
Abstract
Introduction
Biallelic mutations in STUB1, which encodes the E3 ubiquitin ligase CHIP, were originally described in association with SCAR16, a rare autosomal recessive spinocerebellar ataxia, so far reported in 16 kindreds. In the last 2 years, a new form of spinocerebellar ataxia (SCA48), associated with heterozygous mutations in the same gene, has been described in 12 kindreds with autosomal dominant inheritance.
Methods
We reviewed molecular and clinical findings of both SCAR16 and SCA48 described patients.
Results and conclusion
SCAR16 is characterized by early onset spastic ataxia and a wide disease spectrum, including cognitive dysfunction, hyperkinetic disorders, epilepsy, peripheral neuropathy, and hypogonadism. SCA48 is an adult-onset syndrome characterized by ataxia and cognitive-psychiatric features, variably associated with chorea, parkinsonism, dystonia, and urinary symptoms. SCA48, the last dominant ataxia to be described, could emerge as the most frequent among the SCAs due to conventional mutations. The overlap of several clinical signs between SCAR16 and SCA48 indicates the presence of a continuous clinical spectrum among recessively and dominantly inherited mutations of STUB1. Different kinds of mutations, scattered over the three gene domains, have been found in both disorders. Their pathogenesis and the relationship between SCA48 and SCAR16 remain to be clarified.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The STUB1 gene (STIP1 homology and U-box containing protein 1; OMIM, 607207) encodes CHIP, which is an E3 ubiquitin protein ligase. CHIP (C-terminus of HSC70-interacting protein) couples protein folding and proteasome-mediated degradation by interacting with heat shock proteins (HSPs) [1,2,3].
Since its discovery in 1999 [1], CHIP has been shown to play a pivotal role in regulating the cellular balance between protein folding and degradation. CHIP, which is primarily localized to the cytoplasm, is highly expressed in tissues with high metabolic activity and protein turnover [1], and aberrations in its expression and activity are observed in several pathological conditions, especially neurological and oncological diseases [3,4,5].
In 2013, biallelic mutations in STUB1 were associated with the autosomal recessive spinocerebellar ataxia type 16 (SCAR16) [6]. In the following years, SCAR16 has been reported in several families, with a phenotype characterized by early-onset ataxia associated with additional features such as spasticity, cognitive impairment, movement disorders, epilepsy, and occasionally hypogonadism, mimicking Gordon Holmes syndrome [7].
More recently [8], a heterozygous mutation in STUB1 has been associated with a new form of autosomal dominant spinocerebellar ataxia (SCA), named SCA48, in a Spanish kindred presenting with adult-onset ataxia and cognitive and affective symptoms. Interestingly, in SCAR16 families, carriers of one single STUB1 mutation were described as completely asymptomatic. Since the original description in 2018, eleven further SCA48 families have been reported, showing a complex phenotype characterized not only by ataxia and cognitive/behavioral dysfunction but also by a spectrum of movement disorders, encompassing both hypokinetic and hyperkinetic features [9,10,11]. The increasing number of reported patients and of different heterozygous STUB1 mutations (n = 10) supports the concept that SCA48 might be recognized worldwide as a not infrequent form of hereditary ataxia.
Here, we provide a review of the literature of STUB1-associated hereditary ataxias (SCAR16 and SCA48), with a special attention to the lastly described autosomal dominant form. Aims are to better characterize the clinical features of these ataxic disorders and to investigate on genotype-phenotype correlations, modalities of transmission, and possible neurodegeneration mechanisms.
SCAR16 phenotypic characteristics
SCAR16 is a rare autosomal recessive disorder due to homozygous or compound heterozygous mutations in STUB1 gene. The present review included 31 SCAR16 patients (15F, 16M) from 17 families reported in literature (Supplementary Table 1). Three additional patients have been reported [12, 13], but their clinical features have not been fully described, and they have not been included in the phenotype evaluation. Mean age of onset of symptoms was 20.4 ± 13.9 years, ranging from birth to 57 years. Initial symptom was ataxia in 23/31 patients, developmental delays in three, primary or secondary amenorrhea in three, and epilepsy or cognitive impairment in one each. During the disease course, all patients developed progressive cerebellar ataxia, associated with dysmetria and dysarthria. Abnormal ocular movements, mostly nystagmus, were present in 15 patients. Cognitive impairment (intellectual disability and/or deterioration) was reported in 21 patients, while corticospinal signs were present in 19, varying from increased tendon reflexes only (9) to severe tetraspasticity; Babinski signs were positive in six. Movement disorders were frequently described: Indeed, apart from action tremor that could be considered as a part of the cerebellar syndrome, 11 patients presented with hyperkinetic movement disorders, such as chorea (3), athetosis (3), dystonia (3), myoclonus (7), and ballism (2). Hypogonadism, which was a characteristic feature in one of the first SCAR16 reports [7], was observed in five patients from four families. Generalized tonic–clonic seizures (GTCS) were reported in four patients from three families. Psychiatric symptoms were reported only in two cases.
Electrophysiological studies of the central and/or peripheral nervous systems were conducted in 14 patients. Peripheral nerve involvement was found in 4/12 patients, abnormalities of Brainstem Auditory Evoked Potentials (BAEPs) in 3/5, of Visual Evoked Potentials (VEPs) in 3/3, of Motor Evoked Potentials (MEPs) in 3/8, and of Somatosensory Evoked Potentials (SSEPs) in 3/5.
MRI, performed in all but two subjects, showed the constant presence of cerebellar atrophy, sometimes associated with thin corpus callosum (3), atrophy of the pons or midbrain (5), or of the cerebral cortex (2).
A post-mortem study has been performed in one patient only. Neuropathological analysis showed severe loss of Purkinje cells and neurons of the granular layer, accompanied by reactive Bergmann gliosis. Staining with CHIP and HSP70 antibodies showed diffuse reactivity in neurons and astrocytes [14].
It is worthy of note that a patient with late age at onset (54 years) harbored two variants in STUB1: a small deletion leading to frameshift (c.678_679delCA, p.Ile227PhefsTer11), resulting in a prematurely truncated protein with defective or absent ubiquitin ligase activity, and a c.103C>A, p.Arg35Ser, a variant of uncertain significance upon in silico predictions [15]. Although family history was not informative, the global clinical phenotype in that patient, including cerebellar ataxia, cognitive deterioration, behavior abnormalities, generalized chorea, and absence of corticospinal signs, appears to be more similar to SCA48 than SCAR16.
SCA48 phenotypic characteristics
SCA48, the most recently described autosomal dominant spinocerebellar ataxia, is due to heterozygous mutations in the STUB1 gene. We reviewed the clinical and genetic characteristics of the reported 26 patients (18F, 8M), originating from one Spanish [8], ten Italian [9, 10], and one Turkish [11] kindreds (Supplementary Table 2). Mean age of onset was 42.3 ± 8.8 years, ranging from 22 to 56 years. Epilepsy cannot be included with certainty in SCA48 phenotype; therefore, to calculate age at onset, we did not consider as first symptom GTCS, which appeared in childhood and disappeared in adulthood in two patients from the same family (Family C). Initial symptoms were as follows: cerebellar dysfunction (ataxia and/or dysarthria) in 20/26 patients, cognitive and/or psychiatric dysfunction in nine, and choreic movements in one. Over the course of the disease, all patients presented cerebellar symptoms, while all but three showed cognitive impairment. Eye movement abnormalities, mostly broken smooth pursuit, were found in nine subjects and dysphagia in seven. Psychiatric features, mainly anxiety (n = 11), apathy (n = 6), and depression (n = 6), were present in 22 patients. Both hypokinetic and hyperkinetic movement disorders were common and often coexisted: Ten patients had mild parkinsonism, while 13 had chorea and eight dystonia. Hyperreflexia was reported in 12 patients but only in two cases a positive Babinski sign was present. Twelve patients had urge incontinence or other urinary tract symptoms. Three patients from the same family had a positive history for a mild epileptic syndrome (GTCS), for which they did not take any treatment. Hypogonadism was reported only in one patient, affected also by Addison’s disease and Hashimoto’s thyroiditis.
Electrophysiological studies of the central and/or peripheral nervous systems were conducted in 12 patients. Electromyography and peripheral nerve conduction studies did not show any abnormalities in 12 patients, as well as VEPs (n = 4), BAEPs (n = 4), and MEPs (n = 6). SSEPs were absent at lower limbs in two patients, normal in the remaining seven.
MRI was performed in 24 patients, and all showed vermian and hemispheric cerebellar atrophy, more pronounced at the level of the posterior areas [8]. Mild cortical atrophy was observed in 7 patients. A T2-weighted hyperintensity affecting the dentate nuclei and extending to the middle cerebellar peduncles has been demonstrated in seven patients [9, 10]. Diffusion tensor imaging in one patient revealed cerebello-frontal disconnection and dentate nuclei involvement [11].
FDG-PET was performed in six patients and showed glucose hypometabolism, marked in the cerebellum and less severe in the cerebral cortex and striata [9, 10]. 99mTc-HMPAO SPECT imaging showed cerebellar and cerebral cortical hypoperfusion in three studied patients [8, 11]. DaTSCAN detected a slightly reduced striatal uptake in two patients and was normal in one [9, 10].
Post-mortem studies are so far not available in SCA48 (see Addendum).
SCA48 prevalence, diffusion, and penetrance
It is interesting to note that, although SCA48 has been identified only recently, we could collect already clinical and molecular information from 12 published families. One study screened 235 Italian adult-onset patients (218 sporadic, 17 with autosomal dominant inheritance) with a cerebellar ataxia syndrome of unknown etiology [10]. All had tested negative for mutations in the most common ataxia genes (FRDA; SCA types 1, 2, 3, 6, 7, and 17; and FXTAS). The study identified eight unrelated probands (four with a positive family history and four sporadic) affected with SCA48, determining an overall frequency of 3.4%, which rose to 23.5% among familial cases. Considering that cerebellar ataxias of unknown etiology might represent about 48% of dominant ataxia cases [16], SCA48 could represent a significant proportion of all SCAs. A recent NGS study on autosomal dominant ataxias of unknown etiology, performed before SCA48 identification, found relevant genetic variants in 14% of cases, being mutations in CACNA1A the most frequent and accounting for 4% of them [12]. Therefore, SCA48 is likely the most frequent among the SCAs due to conventional mutations.
Another noteworthy point is that, among the families reported so far, only the mutation c.823_824delCT recurred in three families (A, K, M), whereas the remaining kindreds harbored private mutations. Lack of evidence of founder mutations in the described families supports the hypothesis that SCA48 might be identified worldwide.
Among the 12 reported index cases, occurrence of the disease was sporadic in four. This might suggest a reduced penetrance of some mutations or, most likely, missed diagnosis in relatives, considering the late-onset, the mild progression, and the variable presentation which could lead to the wrong diagnosis of dementia, psychiatric disorder, Parkinson’s disease, or chorea.
Comparison between SCA48 and SCAR16
Clinical and laboratory findings are compared in Table 1. As expected for autosomal dominant and recessive disorders, SCAR16 has an earlier onset compared with SCA48 (20.4 ± 13.9 vs 42.3 ± 8.8 years; p < 0.001). The female/male ratio was 0.94 in SCAR16 and 2.25 in SCA48, but this difference was not statistically significant. In both disorders, ataxia was the most common first symptom and subsequently developed in all patients. Cognitive dysfunction was the second most frequent feature either in SCAR16 (68%), often classified as intellectual disability, or SCA48 (88%), always described as cognitive decline. It seems somewhat surprising the low frequency of behavioral abnormalities reported in SCAR16 in comparison with SCA48 (6 vs 85%; p < 0.001), also in view of the well-known association between cognitive and psychiatric dysfunction in children and adolescents. Indeed, the prevalence of psychiatric disorders in young people with intellectual disability ranged from 31 to 72% [17, 18]. Probably, that is why the presence of behavior abnormalities has been just assumed and not mentioned explicitly in most SCAR16 papers.
Mild parkinsonism, not requiring a pharmacological treatment, was present in SCA48 only (38%), whereas hyperkinetic movement disorders were slightly and not significantly more frequent in SCA48 than SCAR16 (54 vs 35%). Myoclonus, however, seems to be specific of the autosomal recessive form.
Upper motor neuron syndrome is present in 61% of SCAR16 patients constituting an important element of the clinical picture. In SCA48, hyperreflexia was present in 46% of patients, but it was associated to Babinski sign in only one patient (4%), while another patient showed isolated Babinski sign (Supplementary Table 2). These findings suggest that corticospinal tract involvement in SCA48 is less relevant than in SCAR16.
Hypogonadism and epilepsy, occurring in four and three families respectively, are definite, although not frequent features of SCAR16. Conversely, their occurrence is sporadic (one family each) in SCA48, and their belonging to the disease spectrum needs to be confirmed by other reports. Neurophysiological studies were often not reported in the reviewed papers; however, evidence of peripheral neuropathy is present in 33% of SCAR16 patients and in none of the SCA48 patients. Similarly, neurophysiological evidence of central nervous system is often found in SCAR16 only.
MRI showed cerebellar atrophy in all investigated patients; therefore, it appears an obligatory feature of STUB1-related disorders. Cerebellar atrophy was found also in three SCA48 presymptomatic individuals [8], and likely it represents an early disease feature. MRI showed a T2-weighted hyperintense signal extending from the dentate nuclei to the middle cerebellar peduncles in seven SCA48 patients [9, 10]. This finding has not been reported in SCAR16 and, even though its evidence is limited to a small number of cases, could be a peculiar imaging biomarker of the dominant form.
In SCA48, PET and SPECT imaging revealed a pattern of hypometabolism/hypoperfusion involving not only the cerebellum but also, to a lesser extent, the cerebral cortex and the striatum. The pre-synaptic dopaminergic system was normal or only minimally affected. Functional imaging studies are lacking in SCAR16.
SCA48 and the cerebellar cognitive affective syndrome
Signs of cognitive impairment were present in the majority of SCA48 patients, ranging from a mild or moderate dysexecutive syndrome to a marked and diffuse deterioration. The multi-domain cognitive impairment, which included also language impairment, disorder of spatial cognition, and behavioral changes, might suggest a cerebellar cognitive affective syndrome (CCAS). This condition has been first described by Schmahmann and Sherman on 20 patients with diseases confined to the cerebellum [19]. The pattern of abnormalities in CCAS involves executive function, visual-spatial cognition, linguistic performance, and personality change with blunting of affect or disinhibited and inappropriate behavior. The syndrome was clinically prominent in patients with lesions involving the posterior lobe of the cerebellum. CCAS may be due to disruption of neural circuitry linking the posterior cerebellar cortex with cerebral cortical association areas and paralimbic regions involved in higher order cognitive processing [20].
The pattern of cognitive and psychiatric changes seen in SCA48 patients might resemble CCAS, although anosognosia, memory loss [8,9,10], and palilalia [11], which are not part of the CCAS, have been described. The more pronounced involvement of cerebellar posterior areas [8] and cerebello-frontal disconnection seen by DTI studies [11] supports the hypothesis that CCAS might match SCA48 cognitive dysfunction. On the other side, several individuals affected with SCA48 showed no cognitive decline up to 30 years after disease onset, and the original description of CCAS refers to pure cerebellar lesions, whereas SCA48 appears to be a widespread neurodegenerative disorder, encompassing cortical, subcortical, and infratentorial areas. Therefore, disruption of the frontostriatal circuitry, and degeneration of the cholinergic basal forebrain nuclei, as well of the neurons of the association cortical areas themselves, may be also implicated in the genesis of cognitive dysfunctions [20]. In our opinion, the relationship between the cerebellum and cognitive dysfunction in SCA48 deserves further studies.
SCA48 and SCA17
Cognitive deficits are generally uncommon and late in the most common SCAs due to expanded polyglutamine tracts [20]. Their frequency has been estimated to be 5–19% in SCA2 and lower in SCA1, SCA3, and SCA6. On the contrary, dementia is an important aspect of the clinical picture in SCA17 and DRPLA, two other polyglutamine-related ataxias.
With regard to movement disorders, they are not rare in SCAs. Their prevalence during overall disease course ranged from 5 to 24% for parkinsonism, from 1 to 9% for chorea, and from 4 to 24% for dystonia in SCA1, SCA2, SCA3, and SCA6 [21]. However, excluding DRPLA, which is a very rare disease in Western countries, SCA17 is the form more often associated with parkinsonism (79%), chorea (48%), and dystonia (53%).
SCA17 is a neurodegenerative disease caused by the expansion above 43–45 units of a CAG/CAA repeat in the coding region of the TATA box-binding protein (TBP) gene [22]. SCA17 shows a complex and variable clinical phenotype, in some cases overlapping that of Huntington’s disease and, for this reason, it is also called Huntington’s disease-like 4 (HDL-4). Disease onset is typically in the third or fourth decade; the presenting symptoms may be either psychiatric disturbances, such as behavioral changes and mood alterations, or neurological features, including ataxia and movement disorders [23]. Cognitive impairment is a frequent (77%) and early complain [24]. The clinical triad of ataxia, movement disorders, and cognitive/affective symptoms underpins both SCA48 and SCA17; however, the latter seems to be a more severe condition, with earlier onset (34.6 ± 13.2 years), more frequent corticospinal signs (57%), and epilepsy (35%) [24]. Indeed, it is already known that compared with polyglutamine ataxias, the forms caused by conventional mutations are more slowly progressive, have a longer survival, and lead to less severe disability [25].
Here we would emphasize the similarity between SCA48 and SCA17, and we propose that, for its association with dementia, psychiatric symptoms, and sometimes chorea, also SCA48 may fall within HD-like syndromes.
STUB1 and the encoded CHIP protein: functions and molecular pathways
STUB1 encodes CHIP, a 35 kDa enzyme involved in protein quality control that acts as ubiquitin ligase, chaperone, and cochaperone binding heat shock proteins [1, 2, 26]. In human, CHIP is mainly expressed in the striated muscle, pancreas, brain, and cerebellum, while minor expression is found in other tissues. Similarly, STUB1 is widely expressed in mouse brain including Purkinje cells and cerebellum [6].
CHIP is composed by three functional domains: an N-terminal tetratricopeptide repeat (TPR) domain for cochaperone activity, a C-terminal U-box domain for ubiquitination activity as E3 ligase, and an intermediate coiled-coil (CC) domain required for CHIP dimerization [4, 27]. TPR domain mainly interacts with heat shock proteins HSC70, HSP70, and HSP90, binding and guiding them to support the refolding of misfolded proteins, whereas U-box domain binds an E2 ubiquitin-conjugating enzyme that ubiquitinates unfolded proteins leading them to proteasome-driven degradation [28, 29].
CHIP hence plays a protective role due to its chaperone, cochaperone, and ubiquitination activity that aims to prevent the neurodegeneration caused by abnormal unfolded protein accumulation [30]. Indeed, CHIP upregulation in vivo displayed an attenuation of tau aggregation [31], and CHIP deficiency has shown to lead early aging in mice [32].
Aggregation of misfolded proteins and defects in protein quality control system are a common and well-known theme in neurodegenerative disorders [33, 34] and nevertheless in hereditary ataxias [35]. For instance, mutations in RNF216 and OTUD4, coding an E3 ubiquitin ligase and a deubiquitinase, respectively, were found to cause, similarly to STUB1, ataxia and hypogonadism [36], likewise later confirmed by other reports which described recessive mutations in RNF216 [37,38,39,40,41,42]. Nonetheless, CHIP was found to control the decay of expanded proteins like ataxin-1 [43] and ataxin-3 [44], involved in SCA1 and SCA3 [45, 46]. Furthermore, other proteins involved in neurodegenerative diseases interact with CHIP. For instance, parkin and α-synuclein, involved in Parkinson’s disease, were found to be targets of CHIP [47].
Impairment of protein quality control caused by CHIP deficiency was investigated through generation of a Stub1−/− knock-out (KO) mouse. Early work in KO mice, which show accelerated cellular senescence, increased oxidative stress, toxic accumulation of proteins, and deterioration of proteasome activity, demonstrated that CHIP is an essential regulator of mammalian longevity via the regulation of protein quality control [32]. Subsequently, it was shown that Stub1−/− mice present severe impairments in motor, sensory, and cognitive functions, in particular in tasks related to cerebellar function [7]. Indeed, complete loss of CHIP resulted in degeneration of Purkinje cells. Moreover, evident defects in behavioral and reproductive functions were noticed, hence suggesting that Stub1−/− mice well represent the human SCAR16 phenotype [7].
Interestingly, motor impairments were found in Stub1+/- mice [48] together with an altered subset of behavior, hence providing that CHIP underexpression leads to defects in neuronal circuitry. Noteworthy, the milder phenotype of Stub1+/- mice could reflect the differences between SCA48 and the more severe SCAR16.
CHIP is also involved in mitophagy [49] and autophagy [50]. The impairment of the autophagic process might represent a potential pathogenetic mechanism by regulating the activity of specific transcription factors and/or mitochondrial biogenesis [51, 52], a feature likewise observed in KO mice [53]. Nonetheless, also the CHIP-mutated driven impairment of the endoplasmic reticulum protein quality control [54], significant in the homeostasis of the autophagy-lysosome pathway, might have a role in the etiology of STUB1-related disorders.
STUB1 mutations and patterns of inheritance
Pakdaman and collaborators analyzed in a heterologous system some of the missense mutations associated with SCAR16 and found that those variants can cause defect in both protein structure and stability and ubiquitination activity, as well as alter CHIP dimerization [55]. Similarly, Kanack and collaborators showed that point mutations causing SCAR16 cause several and variable defects in CHIP functions, often correlated with the corresponding domain where the mutation belongs to, despite this is not a mandatory correlation [56]. For instance, they found that mutations in TPR domain hamper the ability to bind chaperones as expected, but in a variable severity level and, in some cases, also hindering the ubiquitination activity at the same time [56]. This could be explained by the fact that cochaperone binding is a required signal to trigger ubiquitination activity. Besides these defects, they revealed an increased tendency to form soluble oligomers, a lower thermal stability, and a reduction of protein expression, thence confirming that, in addition to nonsense and frameshift mutations, also missense mutations can induce a reduction, although variable, of CHIP levels and impair the activity of residual protein [56]. Interestingly, they also noticed that cellular thermodynamic conditions may have a pivotal role in CHIP function and activity, suggesting that specific CHIP functions are active or inactive depending on the cellular stress condition [56]. Even more recently, Madrigal and collaborators investigated how functional properties of STUB1 missense correlate with different clinical features [57]. They found that mutations in U-box domain have severe effects on the overall CHIP functions and were tightly related to cognitive dysfunction in SCAR16 patients, hence proposing that mutations in certain CHIP domains could bring to specific phenotypic manifestations [57]. The limited number of monoallelic missense variants and the lack of functional investigations on protein function and structural features prevented similar speculations on genotype-phenotype correlations in SCA48.
How STUB1-related ataxias can act through different pattern of transmission is yet to be clarified, as well as why carrier parents in SCAR16 families result, to our knowledge, to be healthy. However, this is not a novel issue that research worldwide must deal with. Indeed, genes with both autosomal dominant and recessive inheritance have been recorded, particularly in neurodegenerative diseases. In fact, this feature characterizes several genes known to be cause of at least one specific type of SCA/SCAR or hereditary spastic paraplegia, as well as genes known to be cause of disorders in which ataxia and/or spasticity are listed among main features (Supplementary Table 3), thus suggesting that zygosity should not be anymore a limit in the definition of a molecular diagnosis. Integration of multiomic data could serve for improved understanding of genotype-phenotype correlations.
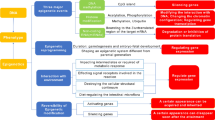
To date, 13 STUB1 mutations have been described in SCA48 and 30 in SCAR16. All types of mutations have been reported, and all widespread along the three functional domains, though only one SCA48 mutation (p.R225*) is placed in the coiled-coil domain, just at the border with the U-box domain (Fig. 1). Despite hitherto clinical reports and both in vitro and in vivo functional studies all converge to loss-of-function mechanisms underlying SCAR16, the processes that might explain the dominant inheritance are yet to be explained, and functional studies are required to clarify this issue. A potential CHIP gain of function may be considered, as well as dominant negative effect, given the fact that CHIP acts as dimer. Anyhow, work in STUB1+/- mice [48] suggests that haploinsufficiency could be the mechanism underlying SCA48.

Scheme of STUB1 gene and CHIP protein with its domains. SCAR16 mutations are reported in black, SCA48 mutations in red. #, p.Y230Cfs9* has been found in both SCA48 and SCAR16
Conclusion
STUB1 encodes for CHIP, a ubiquitin ligase involved in protein quality control, mitophagy, and autophagy, and has a protective role in preventing neurodegeneration related to abnormal unfolded protein accumulation.
Monoallelic mutations in STUB1 lead to SCA48, the last described autosomal dominant form, a complex adult-onset syndrome characterized by ataxia and cognitive-psychiatric disorder, variably associated with chorea, parkinsonism, dystonia, and urinary symptoms. To this multifaceted phenotype corresponds a widespread degeneration pattern, with involvement of the cerebral cortex, the basal ganglia, and the cerebellum. Given the different modalities of presentation of SCA48, the diagnosis may be missed, especially in sporadic cases. The clinical presentation of SCA48 is somewhat close to that of SCA17, since in both ataxia is associated with cognitive, psychiatric, and movement disorders.
Mutations in STUB1 can also cause SCAR16, an autosomal recessive spastic ataxia with onset in children or young adults, and a wide disease spectrum, including cognitive dysfunction, hyperkinetic disorders, epilepsy, peripheral neuropathy, and hypogonadism. The overlap of several clinical signs of SCAR16 and SCA48 indicates the presence of a continuous clinical spectrum among recessively and dominantly inherited mutations (Fig. 2).

Overlap of clinical features in SCAR16 and SCA48
In both SCA48 and SCAR16, different kinds of mutations have been found, and mutations are scattered throughout the entire gene, although only one SCA48 mutation is placed in the coiled-coil domain. The clear pathogenesis of these disorders, the relationship between them, and how the type and location of STUB1 mutations contribute to the clinical expression remain to be clarified.
Data availability
The data supporting the findings of this study are available within the article and its supplementary materials.
References
Ballinger CA, Connell P, Wu Y, Hu Z, Thompson LJ, Yin LY, Patterson C (1999) Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol 19:4535–4545. https://doi.org/10.1128/mcb.19.6.4535
Jiang J, Ballinger CA, Wu Y, Dai Q, Cyr DM, Höhfeld J, Patterson C (2001) CHIP is a U-box-dependent E3 ubiquitin ligase: identification of Hsc70 as a target for ubiquitylation. J Biol Chem 276:42938–42944. https://doi.org/10.1074/jbc.M101968200
Heimdal K, Sanchez-Guixé M, Aukrust I, Bollerslev J, Bruland O, Jablonski GE, Erichsen AK, Gude E, Koht JA, Erdal S, Fiskerstrand T, Haukanes BI, Boman H, Bjørkhaug L, Tallaksen CM, Knappskog PM, Johansson S (2014) STUB1 mutations in autosomal recessive ataxias - evidence for mutation-specific clinical heterogeneity. Orphanet J Rare Dis 9:146. https://doi.org/10.1186/s13023-014-0146-0
Nikolay R, Wiederkehr T, Rist W, Kramer G, Mayer MP, Bukau B (2004) Dimerization of the human E3 ligase CHIP via a coiled-coil domain is essential for its activity. J Biol Chem 279:2673–2678. https://doi.org/10.1074/jbc.M311112200
Cao Z, Li G, Shao Q, Yang G, Zheng L, Zhang T, Zhao Y (2016) CHIP: a new modulator of human malignant disorders. Oncotarget 7:29864–29874. https://doi.org/10.18632/oncotarget.8219
Shi Y, Wang J, Li JD, Ren H, Guan W, He M, Yan W, Zhou Y, Hu Z, Zhang J, Xiao J, Su Z, Dai M, Wang J, Jiang H, Guo J, Zhou Y, Zhang F, Li N, Du J, Xu Q, Hu Y, Pan Q, Shen L, Wang G, Xia K, Zhang Z, Tang B (2013) Identification of CHIP as a novel causative gene for autosomal recessive cerebellar ataxia. PLoS One 8:e81884. https://doi.org/10.1371/journal.pone.0081884
Shi CH, Schisler JC, Rubel CE, Tan S, Song B, McDonough H, Xu L, Portbury AL, Mao CY, True C, Wang RH, Wang QZ, Sun SL, Seminara SB, Patterson C, Xu YM (2014) Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP. Hum Mol Genet 23:1013–1024. https://doi.org/10.1093/hmg/ddt497
Genis D, Ortega-Cubero S, San Nicolás H, Corral J, Gardenyes J, de Jorge L, López E, Campos B, Lorenzo E, Tonda R, Beltran S, Negre M, Obón M, Beltran B, Fàbregas L, Alemany B, Márquez F, Ramió-Torrentà L, Gich J, Volpini V, Pastor P (2018) Heterozygous STUB1 mutation causes familial ataxia with cognitive affective syndrome (SCA48). Neurology. 91:e1988–e1998. https://doi.org/10.1212/WNL.0000000000006550
De Michele G, Lieto M, Galatolo D, Salvatore E, Cocozza S, Barghigiani M, Tessa A, Baldacci J, Pappatà S, Filla A, De Michele G, Santorelli FM (2019) Spinocerebellar ataxia 48 presenting with ataxia associated with cognitive, psychiatric, and extrapyramidal features: a report of two Italian families. Parkinsonism Relat Disord 65:91–96. https://doi.org/10.1016/j.parkreldis.2019.05.001
Lieto M, Riso V, Galatolo D, De Michele G, Rossi S, Barghigiani M, Cocozza S, Pontillo G, Trovato R, Saccà F, Salvatore E, Tessa A, Filla A, Santorelli FM, De Michele G, Silvestri G (2019) The complex phenotype of spinocerebellar ataxia type 48 in eight unrelated Italian families. Eur J Neurol 65:91–96. https://doi.org/10.1111/ene.14094
Palvadeau R, Kaya-Güleç ZE, Şimşir G, Vural A, Öztop-Çakmak Ö, Genç G, Aygün MS, Falay O, Başak AN, Ertan S (2019) Cerebellar cognitive-affective syndrome preceding ataxia associated with complex extrapyramidal features in a Turkish SCA48 family. Neurogenetics. 21:51–58. https://doi.org/10.1007/s10048-019-00595-0
Coutelier M, Coarelli G, Monin ML, Konop J, Davoine CS, Tesson C, Valter R, Anheim M, Behin A, Castelnovo G, Charles P, David A, Ewenczyk C, Fradin M, Goizet C, Hannequin D, Labauge P, Riant F, Sarda P, Sznajer Y, Tison F, Ullmann U, Van Maldergem L, Mochel F, Brice A, Stevanin G, Durr A, SPATAX network (2017) A panel study on patients with dominant cerebellar ataxia highlights the frequency of channelopathies. Brain. 140:1579–1594. https://doi.org/10.1093/brain/awx081
Sun M, Johnson AK, Nelakuditi V, Guidugli L, Fischer D, Arndt K, Ma L, Sandford E, Shakkottai V, Boycott K, Chardon JW, Li Z, Del Gaudio D, Burmeister M, Gomez CM, Waggoner DJ, Das S (2019) Targeted exome analysis identifies the genetic basis of disease in over 50% of patients with a wide range of ataxia-related phenotypes. Genet Med 21:195–206. https://doi.org/10.1038/s41436-018-0007-7
Bettencourt C, de Yébenes JG, López-Sendón JL, Shomroni O, Zhang X, Qian SB, Bakker IM, Heetveld S, Ros R, Quintáns B, Sobrido MJ, Bevova MR, Jain S, Bugiani M, Heutink P, Rizzu P (2015) Clinical and neuropathological features of spastic ataxia in a Spanish family with novel compound heterozygous mutations in STUB1. Cerebellum. 14:378–381. https://doi.org/10.1007/s12311-014-0643-7
Gazulla J, Izquierdo-Alvarez S, Sierra-Martínez E, Marta-Moreno ME, Alvarez S (2018) Inaugural cognitive decline, late disease onset and novel STUB1 variants in SCAR16. Neurol Sci 39:2231–2233. https://doi.org/10.1007/s10072-018-3545-5
Durr A (2010) Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol 9:885–894. https://doi.org/10.1016/S1474-4422(10)70183-6
Einfeld SL, Piccinin AM, Mackinnon A, Hofer SM, Taffe J, Gray KM, Bontempo DE, Hoffman LR, Parmenter T, Tonge BJ (2006) Psychopathology in young people with intellectual disability. JAMA. 296:1981–1989. https://doi.org/10.1001/jama.296.16.1981
Theodoratos O, McPherson L, Franklin C, Tonge B, Einfeld S, Lennox N, Ware RS (2017) Psychopathology of adolescents with an intellectual disability who present to general hospital services. Australas Psychiatry 25:481–485. https://doi.org/10.1177/1039856217706820
Schmahmann JD, Sherman JC (1998) The cerebellar cognitive affective syndrome. Brain. 121:561–579. https://doi.org/10.1093/brain/121.4.561
Lindsay E, Storey E (2017) Cognitive changes in the spinocerebellar ataxias due to expanded polyglutamine tracts: a survey of the literature. Brain Sci 7:83. https://doi.org/10.3390/brainsci7070083
Rossi M, Perez-Lloret S, Cerquetti D, Merello M (2014) Movement disorders in autosomal dominant cerebellar ataxias: a systematic review. Mov Disord Clin Pract 1:154–160. https://doi.org/10.1002/mdc3.12042
Nakamura K, Jeong SY, Uchihara T, Anno M, Nagashima K, Nagashima T, Ikeda S, Tsuji S, Kanazawa I (2001) SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet 10:1441–1448. https://doi.org/10.1093/hmg/10.14.1441
De Michele G, Maltecca F, Carella M, Volpe G, Orio M, De Falco A, Gombia S, Servadio A, Casari G, Filla A, Bruni A (2003) Dementia, ataxia, extrapyramidal features, and epilepsy: phenotype spectrum in two Italian families with spinocerebellar ataxia type 17. Neurol Sci 24:166–167. https://doi.org/10.1007/s10072-003-0112-4
Stevanin G, Brice A (2008) Spinocerebellar ataxia 17 (SCA17) and Huntington's disease-like 4 (HDL4). Cerebellum. 7:170–178. https://doi.org/10.1007/s12311-008-0016-1
Monin ML, Tezenas du Montcel S, Marelli C, Cazeneuve C, Charles P, Tallaksen C, Forlani S, Stevanin G, Brice A, Durr A (2015) Survival and severity in dominant cerebellar ataxias. Ann Clin Transl Neurol 2:202–207. https://doi.org/10.1002/acn3.156
Rosser MF, Washburn E, Muchowski PJ, Patterson C, Cyr DM (2007) Chaperone functions of the E3 ubiquitin ligase CHIP. J Biol Chem 282:22267–22277. https://doi.org/10.1074/jbc.M700513200
Ronnebaum SM, Patterson C, Schisler JC (2014) Emerging evidence of coding mutations in the ubiquitin-proteasome system associated with cerebellar ataxias. Hum Genome 1:14018. https://doi.org/10.1038/hgv.2014.18
Shin Y, Klucken J, Patterson C, Hyman BT, McLean PJ (2005) The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J Biol Chem 280:23727–23734. https://doi.org/10.1074/jbc.M503326200
Qian SB, McDonough H, Boellmann F, Cyr DM, Patterson C (2006) CHIP-mediated stress recovery by sequential ubiquitination of substrates and Hsp70. Nature. 440:551–555. https://doi.org/10.1038/nature04600
Miller VM, Nelson RF, Gouvion CM, Williams A, Rodriguez-Lebron E, Harper SQ, Davidson BL, Rebagliati MR, Paulson HL (2005) CHIP suppresses polyglutamine aggregation and toxicity in vitro and in vivo. J Neurosci 25:9152–9161. https://doi.org/10.1523/JNEUROSCI.3001-05.2005
Sahara N, Murayama M, Mizoroki T, Urushitani M, Imai Y, Takahashi R, Murata S, Tanaka K, Takashima A (2005) In vivo evidence of CHIP up-regulation attenuating tau aggregation. J Neurochem 94:1254–1263. https://doi.org/10.1111/j.1471-4159.2005.03272.x
Min JN, Whaley RA, Sharpless NE, Lockyer P, Portbury AL, Patterson C (2008) CHIP deficiency decreases longevity, with accelerated aging phenotypes accompanied by altered protein quality control. Mol Cell Biol 28:4018–4025. https://doi.org/10.1128/MCB.00296-08
Ross CA, Poirier MA (2004) Protein aggregation and neurodegenerative disease. Nat Med 10(Suppl):10–17. https://doi.org/10.1016/j.ejmech.2016.07.054
Williams AJ, Paulson HL (2008) Polyglutamine neurodegeneration: protein misfolding revisited. Trends Neurosci 31:521–528. https://doi.org/10.1016/j.tins.2008.07.004
Synofzik M, Puccio H, Mochel F, Schöls L (2019) Autosomal recessive cerebellar ataxias: paving the way toward targeted molecular therapies. Neuron. 101:560–583. https://doi.org/10.1016/j.neuron.2019.01.049
Margolin DH, Kousi M, Chan YM, Lim ET, Schmahmann JD, Hadjivassiliou M, Hall JE, Adam I, Dwyer A, Plummer L, Aldrin SV, O'Rourke J, Kirby A, Lage K, Milunsky A, Milunsky JM, Chan J, Hedley-Whyte ET, Daly MJ, Katsanis N, Seminara SB (2013) Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N Engl J Med 368:1992–2003. https://doi.org/10.1056/NEJMoa1215993
Sawyer SL, Schwartzentruber J, Beaulieu CL, Dyment D, Smith A, Warman Chardon J, Yoon G, Rouleau GA, Suchowersky O, Siu V, Murphy L, Hegele RA, Marshall CR; FORGE Canada Consortium, Bulman DE, Majewski J, Tarnopolsky M, Boycott KM (2014) Exome sequencing as a diagnostic tool for pediatric-onset ataxia. Hum Mutat 35:45–49. https://doi.org/10.1002/humu.22451
Santens P, Van Damme T, Steyaert W, Willaert A, Sablonnière B, De Paepe A, Coucke PJ, Dermaut B (2015) RNF216 mutations as a novel cause of autosomal recessive Huntington-like disorder. Neurology. 84:1760–1766. https://doi.org/10.1212/WNL.0000000000001521
Ganos C, Hersheson J, Adams M, Bhatia KP, Houlden H (2015) The 4H syndrome due to RNF216 mutation. Parkinsonism Relat Disord 21:1122–1123. https://doi.org/10.1016/j.parkreldis.2015.07.012
Alqwaifly M, Bohlega S (2016) Ataxia and hypogonadotropic hypogonadism with Intrafamilial variability caused by RNF216 mutation. Neurol Int 8:6444. https://doi.org/10.4081/ni.2016.6444
Calandra CR, Mocarbel Y, Vishnopolska SA, Toneguzzo V, Oliveri J, Cazado EC, Biagioli G, Turjanksi AG, Marti M (2019) Gordon Holmes Syndrome caused by RNF216 novel mutation in 2 Argentinean siblings. Mov Disord Clin Pract 6:259–262. https://doi.org/10.1002/mdc3.12721
Lieto M, Galatolo D, Roca A, Cocozza S, Pontillo G, Fico T, Pane C, Saccà F, De Michele G, Santorelli FM, Filla A (2019) Overt Hypogonadism may not be a sentinel sign of RING finger protein 216: two novel mutations associated with ataxia, chorea, and fertility. Mov Disord Clin Pract 6:724–726. https://doi.org/10.1002/mdc3.12839
Al-Ramahi I, Lam YC, Chen HK, de Gouyon B, Zhang M, Pérez AM, Branco J, de Haro M, Patterson C, Zoghbi HY, Botas J (2006) CHIP protects from the neurotoxicity of expanded and wild-type ataxin-1 and promotes their ubiquitination and degradation. J Biol Chem 281:26714–26724
Jana NR, Dikshit P, Goswami A, Kotliarova S, Murata S, Tanaka K, Nukina N (2005) Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J Biol Chem 280:11635–11640. https://doi.org/10.1074/jbc.M412042200
Orr HT, Chung MY, Banfi S, Kwiatkowski TJ Jr, Servadio A, Beaudet AL, McCall AE, Duvick LA, Ranum LP, Zoghbi HY (1993) Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet 4:221–226. https://doi.org/10.1038/ng0793-221
Takiyama Y, Sakoe K, Soutome M, Namekawa M, Ogawa T, Nakano I, Igarashi S, Oyake M, Tanaka H, Tsuji S, Nishizawa M (1997) Single sperm analysis of the CAG repeats in the gene for Machado-Joseph disease (MJD1): evidence for non-Mendelian transmission of the MJD1 gene and for the effect of the intragenic CGG/GGG polymorphism on the intergenerational instability. Hum Mol Genet 6:1063–1068. https://doi.org/10.1093/hmg/6.7.1063
Dickey CA, Patterson C, Dickson D, Petrucelli L (2007) Brain CHIP: removing the culprits in neurodegenerative disease. Trends Mol Med 13:32–38. https://doi.org/10.1016/j.molmed.2006.11.003
McLaughlin B, Buendia MA, Saborido TP, Palubinsky AM, Stankowski JN, Stanwood GD (2012) Haploinsufficiency of the E3 ubiquitin ligase C-terminus of heat shock cognate 70 interacting protein (CHIP) produces specific behavioral impairments. PLoS One 7:e36340. https://doi.org/10.1371/journal.pone.0036340
Lizama BN, Palubinsky AM, Raveendran VA, Moore AM, Federspiel JD, Codreanu SG, Liebler DC, McLaughlin B (2018) Neuronal preconditioning requires the mitophagic activity of C-terminus of HSC70-interacting protein. J Neurosci 38:6825–6840. https://doi.org/10.1523/JNEUROSCI.0699-18.2018
Guo D, Ying Z, Wang H, Chen D, Gao F, Ren H, Wang G (2015) Regulation of autophagic flux by CHIP. Neurosci Bull 31:469–479. https://doi.org/10.1007/s12264-015-1543-7
Rao L, Sha Y, Eissa NT (2017) The E3 ubiquitin ligase STUB1 regulates autophagy and mitochondrial biogenesis by modulating TFEB activity. Mol Cell Oncol 4(6):e1372867. https://doi.org/10.1080/23723556.2017.1372867
Sha Y, Rao L, Settembre C, Ballabio A, Eissa NT (2017) STUB1 regulates TFEB-induced autophagy-lysosome pathway. EMBO J 36:2544–2552. https://doi.org/10.15252/embj.201796699
Wei Q, Sha Y, Bhattacharya A, Abdel Fattah E, Bonilla D, Jyothula SS, Pandit L, Khurana Hershey GK, Eissa NT (2014) Regulation of IL-4 receptor signaling by STUB1 in lung inflammation. Am J Respir Crit Care Med 189:16–29. https://doi.org/10.1164/rccm.201305-0874OC
Matsumura Y, Sakai J, Skach WR (2013) Endoplasmic reticulum protein quality control is determined by cooperative interactions between Hsp/c70 protein and the CHIP E3 ligase. J Biol Chem 288:31069–31079. https://doi.org/10.1074/jbc.M113.479345
Pakdaman Y, Sanchez-Guixé M, Kleppe R, Erdal S, Bustad HJ, Bjørkhaug L, Haugarvoll K, Tzoulis C, Heimdal K, Knappskog PM, Johansson S, Aukrust I (2017) In vitro characterization of six STUB1 variants in spinocerebellar ataxia 16 reveals altered structural properties for the encoded CHIP proteins. Biosci Rep 37. https://doi.org/10.1042/BSR20170251
Kanack AJ, Newsom OJ, Scaglione KM (2018) Most mutations that cause spinocerebellar ataxia autosomal recessive type 16 (SCAR16) destabilize the protein quality-control E3 ligase CHIP. J Biol Chem 293:2735–2743. https://doi.org/10.1074/jbc.RA117.000477
Madrigal SC, McNeil Z, Sanchez-Hodge R, Shi CH, Patterson C, Scaglione KM, Schisler JC (2019) Changes in protein function underlie the disease spectrum in patients with CHIP mutations. J Biol Chem 294:19236–19245. https://doi.org/10.1074/jbc.RA119.011
Chen DH, Latimer C, Yagi M, Ndugga-Kabuye MK, Heigham E, Jayadev S, Meabon JS, Gomez CM, Keene CD, Cook DG, Raskind WH, Bird TD (2020) Heterozygous STUB1 missense variants cause ataxia, cognitive decline, and STUB1 mislocalization. Neurol Genet 6:1–13. https://doi.org/10.1212/NXG.0000000000000397
Mol MO, van Rooij JGJ, Brusse E, Verkerk AJMH, Melhem S, den Dunnen WFA, Rizzu P, Cupidi C, van Swieten JC, Donker Kaat L (2020) Clinical and pathologic phenotype of a large family with heterozygous STUB1 mutation. Neurol Genet 6:e417. https://doi.org/10.1212/NXG.0000000000000417
Synofzik M, Schüle R, Schulze M, Gburek-Augustat J, Schweizer R, Schirmacher A, Krägeloh-Mann I, Gonzalez M, Young P, Züchner S, Schöls L, Bauer P (2014) Phenotype and frequency of STUB1 mutations: next-generation screenings in Caucasian ataxia and spastic paraplegia cohorts. Orphanet J Rare Dis 9:57. https://doi.org/10.1186/1750-1172-9-57
Depondt C, Donatello S, Simonis N, Rai M, van Heurck R, Abramowicz M, D'Hooghe M, Pandolfo M (2014) Autosomal recessive cerebellar ataxia of adult onset due to STUB1 mutations. Neurology. 82:1749–1750. https://doi.org/10.1212/WNL.0000000000000416
Cordoba M, Rodriguez-Quiroga S, Gatto EM, Alurralde A, Kauffman MA (2014) Ataxia plus myoclonus in a 23-year-old patient due to STUB1 mutations. Neurology. 83:287–288. https://doi.org/10.1212/WNL.0000000000000600
Kawarai T, Miyamoto R, Shimatani Y, Orlacchio A, Kaji R (2016) Choreoathetosis, dystonia, and myoclonus in 3 siblings with autosomal recessive spinocerebellar ataxia type 16. JAMA Neurol 73:888–890. https://doi.org/10.1001/jamaneurol.2016.0647
Hayer SN, Deconinck T, Bender B, Smets K, Züchner S, Reich S, Schöls L, Schüle R, De Jonghe P, Baets J, Synofzik M (2017) STUB1/CHIP mutations cause Gordon Holmes syndrome as part of a widespread multisystemic neurodegeneration: evidence from four novel mutations. Orphanet J Rare Dis 12:31. https://doi.org/10.1186/s13023-017-0580-x
Turkgenc B, Sanlidag B, Eker A, Giray A, Kutuk O, Yakicier C, Tolun A, Temel SG (2018) STUB1 polyadenylation signal variant AACAAA does not affect polyadenylation but decreases STUB1 translation causing SCAR16. Hum Mutat 39:1344–1348. https://doi.org/10.1002/humu.23601
Acknowledgements
The study was partially supported by a grant from the Italian Association for Ataxic Syndromes (AISA) to A.F.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
We have no financial interests in this manuscript.
Ethical approval
None.
Human and animal rights and informed consent
This article is a review of the literature; therefore, it did not involve human participants and did not need informed consent.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Giovanna De Michele and Daniele Galatolo shared the first position.
Appendix
Appendix
Addendum
After submission, two articles have been published, describing three novel SCA48 families, two from the USA [58] and one from the Netherlands [59]. The papers confirm the complex phenotype of the disease and add valuable information on its neuropathology. Both studies describe marked loss of Purkinje cells (PC) with Bergmann gliosis, and both did not find TDP-43 or alpha-synuclein positive inclusions. The Dutch study also revealed severe neuronal loss in the mesencephalon and medulla oblongata. Aberrant STUB1 localization in the distal PC dendritic arbors [58] and ubiquitin/p62-positive neuronal inclusions in the cerebellum, neocortex, and brainstem [59] have been also described.
Rights and permissions
About this article

Cite this article
De Michele, G., Galatolo, D., Barghigiani, M. et al. Spinocerebellar ataxia type 48: last but not least. Neurol Sci 41, 2423–2432 (2020). https://doi.org/10.1007/s10072-020-04408-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-020-04408-3