blue plot). Capsid-free DNA forms the blue peak labeled D; a DNA–NHD capsid complex (DNA outside of the capsid) forms the blue peak labeled D-C. The arrow indicates a variant of NLD capsid II
NLD and NHD capsid II are obtained from lysates of wild type T3- or T7-infected Escherichia coli. NLD capsid II is the same as Metrizamide low-density capsid II, first isolated by Serwer (1980). Nycodenz replaces Metrizamide commercially. Both NLD and NHD capsid II are generated during DNA packaging in wild type T3- and T7-infected cells, but these capsids detach from DNA during or after cellular lysis (Serwer 1980).
Finally, we note that, empirically, preparative Nycodenz density gradients are usually not at equilibrium and are non-linear when appropriate for BDU (Rickwood et al. 1982). None of the previous studies of T3/T7 capsids used equilibrium density gradients. Simulated results at equilibrium are shown in Fig. 7 for centrifugation speeds of 40,000 rpm (red), 50,000 rpm (green), and 60,000 rpm (blue), and centrifugation time more than sufficient to form bands of T3 capsids and phage. Via UltraScan software (Demeler and Gorbet 2016; Demeler et al. 2017), we are working on algorithms for optimizing fractionations and obtaining Nycodenz densities after analytical BDU.

Schematic view of the effect of centrifugation speed on the density gradient. The gradient-forming material (unitary loading concentration, horizontal black line) changes its distribution in the direction of a steeper gradient when the centrifugation speed increases (red: 40,000 rpm; green: 50,000 rpm; blue: 60,000 rpm). The banding position of the particle changes in response (points A, B, and C). The gray line represents the initial density of the Nycodenz solution; the purple line represents the density of a particle being fractionated. The purple line is a first-order approximation in that particles will typically have a density that varies with water activity (and, therefore, Nycodenz concentration) and hydrostatic pressure. The simulation used a Nycodenz molar mass of 821 g/mol and a Nycodenz partial specific volume of 0.483 mL/g
Results relevant to DNA packaging
BDU in Nycodenz density gradients reveals that some NLD capsid II particles have an altered gp10 shell. In one study, phage T3 was first adapted to propagation in medium made to be cell shrinking by adding 0.94 M sucrose. Before fractionation in a Nycodenz density gradient, capsid II was isolated with the final step being BDU in a cesium chloride density gradient (Fig. 8a). The capsid II of Fig. 8a (labeled CII, next to density in g/mL) was then subjected to BDU in a Nycodenz density gradient. Both NHD capsid II (NHD in Fig. 8b) and NLD capsid II (called U-NLD in Fig. 8b for reasons given below) were observed. U-NLD capsid II floats to the upper surface of the Nycodenz density gradient (light scattering detection: Fig. 8b). In contrast, wild type NLD capsid II, in a separate experiment, forms a band at the position marked WT-NLD in Fig. 8b. Densities (g/mL) are next to these labels in Fig. 8b.

Preparative fractionation of particles produced by infection with a T3 mutant. A T3 mutant was obtained by adaptation to propagation in the presence of 0.94 M sucrose (T3Su-1; Serwer and Wright 2016). Escherichia coli BB/1 cells were infected with T3Su-1 in medium with 0.94 M sucrose. The infected cell lysate was processed and subjected to centrifugation in a cesium chloride step gradient. The capsid band of the step gradient was subjected to buoyant density centrifugation in a cesium chloride step gradient, all as described by Serwer and Wright (2016). Light scattering from T3Su-1 capsids is shown after: a the buoyant density centrifugation in a cesium chloride density gradient and b a subsequent buoyant density centrifugation in a Nycodenz density gradient (Serwer and Wright 2016) of particles from the capsid II (CII) band region of the cesium chloride density gradient. M, meniscus; B, bottom of centrifuge tube. The numbers at the right of a centrifuge tube indicate the density in g/mL
The floating of the mutant U-NLD capsid II indicates ultra-low density, < 1.06 g/mL. This is 0.02–0.04 g/mL lower than the density of wild type NLD capsid II. This ultra-low density is the reason for the U-NLD nomenclature. By Eq. 1, the U-NLD status indicates an average capsid radius at least 8% larger than the radius of wild type capsid II. Close examination reveals that the U-NLD capsid II band is also unusual in having relatively high granularity and slightly green tint. Capsid bands are typically blue-tinted. Both of these characteristics imply aggregation of the capsids. The green tint occurs because of multiple (Mie) scattering of a light quantum (Ye et al. 2009). It does not occur because of contamination.
In confirmation of visual analysis, EM of U-NLD capsid II confirms aggregation (Fig. 9; Serwer and Wright 2016). In Serwer and Wright (2016), (1) the protein composition is found to be that of T3 capsid II, (2) analysis of the EM images confirms the hyper-expansion and low permeability, and (3) the EM also reveals some contracted capsid II, as also seen in Fig. 9. A single particle of conventional capsid II is indicated by C-CII in Fig. 9. No DNA was present in U-NLD capsid II, in analogy with wild type NLD capsid II. The DNA being packaged had been lost during either cell lysis or subsequent purification. Visual analysis of the color and texture of bands, such as the U-NLD band in Fig. 8b, provides a guide for subsequent more detailed analysis. This guide is especially useful when fractionating intermediates, because of the variability of potential results.

EM of U-NLD capsid II. The specimen was prepared by negative staining with 1% sodium phosphotungstate, pH 8.6, as described by Serwer and Wright (2016). The arrow marked C-CII is a rare particle of conventional capsid II, i.e., a particle indistinguishable from the capsid II obtained from lysates of wild type T3-infected cells. The unusually high electron transparency of other particles is caused by impermeability to the negative stain. All particles are capsid II. Most particles appearing either larger or smaller than conventional capsid II really are larger or smaller, as discussed in detail by Serwer and Wright (2016). [Image reproduced from Serwer and Wright (2016) under open access licensing]
For U-NLD capsid II, both the aggregation and the loss of DNA are limitations to further analysis. However, NLD capsid II from another directed evolution-derived T3 mutant is isolated un-aggregated (Serwer and Wright 2017). This NLD capsid II also has ipDNA (ipDNA-capsid II), as judged by gel electrophoresis after expelling DNA. Hyper-expanded ipDNA-capsid II particles are used for the study of the effects of magnesium ATP, as discussed at the beginning of this section (Serwer and Wright 2017).
To advance the usefulness of the above procedures, we are further investigating (1) the (complex) details of how the density of NLD ipDNA-capsid II depends on the length of ipDNA in Nycodenz density gradients and (2) the use of alternative density gradients that identify impermeable capsids, while also providing a means to use density to determine the length of ipDNA.
Discussion
Related past data from several phages
Given the recent data on T3 capsid II flexibility, one naturally asks whether related past data exist for T3/T7 and other phages. Evidence of mature shell (in contrast to procapsid or intermediate shell) flexibility for phage HK97 is the finding by cryo-EM that, after loss of DNA, the capsid’s shell “bows out” more than it does before loss of DNA (Duda et al. 2009). This observation is similar to the observation that purified wild type T7 NLD capsid II is 1.4% hyper-expanded relative to the gp10 shell capsid of a T7 phage particle (Guo et al. 2014). Similar hyper-expansion is observed for wild type T7 capsid II captured directly from a lysate using an affinity support. The affinity is from anti-T7 phage serum (Yu et al. 2014). For the following reason, intermediate status for NLD capsid II (Serwer 1980) raises the possibility that energy stored in the shell is released during DNA packaging. Work must be done against the internal DNA pressure to go from DNA un-packaged to DNA packaged state. The logic of this consideration is independent of the observation of the effects, described above, of ATP on T3 capsid shells. Finally, evidence exists for ATPase activity of capsid I-associated T3 gp10 (Hamada et al. 1987) and the major shell protein of phage T4 (Rao and Black 1985b).
Early studies of phage T4 include EM of thin sections to characterize DNA packaging intermediates. This technique has potential for the investigation of shell flexibility in the future. One lesson from the early studies is that EM of thin sections can reveal intracellular capsids that are not found during isolation/characterization (Kellenberger et al. 1967).
A second lesson is that ipDNA-containing capsids are detectable by EM of thin sections. In a study of phage T4, ipDNA-containing capsids are observed as the “grizzled” particles of Schaerli and Kellenberger (1980), for example. These particles appear to be intermediates of DNA packaging and are potential targets of future, more advanced studies.
An essential foundation for these and future studies is past investigation of specimen alterations (artifacts) generated by dehydration/embedding and thin-sectioning for EM. Compression effects during thin-sectioning occur. Lickfeld et al. (1977) demonstrated that the radius of Epon-embedded particles before compression is the radius observed by EM in a direction perpendicular to the direction of the compression. Based on comparison of dimensions of phage particles in solution to the latter dimension in thin sections, a ~ 12% shrinkage in dimensions occurs during dehydration/embedding (Lickfeld et al. 1977).
Finally, the observed radius of capsid shells is not distorted by non-inclusion in a section of a capsid’s equator. The reason for this is that shells are not seen with clarity unless the section includes the equator (Lickfeld et al. 1977). Thus, although artifacts occur, relative dimensions can be interpreted realistically.
A more recently developed, less artifact-prone alternative to thin-sectioning is cryo-EM of intracellular phage and capsids. This procedure can be integrated with tomography to obtain images sufficient to determine shell radius (Chaikeeratisak et al. 2017; Dai et al. 2013). The latter reference enhanced contrast with a phase plate and achieved 50–70 Å resolution during 3D reconstruction of intracellular cyanophage Syn5, its procapsid, capsid II homolog, and heads. Phase plates became more generally available when their production was developed via a beam-induced (Volta) potential on a thin film (Danev et al. 2014). Additional advances are expected in this area in the future.
Obtaining and using assembly intermediates
The obtaining of a complete set of assembly intermediates is a prerequisite for understanding mechanisms of any assembly process, including phage DNA packaging. However, the following conundrum exists. We do not know how to isolate the needed intermediates until we isolate them. Likely limitations to any isolation are (1) instability of intermediates and (2) short life of intermediates during assembly. One way to resolve this conundrum is to use current data to design and pursue novel isolation procedures. Several extensions of the previous procedure can be used.
The first extension is the introduction of more discriminating, gentler fractionation. Hydration-based BDU (i.e., the use of relatively large, dense compounds for density gradient formation) is one possibility, as illustrated here. Hydration-based BDU has the advantage of sometimes fractionating by permeability. In general, impermeable particles are likely to have been functional when intracellular, although possibly altered during isolation. This conclusion derives from the perfection of sealing required for impermeability. This sealing is not likely to be accidental because of the complexity of gp10 interactions in the shell (Guo et al. 2014). Sealing perfection, therefore, is likely to have been generated by selection during evolution. Selection during evolution implies function. Specifically, retention of the NLD characteristic by hyper-expanded T3 capsid II implies the likelihood of function for the hyper-expansion.
Characteristics lost during isolation can include the presence of a loosely attached enzyme. Thus, as isolated, an intermediate may not progress in the assembly pathway in vitro, as a native intermediate would. Indeed, isolated (DNA-free) wild type T7 capsid II does not have gp19 terminase (Serwer 1980; Serwer and Wright 2017) and does not package DNA in vitro, although co-fractionated capsid I does package DNA in vitro (Masker and Serwer 1982). Nonetheless, capsid II is an intermediate, based on kinetics of appearance (Serwer 1980). Indeed, the T4 equivalent of T3/T7 capsid II does package DNA in vitro (Rao and Black 1985a). The loss of DNA packaging capacity occurs either during cellular lysis or during subsequent fractionation. Loss of packaging capacity by isolated intermediates blocks the analysis of these intermediates by in vitro DNA packaging, but does not negate intermediate status.
Coupling of hydration-based BDU with the use of a multi-site T3 mutant yields ipDNA-capsid II particles. Some ipDNA-capsid II particles have the NLD characteristic (Serwer and Wright 2017), which supports assumption of intermediate status. These particles are a step closer to the functional, native in vivo intermediate. However, the ipDNA-capsid II particles have the limitations that gp19 terminase is not present and that the DNA substrate is cleaved (prematurely). Obtaining intermediates without these two limitations is the next objective. The mutant used to obtain ipDNA-capsids is a product of directed evolution (Serwer and Wright 2017).
The use of directed evolution-derived mutants is a second extension of conventional procedure. This extension has, in the past, been limited by impracticality of thorough genomic characterization of multi-site mutants. Complete genomic characterization has been made practical, within the last ~ 5 years, by advances in whole-genome sequencing technology, as illustrated by Serwer et al. (2014a).
The third extension is the use of atypical propagation conditions, including conditions closer to those in the wild. Examples of the latter conditions are lowered water activity and the presence of polymers. In Serwer and Wright (2016), this extension is integrated with directed evolution and hydration-based BDU to obtain the U-NLD capsid II particles. We suspect that more advanced versions of the above procedural extensions will be needed to obtain DNA packaging intermediates less altered than our current ipDNA-capsids.
Variations of the above extensions are likely to apply to motors other than DNA packaging motors. In general, phage DNA injection and packaging provide models for developing both concepts and fractionation/characterization technology for understanding other bio-nanomachines. Our current work implies, at the least, that inert shell models should not be used uncritically for any virus.
Identifying and ordering assembly intermediates
Kinetics of appearance are sometimes used for obtaining the first rendition of (1) what the intermediates are and (2) temporal ordering of the intermediates. After an intermediate first appears post-infection, the amount of the intermediate increases as it is progressively generated from a precursor. Then, the amount decreases as the intermediate is converted to a successor in the production of the final product. The final product is a phage particle in our case. Phage particles progressively increase in amount with time, as do abortive end products of assembly.
Previously observed abortive end products include hollow, cylindrical mis-assemblies of T7 gp10. These mis-assemblies are sometimes called polycapsids. The polycapsids have gp10 shell gaps of at least 2 nm and do not display impermeability to Metrizamide, i.e., they have NHD status (Serwer 1980). NHD status is likely to occur for all end products made abortive by gp10 organization.
Reasons exist to further test a temporal order deduced above. For example, intermediates have kinetics that are also kinetics of particles that undergo disintegration (either intracellular or extracellular) after being produced. The following reason exists for assuming disintegration to be only a remote possibility for capsid II and equivalent capsids for other phages. These capsids resemble mature phage capsids in their stability in vitro (see Serwer et al. 1982 for T3/T7). Furthermore, in vivo phage capsid disintegration has never been reported, to our knowledge. Nonetheless, the next section describes further tests.
Pathway analysis: modification for DNA packaging
The following is the classical (and successful) test used for ordering step-by-step pathways of the addition of proteins to form phage tails and tail fibers. First, conditional lethal mutants are obtained. Each mutant specifically blocks synthesis of one needed protein. Then, for each protein, a candidate is obtained for substrate on which the protein will assemble. One does this by observing what accumulates when the protein is missing. A candidate is confirmed if it progresses in the pathway when the missing protein is provided in vitro. Provision of the missing protein was originally done by the use of extracts of mutant-infected cells.
Finally, the substrates (intermediates) are ordered in the direction of increasing assembly (original studies: Edgar and Wood 1966; Wood et al. 1968). In more recent studies of the T4 tail, purified, recombinant proteins are used to provide missing proteins; 3D structures are determined by cryo-EM (for reviews, see Leiman et al. 2010; Arisaka et al. 2016).
This strategy, however, does not work with DNA packaging. The reason for this is that DNA packaging pathways typically have numerous intermediates that do not progress via the addition of a protein. DNA packaging proteins are pre-assembled. Progress involves coordinated, cyclical changes in the pre-assembled motor.
Thus, phage DNA packaging is typically analyzed with details obscured. An instructive analogy is embedded in the following question. If a visitor from another planet does not know Earth-based engine technology, could this visitor learn the mechanisms of an automobile engine by the analysis of an automobile with the engine obscured by a closed hood? Visual inspection will not provide much information.
The visitor could obtain constraints by placing a mechanical sensor on the hood and characterizing motor vibrations. “Sensor-on-hood” strategy is analogous to nanometry-based strategy with DNA packaging. However, in both cases, more than one explanation will exist for each vibration pattern. Thus, these strategies are misleading if too much reliance is placed on them.
In addition, one might try to deduce the mechanism by (1) maiming the automobile engine via the launching of various physical and electromagnetic projectiles at the automobile and (2) observing changes in motor function. This approach has some analogy with extending traditional genetic strategy to DNA packaging motors. One is likely to get interesting data. But, again, the data will have more than one explanation. Imagine, for example, that the automobile has a hybrid electrical/internal combustion engine and, therefore, has more than one cycle.
We contend that a naïve investigator has to (1) look under the hood and beneath the engine (cylinder) head, and (2) watch the engine undergo a cycle(s), in order to understand the motor that moves an automobile. The strategy discussed here is one attempt at analogous biochemistry-based analysis for DNA packaging motors. The obscuring effect of the car hood is analogous to the obscuring effect of not knowing the DNA packaging intermediates. Intermediates-based strategy also can produce opportunities for practical uses of isolated intermediates (for example, see Serwer et al. 2014b). This strategy is projected to complement (not to replace) the various procedures of real-time single-molecule analysis.
The information from intermediates-based strategy is increased when an intermediate is active in vitro in conditions compatible with the detection of progression of the intermediate toward the end product. Even more information is obtained if the progression can be monitored in real time at nanometer or better resolution. To accomplish the latter objective, one recent, promising development is high-speed (~ 10 images per second), tapping mode atomic force microscopy (AFM; reviewed by Li et al. 2017). In this procedure, a very fine probe is raster-scanned across the specimen and the specimen’s effect on the probe is measured via the effect on a probe-attached cantilever. AFM requires the attachment of the specimen to a solid surface, typically mica. If the intermediate remains active after attachment, then high-speed AFM should be capable of detecting transitions in the dimensions and surface properties of the shells of intermediates. Relevant transitions include the ATP-induced shell transitions of Serwer and Wright (2017). Thus, high-speed AFM is one possible technical advance for the future.
References
Agirrezabala X, Martín-Benito J, Castón JR, Miranda R, Valpuesta JM, Carrascosa JL (2005) Maturation of phage T7 involves structural modification of both shell and inner core components. EMBO J 24:3820–3829. https://doi.org/10.1038/sj.emboj.7600840
Arisaka F, Yap ML, Kanamaru S, Rossmann MG (2016) Molecular assembly and structure of the bacteriophage T4 tail. Biophys Rev 8:385–396. https://doi.org/10.1007/s12551-016-0230-x
Black LW, Thomas JA (2012) Condensed genome structure. Adv Exp Med Biol 726:469–487. https://doi.org/10.1007/978-1-4614-0980-9_21
Center MS, Studier FW, Richardson CC (1970) The structural gene for a T7 endonuclease essential for phage DNA synthesis. Proc Natl Acad Sci U S A 65:242–248
Chaikeeratisak V, Nguyen K, Khanna K, Brilot AF, Erb ML, Coker JKC, Vavilina A, Newton GL, Buschauer R, Pogliano K, Villa E, Agard DA, Pogliano J (2017) Assembly of a nucleus-like structure during viral replication in bacteria. Science 355:194–197. https://doi.org/10.1126/science.aal2130
Chemla YR, Smith DE (2012) Single-molecule studies of viral DNA packaging. Adv Exp Med Biol 726:549–584. https://doi.org/10.1007/978-1-4614-0980-9_24
Costello RC, Baldwin RL (1972) The net hydration of phage lambda. Biopolymers 11:2147–2169. https://doi.org/10.1002/bip.1972.360111015
Dai W, Fu C, Raytcheva D, Flanagan J, Khant HA, Liu X, Rochat RH, Haase-Pettingell C, Piret J, Ludtke SJ, Nagayama K, Schmid MF, King JA, Chiu W (2013) Visualizing virus assembly intermediates inside marine cyanobacteria. Nature 502:707–710. https://doi.org/10.1038/nature12604
Danev R, Buijsse B, Khoshouei M, Plitzko JM, Baumeister W (2014) Volta potential phase plate for in-focus phase contrast transmission electron microscopy. Proc Natl Acad Sci U S A 111:15635–15640. https://doi.org/10.1073/pnas.1418377111
Demeler B, Gorbet GE (2016) Analytical ultracentrifugation data analysis with UltraScan-III. In: Uchiyama S, Arisaka F, Stafford WF, Laue T (eds) Analytical ultracentrifugation: instrumentation, software, and applications. Springer, New York, pp 119–143
Demeler B, Gorbet G, Zollars D, Dubbs B, Brookes E, Cao W (2017) UltraScan-III version 3.5: a comprehensive data analysis software package for analytical ultracentrifugation experiments. http://www.ultrascan3.uthscsa.edu. Accessed 26 Oct 2017
Duda RL, Ross PD, Cheng N, Firek BA, Hendrix RW, Conway JF, Steven AC (2009) Structure and energetics of encapsidated DNA in bacteriophage HK97 studied by scanning calorimetry and cryo-electron microscopy. J Mol Biol 391:471–483. https://doi.org/10.1016/j.jmb.2009.06.035
Edgar RS, Wood WB (1966) Morphogenesis of bacteriophage T4 in extracts of mutant-infected cells. Proc Natl Acad Sci U S A 55:498–505
Fokine A, Rossmann MG (2014) Molecular architecture of tailed double-stranded DNA phages. Bacteriophage 4:e28281. https://doi.org/10.4161/bact.28281
Forrey C, Muthukumar M (2006) Langevin dynamics simulations of genome packing in bacteriophage. Biophys J 91:25–41. https://doi.org/10.1529/biophysj.105.073429
Fujisawa H, Morita M (1997) Phage DNA packaging. Genes Cells 2:537–545
González-García VA, Bocanegra R, Pulido-Cid M, Martín-Benito J, Cuervo A, Carrascosa JL (2015) Characterization of the initial steps in the T7 DNA ejection process. Bacteriophage 5:e1056904. https://doi.org/10.1080/21597081.2015.1056904
Guo F, Liu Z, Vago F, Ren Y, Wu W, Wright ET, Serwer P, Jiang W (2013) Visualization of uncorrelated, tandem symmetry mismatches in the internal genome packaging apparatus of bacteriophage T7. Proc Natl Acad Sci U S A 110:6811–6816. https://doi.org/10.1073/pnas.1215563110
Guo F, Liu Z, Fang PA, Zhang Q, Wright ET, Wu W, Zhang C, Vago F, Ren Y, Jakana J, Chiu W, Serwer P, Jiang W (2014) Capsid expansion mechanism of bacteriophage T7 revealed by multistate atomic models derived from cryo-EM reconstructions. Proc Natl Acad Sci U S A 111:E4606–E4614. https://doi.org/10.1073/pnas.140702011
Hamada K, Fujisawa H, Minagawa T (1987) Characterization of ATPase activity of a defined in vitro system for packaging of bacteriophage T3 DNA. Virology 159:244–249. https://doi.org/10.1016/0042-6822(87)90461-2
Harvey SC, Petrov AS, Devkota B, Boz MB (2009) Viral assembly: a molecular modeling perspective. Phys Chem Chem Phys 11:10553–10564. https://doi.org/10.1039/b912884k
Hearst JE, Vinograd J (1961a) The net hydration of T-4 bacteriophage deoxyribonucleic acid and the effect of hydration on buoyant behavior in a density gradient at equilibrium in the ultracentrifuge. Proc Natl Acad Sci U S A 47:1005–1014
Hearst JE, Vinograd J (1961b) A three-component theory of sedimentation equilibrium in a density gradient. Proc Natl Acad Sci U S A 47(7):999–1004
Hu B, Margolin W, Molineux IJ, Liu J (2013) The bacteriophage T7 virion undergoes extensive structural remodeling during infection. Science 339:576–579. https://doi.org/10.1126/science.1231887
Kellenberger E, Eiserling FA, Boy de la Tour E (1967) Studies on the morphopoiesis of the head of phage T-even III. The cores of head-related structures. J Ultrastruct Res 21:335–360. https://doi.org/10.1016/S0022-5320(67)80099-6
Khan SA, Griess GA, Serwer P (1992) Assembly-associated structural changes of bacteriophage T7 capsids. Detection by use of a protein-specific probe. Biophys J 63:1286–1292. https://doi.org/10.1016/S0006-3495(92)81724-1
Leiman PG, Arisaka F, van Raaij MJ, Kostyuchenko VA, Aksyuk AA, Kanamaru S, Rossmann MG (2010) Morphogenesis of the T4 tail and tail fibers. Virol J 7:355. https://doi.org/10.1186/1743-422X-7-355
Leptihn S, Gottschalk J, Kuhn A (2016) T7 ejectosome assembly: a story unfolds. Bacteriophage 6:e1128513. https://doi.org/10.1080/21597081.2015.1128513
Li M, Dang D, Liu L, Xi N, Wang Y (2017) Imaging and force recognition of single molecular behaviors using atomic force microscopy. Sensors (Basel) 17:200. https://doi.org/10.3390/s17010200
Lickfeld KG, Menge B, Wunderli H, van den Broek J, Kellenberger E (1977) The interpretation and quantitation of sliced intracellular bacteriophages and phage-related particles. J Ultrastruct Res 60:148–168
Liu S, Chistol G, Hetherington CL, Tafoya S, Aathavan K, Schnitzbauer J, Grimes S, Jardine PJ, Bustamante C (2014) A viral packaging motor varies its DNA rotation and step size to preserve subunit coordination as the capsid fills. Cell 157:702–713
Masker WE, Serwer P (1982) DNA packaging in vitro by an isolated bacteriophage T7 procapsid. J Virol 43:1138–1142
Molineux IJ (2001) No syringes please, ejection of phage T7 DNA from the virion is enzyme driven. Mol Microbiol 40:1–8
Pajunen MI, Elizondo MR, Skurnik M, Kieleczawa J, Molineux IJ (2002) Complete nucleotide sequence and likely recombinatorial origin of bacteriophage T3. J Mol Biol 319:1115–1132. https://doi.org/10.1016/S0022-2836(02)00384-4
Pao CC, Speyer JF (1973) Order of injection of T7 bacteriophage DNA. J Virol 11:1024–1026
Petrov AS, Harvey SC (2007) Structural and thermodynamic principles of viral packaging. Structure 15:21–27. https://doi.org/10.1016/j.str.2006.11.013
Popa MP, Mckelvey TA, Hempel J, Hendrix RW (1991) Bacteriophage HK97 structure: wholesale covalent cross-linking between the major head shell subunits. J Virol 65:3227–3237
Rao VB, Black LW (1985a) DNA packaging of bacteriophage T4 proheads in vitro evidence that prohead expansion is not coupled to DNA packaging. J Mol Biol 185:565–578
Rao VB, Black LW (1985b) Evidence that a phage T4 DNA packaging enzyme is a processed form of the major capsid gene product. Cell 42:967–977
Rao VB, Feiss M (2015) Mechanisms of DNA packaging by large double-stranded DNA viruses. Annu Rev Virol 2:351–378. https://doi.org/10.1146/annurev-virology-100114-055212
Rickwood D, Birnie GD (1975) Metrizamide, a new density-gradient medium. FEBS Lett 50:102–110
Rickwood D, Ford T, Graham J (1982) Nycodenz: a new nonionic iodinated gradient medium. Anal Biochem 123:23–31
Rixon FJ, Schmid MF (2014) Structural similarities in DNA packaging and delivery apparatuses in Herpesvirus and dsDNA bacteriophages. Curr Opin Virol 5:105–110. https://doi.org/10.1016/j.coviro.2014.02.003
Sadowski PD, Kerr C (1970) Degradation of Escherichia coli B deoxyribonucleic acid after infection with deoxyribonucleic acid-defective amber mutants of bacteriophage T7. J Virol 6:149–155
Schaerli C, Kellenberger E (1980) Head maturation pathway of Bacteriophages T4 and T2. V. Maturable ε-particle accumulating an acridine-treated bacteriophage T4-infected cells. J Virol 33:830–844
Schweiger M, Herrlich P, Scherzinger E, Rahmsdorf HJ (1972) Negative control of protein synthesis after infection with bacteriophage T7. Proc Natl Acad Sci U S A 69:2203–2207
Serwer P (1975) Buoyant density sedimentation of macromolecules in sodium iothalamate density gradients. J Mol Biol 92:433–448
Serwer P (1978) Observation of DNA by negative staining: phage T7 DNA–capsid complexes. In: Proceedings of the ninth international congress on electron microscopy, Toronto, Canada, August 1978, vol II, pp 228–229
Serwer P (1980) A metrizamide-impermeable capsid in the DNA packaging pathway of bacteriophage T7. J Mol Biol 138:65–91
Serwer P, Jiang W (2012) Dualities in the analysis of phage DNA packaging motors. Bacteriophage 2:239–255. https://doi.org/10.4161/bact.23829
Serwer P, Watson RH (1981) Capsid–DNA complexes in the DNA packaging pathway of bacteriophage T7: characterization of the capsids bound to monomeric and concatemeric DNA. Virology 108:164–176
Serwer P, Wright ET (2016) Testing a proposed paradigm shift in analysis of phage DNA packaging. Bacteriophage 6:e1268664. https://doi.org/10.1080/21597081.2016.1268664
Serwer P, Wright ET (2017) ATP-driven contraction of phage T3 capsids with DNA incompletely packaged in vivo. Viruses 9:119. https://doi.org/10.3390/v9050119
Serwer P, Hayes SJ, Watson RH (1982) The structure of a bacteriophage T7 procapsid and its in vivo conversion product probed by digestion with trypsin. Virology 122:392–401
Serwer P, Watson RH, Hayes SJ (1992) Formation of the right before the left mature DNA end during packaging-cleavage of bacteriophage T7 DNA concatemers. J Mol Biol 226:311–317
Serwer P, Khan SA, Griess GA (1995) Non-denaturing gel electrophoresis of biological nanoparticles: viruses. J Chromatogr A 698:251–261
Serwer P, Wright ET, Hakala KW, Weintraub ST (2008) Evidence for bacteriophage T7 tail extension during DNA injection. BMC Res Notes 1:36. https://doi.org/10.1186/1756-0500-1-36
Serwer P, Wright ET, Liu Z, Jiang W (2014a) Length quantization of DNA partially expelled from heads of a bacteriophage T3 mutant. Virology 456-457:157–170. https://doi.org/10.1016/j.virol.2014.03.016
Serwer P, Wright ET, Chang JT, Liu X (2014b) Enhancing and initiating phage-based therapies. Bacteriophage 4:e961869. https://doi.org/10.4161/21597073.2014.961869
Smith KA (2012) Louis Pasteur, the father of immunology? Front Immunol 3:68. https://doi.org/10.3389/fimmu.2012.00068
Spakowitz AJ, Wang ZG (2005) DNA packaging in bacteriophage: is twist important? Biophys J 88:3912–3923. https://doi.org/10.1529/biophysj.104.052738
Sternberg N, Weisberg R (1975) Packaging of prophage and host DNA by coliphage lambda. Nature 256:97–103
Stroud RM, Serwer P, Ross MJ (1981) Assembly of bacteriophage T7. Dimensions of the bacteriophage and its capsids. Biophys J 36:743–757. https://doi.org/10.1016/S0006-3495(81)84763-7
Studier FW (1979) Relationships among different strains of T7 and among T7-related bacteriophages. Virology 95:70–84
Veesler D, Johnson JE (2012) Virus maturation. Annu Rev Biophys 41:473–496. https://doi.org/10.1146/annurev-biophys-042910-155407
Veesler D, Quispe J, Grigorieff N, Potter CS, Carragher B, Johnson JE (2012) Maturation in action: CryoEM study of a viral capsid caught during expansion. Structure 20:1384–1390. https://doi.org/10.1016/j.str.2012.05.011
Wood WB, Edgar RS, King J, Lielausis I, Henninger M (1968) Bacteriophage assembly. Fed Proc 27:1160–1166
Ye QL, Yoshikawa H, Bandow S, Awaga K (2009) Green magnetite (Fe3O4): unusual optical Mie scattering and magnetic isotropy of submicron-size hollow spheres. Appl Phys Lett 94:063114. https://doi.org/10.1063/1.3079407
Yu G, Vago F, Zhang D, Snyder JE, Yan R, Zhang C, Benjamin C, Jiang X, Kuhn RJ, Serwer P, Thompson DH, Jiang W (2014) Single-step antibody-based affinity cryo-electron microscopy for imaging and structural analysis of macromolecular assemblies. J Struct Biol 187:1–9. https://doi.org/10.1016/j.jsb.2014.04.006
Zavriev SK, Shemyakin MF (1982) RNA polymerase-dependent mechanism for the stepwise T7 phage DNA transport from the virion into E. coli. Nucleic Acids Res 10:1635–1652
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by NIH (R01-GM120600 to BD and R01-AI072035 to WJ) and the Welch Foundation (AQ-764 to PS).
Conflict of interest
Philip Serwer declares that he has no conflict of interest. Elena T. Wright declares that she has no conflict of interest. Borries Demeler declares that he has no conflict of interest. Wen Jiang declares that he has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
This article is part of a Special Issue on ‘Biomolecules to Bio-nanomachines - Fumio Arisaka 70th Birthday’ edited by Damien Hall, Junichi Takagi, and Haruki Nakamura.
Rights and permissions
About this article

Cite this article
Serwer, P., Wright, E.T., Demeler, B. et al. States of phage T3/T7 capsids: buoyant density centrifugation and cryo-EM. Biophys Rev 10, 583–596 (2018). https://doi.org/10.1007/s12551-017-0372-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12551-017-0372-5