
Abstract
The spinocerebellar ataxias (SCA) are a group of rare inherited neurodegenerative diseases characterized by slowly progressive cerebellar ataxia, resulting in unsteady gait, clumsiness, and dysarthria. The disorders are predominantly inherited in an autosomal dominant manner. Mutations in the gene AFG3L2 that encodes a subunit of the mitochondrial m-AAA protease have previously been shown to cause spinocerebellar ataxia type 28 (SCA28). Here, we present the clinical phenotypes of three patients from a family with autosomal dominant cerebellar ataxia and show by molecular genetics and in silico modelling that this is caused by a novel missense mutation in the AFG3L2 gene. Furthermore, we show, for the first time, fluorodeoxyglucose-positron emission tomography (FDG-PET) scans of the brain and selective type I fiber atrophy of skeletal muscle of SCA28 patients indicating non-nervous-system involvement in SCA28 as well.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The spinocerebellar ataxias (SCA) are a group of rare inherited neurodegenerative diseases characterized by slowly progressive cerebellar ataxia, resulting in unsteady gait, clumsiness, and dysarthria. Inheritance is predominantly autosomal dominant (also known as autosomal dominant ataxia (ADCA)), but can be autosomal recessive, X-linked, or mitochondrial [1]. The genetic heterogeneity in ADCA is prominent with more than 37 known loci and 32 identified genes in which mutations are pathogenic. Three different groups of mutations cause the ADCAs: 1. Expansions of coding CAG repeats resulting in polyglutamine expansions, which are by far the most frequent cause, 2. expansions in non-coding regions, or 3. non-repeat mutations. The non-repeat mutations are often private mutations not previously associated with disease, and the interpretation of the pathogenic effect of these is therefore often challenging, especially in small families [2].
The SCA28 subtype belongs to the group of ADCAs caused by non-repeat mutations. The SCA28 locus on chromosome 18p was published in 2006 upon linkage studies of a four-generation family [3] and the associated AFG3L2 gene was published in 2010 [4]. The causative mutations described are either missense mutations or partial deletions [5, 6]. SCA28 comprise approximately 1.5 % of ADCA and only few families have been reported. The clinical phenotype is a slowly progressive unsteady gait, dysarthria, limb ataxia, gaze-evoked nystagmus, and ptosis, and the age of onset is variable from early childhood to 60 years of age. Most patients maintain gait function. Muscle weakness and atrophy have been described in a few patients, but so far the reported muscle biopsies were shown to be normal [3, 6]. Magnetic resonance imaging (MRI) has shown cerebellar atrophy, especially of the vermis [3–9]. One homozygous mutation has been described to result in a more severe phenotype even though this specific mutation did not result in any phenotype in the heterozygous form [10].
AFG3L2 encodes a subunit of the homo- or hetero-hexameric mitochondrial m-AAA protease (ATPases associated with various cellular activities), which is a component of the mitochondrial ATP-dependent metalloprotease that takes part in proteolytic quality control and chaperone-like activities in the mitochondria [11]. The other subunit of the hetero-hexameric form is paraplegin, encoded by SPG7, in which mutations can cause hereditary spastic paraplegia. In silico 3D reconstruction of the AFG3L2 protein has shown that missense mutations found in SCA28 patients decrease the mean interaction energy of the hexamer, which supposedly destabilizes the m-AAA complex, supporting a pathogenic effect of the mutations [5].
Here, we present the clinical phenotypes of three patients from a family with ADCA and show by molecular genetics and in silico modelling that this is caused by a novel missense mutation in the AFG3L2 gene. Furthermore, we show, for the first time, fluorodeoxyglucose-positron emission tomography (FDG-PET) scans of the brain and selective type I fiber atrophy of skeletal muscle of SCA28 patients.
Materials and Methods
The index patient was referred to the Neurogenetics Clinic, Department of Neurology, Rigshospitalet, University of Copenhagen, and subsequently two additional family members were referred (Fig. 1a). They all underwent clinical examination, skin and muscle biopsy as well as MRI and FDG-PET scanning.

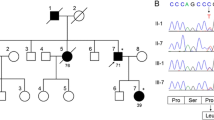
a Pedigree of the family. The arrow denotes the index patient. b Sequences showing part of the DNA sequence of exon 16 and the corresponding protein sequence in different species. The amino acid change (p.P688T) resides at a highly conserved site. The mutation is marked by the red box
For patient II-2 and II-3, a 10-min simultaneous PET/MRI acquisition was performed 40 min after i.v. injection of 200 MBq [18F]-FDG on a fully-integrated PET/MRI (3Tesla) system (Biograph mMR, Siemens Healthcare). Patient III-1 received a [18F]-FDG-PET scan on a Siemens Biograph Truepoint 64 slice PET/CT scanner, and MRI on a 1.5 T Siemens MRI. Standard clinical non-contrast enhanced MRI sequences were performed comprising T1 3D MPRAGE, T2 Turbo Spin Echo (TSE), T2 Fluid Attenuated Inversion Recovery (FLAIR), T2* weighted imaging, and diffusion weighted imaging (DWI). The [18F]-FDG-PET scan was attenuation-corrected using low dose CT scanning [12]. All FDG-PET scans were compared to a database of age-matched healthy subjects (Scenium 3.0, Syngo-Via, Siemens Healthcare).
Muscle biopsies from two affected patients were procured from the vastus lateralis muscle of the thigh and processed for histological evaluation according to standard procedures. Respiratory chain enzyme activity in muscle tissue was determined as described [13] and mDNA was isolated from the muscle biopsies and subjected to deletion analysis by PCR [14]. Incisional skin biopsies were performed from the thigh in all patients. Blood samples were drawn and DNA was extracted using standard methods. In the index patient, repeat expansions in SCA1 (ATXN1), SCA2 (ATXN1), SCA3(ATXN3), SCA6 (CACNA1A), SCA7(ATXN7), SCA10 (ATXN10), SCA12 (PPP2R2B), and SCA17(TDP) were excluded before the SCA11 (TTBK2), SCA13 (KCNC3), SCA14 (PRKCG), SCA15 (ITPR1), SCA18 (IFRD1), SCA23 (PDYN), SCA27 (FGF14), and SCA28 (AFG3L2) genes were Sanger sequenced after PCR amplification using standard techniques. Finally, all 17 exons of the SPG7 gene coding for paraplegin were Sanger sequenced after PCR amplification using standard techniques. All primer sequences and PCR conditions are available upon request.
In silico modelling was performed to analyze protein structure and stability of AFG3L2. Detailed description of the method can be found in the Supplementary information.
The study was approved by the Ethics Committee of the Capital Region of Denmark (H-4-2011-157), and written informed consent was obtained from each participant before enrollment.
Results
The index patient (III-1) belonged to a three-generation family with a pure ADCA phenotype including saccadic pursuit, dysarthria, and limb and gait ataxia. Patient I-1 died at age 77 years. She experienced unsteady gait with late adult onset and stayed ambulant. The phenotypes of the index patient and patients II-2 and II-3 are summarized in Table 1. MRI showed cerebellar atrophy, and FDG-PET showed reduced activity in cerebellum (Fig. 2). The skin biopsies showed normal intradermal nerve fiber density (results not shown). In patients II-3 and III-1, muscle biopsies showed selective type-1 fiber atrophy resulting in increased variability in fiber diameter (Fig. 3) and the atrophied type-1 fibers exhibited reorganized distribution of mitochondria. No mDNA deletions were found and the activity of respiratory chain complexes was normal (results not shown).

Comparison of age matched healthy control (top row (a–c)) to subject III-1 (bottom row (d–f) with sagittal sections through interhemispheric midline co-registered T1 weighted MRI (a, d), [18F]-FDG PET (b, e), and quantitative statistical comparisons of [18F]-FDG PET (c, f). There is severe structural atrophy and reduction of the metabolic activity of the cerebellar vermis (red arrows)

a Myosin heavy-chain staining of type I fibers in skeletal muscle from subject II-3, showing atrophy of type I fibers compared to type II fibers. b Cytochrome c staining showing reorganized mitochondrial content of the atrophied type I fibers in subject II-3
A heterozygous c.2062C>A, p.P688T missense mutation in AFG3L2 (NM_006796) was identified by Sanger sequencing, and it was present in all tested symptomatic family members. It was located in the proteolytic domain of AFG3L2 in region of highly conserved amino acids (Fig. 1b), and the mutation was absent in 2000 ethnically matched control chromosomes [16] as well as from the ExAC database (http://exac.broadinstitute.org/) collecting data on >120,000 alleles [17].
Since AFG3L2 and paraplegin can form a heteromeric form of the m-AAA protease complex [18], we furthermore sequenced the paraplegin coding gene, SPG7, to exclude the possibility that a mutation in SPG7 was the reason for the phenotype in the family. We found no known mutations or other sequence variations in any of the 17 exons of SPG7.
Using molecular dynamics, we imposed an in silico strategy to investigate if the p.P688T mutation affects protein structure or stability. We compared the wild-type protein complex to a protein harboring the new p.P688T mutation as well as to the previously reported p.G671E-mutation that was previously shown to destabilize the structure of the protein complex by increasing the interaction energies between the individual subunits [5]. In our in silico modelling, we confirm that both the p.P688T as well as the p.G671E mutation exhibit a slightly destabilizing effect on the protein complex [5] (see Supplementary Information for details), supporting the hypothesis that this sequence variant is pathogenic.
Discussion
Here, we present the clinical phenotypes of three patients from a family with ADCA and show that the symptoms are most likely caused by a novel missense mutation in the AFG3L2 gene.
The phenotypes of the patients are among the milder reported previously for SCA28, as we found no nystagmus, no ptosis, and no pyramidal signs; while the age at onset was similar to previous reported median age at onset [5]. The MRI scans showed cerebellar atrophy and the youngest and least affected patient (III-1) had localized atrophy of the superior vermis. This suggests that the vermis might be a particularly vulnerable structure in SCA28, since this specific finding has previously been reported in other SCA28 patients [8]. The FDG-PET scans showed the same pattern of reduced metabolism of the vermis in the youngest patient and more globally affected cerebella in the oldest patients, where also slightly reduced cerebral cortical metabolism was observed.
In addition to the FDG-PET scan, we show for the first time a type-1 muscle fiber selective atrophy in muscle biopsies from two patients. Selective type-2 fiber atrophy is a common finding and often associated with disuse of muscle, even in healthy individuals. Selective type-1 fiber atrophy, on the other hand, is unusual and typically associated with specific muscle diseases, such as myotonic dystrophy type 1 and congenital myopathies. The atrophy of type 1 fibers correlates well with the muscle weakness and atrophy that have been reported in some SCA28 patients, although muscle weakness and atrophy was not found in our patients. Muscle weakness in SCA28 patients might be present in other patients with severe ataxia, but it is probably often interpreted as inactivity atrophy due to short walking distance or use of wheelchair, and therefore unrecognized as a direct consequence of the mutations in AFG3L2. While muscle biopsies from SCA28 patients with more severe phenotypes with regard to ophtalmoparesis, ptosis, and slow saccades were previously reported to be normal [3, 6], muscle biopsies from more families with SCA28 are needed to confirm if this phenotype is attributed only to a single family, a specific mutation or if it is a more general feature of SCA28.
The novel and putative disease causing mutation, c.2062C>A, reported in this family is located in the 16th exon of the gene, which is translated into the proteolytic domain in the m-AAA-protease (the Peptidase-M41 domain) [5]. Eleven of the 19 known pathogenic mutations reside in this exon (HGMD professional). The disease causing nature of this mutation is supported by several lines of evidence: Firstly, the mutation segregated with the disease in the family, and the mutation was not found in 1000 matched control subjects, and nor was it found in the ExAC database [17]. Secondly, it has previously been described that exons 15 and 16 of the AFG3L2 gene are hot spots for mutations causative of SCA28, which is further supported by the fact that the proteolytic domain encoded partly by exons 15 and 16 is highly conserved across many species [5]. Finally, in silico analyses of protein complex stability of the m-AAA-protease showed that the mutation destabilized the protein structure. Although our in silico analysis showed a milder impact on protein stability compared to previously published in silico analyses of mutations in this region [5], the changes observed are still at comparable levels and support the impact of the mutation on the protein complex.
In summary, we have reported the phenotypes of a SCA28 family with a novel mutation in AFG3L2. We show that the c.2062C>A mutation is most likely causative of disease as it segregates with an ADCA phenotype and in silico analysis supports a destabilization of the protein by the mutation. Furthermore, we show early involvement of the vermis on FDG-PET and selective type-1 muscle fiber atrophy indicating non-nervous-system involvement in SCA28 as well.
References
Bird TD. Hereditary Ataxia Overview. GeneReviews. 2015.
Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 2010;9:885–94.
Cagnoli C, Mariotti C, Taroni F, Seri M, Brussino A, Michielotto C, et al. SCA28, a novel form of autosomal dominant cerebellar ataxia on chromosome 18p11.22-q11.2. Brain. 2006;129:235–42.
Di Bella D, Lazzaro F, Brusco A, Plumari M, Battaglia G, Pastore A, et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat Genet. 2010;42:313–21.
Cagnoli C, Stevanin G, Brussino A, Barberis M, Mancini C, Margolis RL, et al. Missense mutations in the AFG3L2 proteolytic domain account for ∼1.5% of European autosomal dominant cerebellar ataxias. Hum Mutat. 2010;31:1117–24.
Smets K, Deconinck T, Baets J, Sieben A, Martin J-J, Smouts I, et al. Partial deletion of AFG3L2 causing spinocerebellar ataxia type 28. Neurology. 2014;82:2092–100.
Edener U, Wöllner J, Hehr U, Kohl Z, Schilling S, Kreuz F, et al. Early onset and slow progression of SCA28, a rare dominant ataxia in a large four-generation family with a novel AFG3L2 mutation. Eur J Hum Genet. 2010;18:965–8.
Mariotti C, Brusco A, Di Bella D, Cagnoli C, Seri M, Gellera C, et al. Spinocerebellar ataxia type 28: a novel autosomal dominant cerebellar ataxia characterized by slow progression and ophthalmoparesis. Cerebellum. 2008;7:184–8.
Löbbe AM, Kang J-S, Hilker R, Hackstein H, Müller U, Nolte D. A novel missense mutation in AFG3L2 associated with late onset and slow progression of spinocerebellar ataxia type 28. J Mol Neurosci. 2014;52:493–6.
Pierson TM, Adams D, Bonn F, Martinelli P, Cherukuri PF, Teer JK, et al. Whole-exome sequencing identifies homozygous AFG3L2 mutations in a spastic ataxia-neuropathy syndrome linked to mitochondrial m-AAA proteases. PLoS Genet. 2011;7, e1002325.
Leonhard K, Stiegler A, Neupert W, Langer T. Chaperone-like activity of the AAA domain of the yeast Yme1 AAA protease. Nature. 1999;398:348–51.
Andersen FL, Ladefoged CN, Beyer T, Keller SH, Hansen AE, Højgaard L, et al. Combined PET/MR imaging in neurology: MR-based attenuation correction implies a strong spatial bias when ignoring bone. Neuroimage. 2014;84:206–16.
Larsen S, Nielsen J, Hansen CN, Nielsen LB, Wibrand F, Stride N, et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol. 2012;590:3349–60.
Kleinle S, Wiesmann U, Superti-Furga A, Krähenbühl S, Boltshauser E, Reichen J, et al. Detection and characterization of mitochondrial DNA rearrangements in Pearson and Kearns-Sayre syndromes by long PCR. Hum Genet. 1997;100:643–50.
Schmitz-Hubsch T, du Montcel ST, Baliko L, Berciano J, Boesch S, Depondt C, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology. 2006;66:1717–20.
Lohmueller KE, Sparsø T, Li Q, Andersson E, Korneliussen T, Albrechtsen A, et al. Whole-exome sequencing of 2,000 Danish individuals and the role of rare coding variants in type 2 diabetes. Am J Hum Genet. 2013;93:1072–86.
Exome Aggreg. Consort. (ExAC), Cambridge, MA (URL http://exac.broadinstitute.org). Accessed 07–2015. 2015.
Koppen M, Metodiev MD, Casari G, Rugarli EI, Langer T. Variable and tissue-specific subunit composition of mitochondrial m-AAA protease complexes linked to hereditary spastic paraplegia. Mol Cell Biol. 2007;27:758–67.
Acknowledgments
We thank the family members for participating in this study. We thank Dr. Alfredo Brusco for kindly sharing the in silico structure file describing the m-AAA protease with us. Finally, we thank The Novo Nordisk Foundation, The Aase and Ejnar Danielsen Foundation, Gangstedfonden, and The Lundbeck Foundation for financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The study was approved by the Ethics Committee of the Capital Region of Denmark (H-4-2011-157), and written informed consent was obtained from each participant before enrollment.
Conflict of Interest
The authors declare that they have no competing interests.
Additional information
Kirsten Svenstrup and Troels Tolstrup Nielsen contributed equally to this work.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 4413 kb)
Rights and permissions
About this article

Cite this article
Svenstrup, K., Nielsen, T.T., Aidt, F. et al. SCA28: Novel Mutation in the AFG3L2 Proteolytic Domain Causes a Mild Cerebellar Syndrome with Selective Type-1 Muscle Fiber Atrophy. Cerebellum 16, 62–67 (2017). https://doi.org/10.1007/s12311-016-0765-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12311-016-0765-1