
Abstract
Myeloid and lymphoid neoplasms with eosinophilia (M/Ls-Eo) encompass heterogeneous but aggressive hematopoietic disorders triggered by fusion genes or mutations that typically lead to constitutive overexpression of tyrosine kinase. The occurrence of T-lymphoblastic lymphoma in the setting of M/Ls-Eo has been reported rarely in the literature. Herein, we present an unusual case of a 28-year-old male patient who presented with massive lymphadenopathy and T-lymphoblastic lymphoma in the lymph node occurring concurrently with myeloid hyperplasia, eosinophilia and basophilia in peripheral blood and bone marrow biopsy. The syndrome was associated with a novel complex karyotype involving der(8)t(1;8;10)(p31;q24;q11.2). The FISH study was negative for BCR::ABL1, JAK2, PDGFRA, PDGFRB, and FGFR1 rearrangements. The patient’s clinical course was aggressive and resistant to multiple lines of intensive chemotherapy regimens. Therefore, he underwent allogenic stem cell transplantation with a fully matched donor. A brief review of the occurrence of T-LBL in conjunction with M/Ls-Eo neoplasm was made with a special focus on molecular aspects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
T-Lymphoblastic lymphoma (T-LBL) is an aggressive malignancy that arises from the neoplastic transformation of immature precursor T cells and is characterized by the proliferation of T lymphoblasts in bone marrow, thymus, nodal and extra-anodal sites [1, 2]. On rare occasions, T-LBL occurs in the setting of myeloid and lymphoid neoplasm with eosinophilia (M/Ls-Eo). Such syndromes are highly aggressive and potentially lead to a diagnostic dilemma [3, 4].
Here we describe an unusual case of myeloid and lymphoid neoplasm with massive lymphadenopathy and T-lymphoblastic lymphoma in the lymph node occurring concurrently with myeloid hyperplasia, eosinophilia and basophilia in peripheral blood and bone marrow biopsy. Moreover, it was associated with a novel complex karyotype and aggressive clinical course.
Patient and methods
Case report
A 28-year-old male patient presented to our hospital with a 3-week history of bilateral extensive and massive cervical, axillary and inguinal lymphadenopathy preceded by 3 months of mild sweating and loss of 9 kg of body weight. His past medical and family history were unremarkable. During the physical examination, no skin manifestations or hepatosplenomegaly were observed.
Laboratory investigations revealed a hemoglobin of 12 g/dL, platelet count was 626 × 109/L, and white cell count (WBC) of 74.8 × 109/L, with neutrophils of 42.8 × 109/L, lymphocytes were 6.9 × 109/L, monocytes were 7.9 × 109/L, eosinophils were 11.5 × 109/L and basophils were 5.7 × 109/L. Peripheral blood smear examination showed marked myeloid hyperplasia with left-shifted granulocytes, eosinophilia, basophilia, and monocytosis. The circulating blasts were 2%.
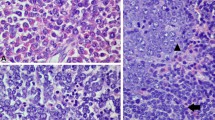
Both bone marrow aspirate and trephine biopsy yielded a markedly hypercellular bone marrow (100% cellularity) with myeloid hyperplasia and prominent eosinophilia (44%) without any significant dysplastic changes. Blasts were 4% with myeloid phenotype and no aggregates of immature-looking or lymphoid cells were detected (Fig. 1A).

Morphology and Immunohistochemistry findings of bone marrow and lymph node biopsy. A BM biopsy is markedly hypercellular with myeloid hyperplasia and prominent eosinophilia. B Lymph node biopsy is diffusely infiltrated with monotonous small to medium immature-looking cells. C IHC stains show strong positivity for CD3 (C) and TDT (D)
A morphological diagnosis of chronic myeloid leukemia in chronic phase (CML-CP) was suspected and the patient was started on a short course of dasatanib 50 mg. However, BCR::ABL1 (p210) mRNA transcript level was evaluated using Xpert BCR::ABL1 ultra Assay (Cepheid) and was negative. The initial morphological suspicion of CML was thus re-evaluated.
A right submandibular lymph node biopsy was performed and revealed diffuse infiltration with monotonous small to medium immature-looking cells (Fig. 1B). The immunohistochemistry stains (GA-R2; Ventana Medical System, Inc.) showed strong positivity for CD1A, CD2, CD3, CD4, CD5, CD7, and TDT while Ki 67 was 90%. (Fig. 1 C and D). They were negative for CD8, CD30, CD156, CD20, CD34, and granzyme B. The final report of lymph node biopsy was consistent with acute T-lymphoblastic lymphoma (T-LBL).
Chromosomal karyotyping was performed on 20 metaphases that demonstrated the presence of complex translocation involving der(8)t(1;8;10)(p31;q24;q11.2) in 100% of the analyzed cells (Fig. 2). An extended panel of fluorescent in situ hybridization (FISH) was carried out on the bone marrow cells and yielded negative results for BCR::ABL1, JAK2, ETV::RUNX1, KMT2A gene, C-MYC gene, TCF3::PBX1, IGH::IL3, TCR gene, PDGFRA, PDGFRB, and FGFR1. Bone marrow specimen was sent out to a reference laboratory for repeating the FISH study for PDGFRA, PDGFRB, and FGFR1 rearrangements, the results of which were within normal limits for all genes.

Complex karyotype with 3 chromosomal aberrations: 46,XY,der(8)t(1;8;10)(p31;q24;q11.2)
CT scan confirmed the presence of widespread lymphadenopathy including mediastinal and abdominal lymph nodes with increased metabolic activity in the PET scan. There was a heterogenous increase in bone marrow tracer uptake as well.
The final report was released as a myeloid and lymphoid neoplasm with T-LBL, eosinophilia, basophilia, and der(8)t(1;8;10)(p31;q24;q11.2) translocation.
Therapy and hospital course
The patient was started on induction treatment according to Dana Farber Consortium Protocol (DFCP) with vincristine, doxorubicin, prednisone, asparaginase, and methotrexate. He had a good response within the first week with complete disappearance of the lymphadenopathy and normalization of the blood counts. Post-induction PET scan showed a complete metabolic response. He received CNS treatment and intensification as well.
Three months later, the patient presented with generalized lymphadenopathy, leukocytosis, eosinophilia, and thrombocytosis. A repeat bone marrow aspirate and trephine biopsy showed marked hypercellularity with granulocytic hyperplasia, eosinophilia (23%), and 11% of T lymphoblasts confirmed immunophenotypically and immunohistochemically. The cytogenetic analysis found the same abnormal translocation in 65% of cells. The PET scan also confirmed the relapsed disease.
He received fludarabine, cytarabine, granulocyte colony-stimulating factor, and idarubicin (FLAG-IDA) chemotherapy followed by complete hematological resolution of the disease. The CT scan confirmed the disease’s resolution. However, cytogenetic analysis detected the persistence of the complex translocation t (1,8,10) in 5% of the analyzed cells indicating the persistence of molecular disease despite morphological remission.
The patient then underwent an allogeneic hematopoietic stem cell transplantation (HSCT). The conditioning regimen included fludarabine, etoposide, and total body irradiation. Additionally, he also received ATG with cyclosporine as graft-versus-host disease (GVHD) prophylaxis. The patient was engrafted on day 17 with 100% chimerism on day 60 post-allogeneic HSCT. In the first few weeks, his post-transplant course was associated with nausea and dry mouth with features of mild gut and liver GVHD. Tapering of cyclosporine was delayed to day 100. He was started on imatinib 100 mg daily, Prednisolone 20 mg and artificial saliva, antiviral, and antifungal prophylaxis.
The patient had one episode of neutropenia and pneumonia requiring hospital admission and GCSF support. PET scan at 12 months post-transplant showed no disease. His immunosuppressive therapy was gradually tapered. His chimerism studies at 12 months post-transplant showed full donor chimerism. His lung function and bone densitometry tests were within normal limits.
Discussion
Myeloid and lymphoid neoplasms with eosinophilia (M/Ls-Eo) encompass heterogeneous but aggressive hematopoietic disorders derived from pluripotent lymphoid/myeloid stem cells. The pathogenesis of M/Ls-Eo is triggered by fusion genes or mutations that typically lead to constitutive overexpression of tyrosine kinases [5]. According to the 5th edition of the WHO classification of hematolymphoid tumors, this category includes M/Ls-Eo with either PDGFRA, PDGFRB, FGFR1, JAK2, FLT3 rearrangement, ETV6, ABL1 fusion gene or with other tyrosine kinase fusion genes [6, 7]. The neoplasm can present as chronic eosinophilic leukemia, myeloproliferative neoplasms (MPN), myelodysplastic syndrome/MPN, and acute myeloid or lymphoid leukemia. Eosinophilia is a constant feature reported in more than 90% of the cases while basophilia is an uncommon finding [8].
Our patient presented with an MPN picture associated with both eosinophilia and basophilia in the bone marrow along with massive lymphadenopathy and T-LBL in lymph nodes simultaneously. Basophilia has been observed more with certain genetic abnormalities such as t(1;8)(q31;p11.2) resulting in TPR-FGFR1 fusion gene and (8;22)(p11.2;q11) resulting in BCR-FGFR1 gene.
The occurrence of T-LBL concurrent with M/Ls-Eo has been reported rarely in the literature [3, 4] Zanelli et.al reported 11 cases of this neoplasm in which T-LBL diagnosis was made in conjunction with (M/Ls-Eo). The majority of the patients (6/11) had shown FGFR1 gene rearrangement with either ZMYM2-FGFR1 or BCR-FGFR1 fusion genes. Two cases of T-LBL were associated with the FIP1L1-PDGFRA fusion gene and one case was associated with PCM1::JAK2 fusion gene. In the same report, two additional cases were confirmed to have MPN with eosinophilia and T-LBL by both bone marrow and lymph node biopsies however, no cytogenetic or molecular abnormalities were detected [3].
Roger et.al described two patients with a syndrome of T-LBL, eosinophilia, and myeloid hyperplasia associated with t(8;13) (p11;q11). Both patients presented with lymphadenopathy and peripheral eosinophilia [4].
In the current case, the syndrome was associated with a novel complex karyotype composed of a derivative of chromosome 8 and (1;8;10)(p31;q24;q11.2) translocation. To the best of our knowledge, no such genetic abnormalities have been reported previously.
The 10q11.2 encodes RET protooncogene, which is a tyrosine kinase surface receptor, that regulates cellular proliferation and survival through activation of the Raf/Mek/ERK1/2 cascade and PI3K/Akt signal transduction pathway respectively [9]. Two tyrosine kinase fusion genes have been recently recognized by the 5th edition of the WHO classification of hematolymphoid tumors under the category of myeloid/lymphoid neoplasms with tyrosine kinase fusion genes (NOS). The fusion genes are FGFR1OP::RET and BCR::RET resulting from t(6;10)(q27;q11), and t(10;22)(q11;q11) balanced translocation, respectively [6, 10]. No evidence of such balanced translocations was detected in the current case despite the presence of a complex karyotype with three chromosomal aberrations. Conventional karyotype revealed that 10q11.2 has translocated and moved to 8q24 along with 1p31. High-throughput techniques such as RNA sequencing are required to elucidate the nature of the potential RET fusion gene resulting from such novel translocation [11].
M/Ls-Eo with FGFR1 rearmament is an aggressive disorder with rapid blastic transformation to either T lymphoid, myeloid, or bi-phenotypic acute leukemia. 8p11 translocation is the genetic defining event involving the gene of FGFR 1 tyrosine kinase and its multiple partner genes and thereby leading to constitutive activation of the oncogenic signal transduction pathway [12]. So far, 14 different reciprocal translocations have been linked to the syndrome, out of which, t(8;13) (p11;q12) resulting in the ZMYM2::FGFR1 fusion gene being the most common genetic abnormality [2, 13, 14]. The TPR-FGFR1 fusion gene arising from t(1;8)(q31;p11.2) is the least common chromosomal translocation reported only in two cases to the best of our knowledge [15,16,17].
Clinically, M/Ls-Eo with T-LBL is characterized by refractory disease with poor outcome even with intensive chemotherapy. The majority of the reported cases had shown poor responses to different chemotherapy regimens [3, 4, 18]. Moreover, the response to the available tyrosine kinase inhibitors is still suboptimal except for PGFRA and PDGFRB rearrangements. Therefore, all-HSCT is the only therapeutic modality to achieve long-term disease-free survival [6].
Similarly, the clinical course of our patient was aggressive and resistant to intensive chemotherapy regimens. The patient relapsed shortly, within 3 months, after the initial response to the DFCP chemotherapy and re-inducted again with the FLAG-IDA regimen. Despite there being a complete hematological and radiological response, the cytogenetic study revealed the persistence of the disease. Therefore, he underwent allogenic stem cell transplantation with a fully matched donor.
Conclusion
The diagnosis of M/Ls-Eo with T-LBL might be quite peculiar and challenging due to the bi-phenotypic nature of the syndrome especially when associated with an aggressive clinical course and novel complex translocation. High throughput NGS technology such as RNA sequencing provides clear insight and a more detailed view of the implicated gene rearrangement. Furthermore, additional scientific reports might help to identify the resulting fusion gene from such a novel and complex translocation.
Data Availability
The data availability and consent for publication are provided to the journal.
References
Litzow MR, Ferrando AA (2015) How I treat T-cell acute lymphoblastic leukemia in adults. Blood 126(7):833–841
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J (eds) (2017) World Health Organization classification of tumours of haematopoietic and lymphoid tissues. Revised 4th ed. Lyon: IARC
Zanelli M, Loscocco GG, Sabattini E et al (2021) T-Cell lymphoblastic lymphoma arising in the setting of myeloid/lymphoid neoplasms with eosinophilia: LMO2 immunohistochemistry as a potentially useful diagnostic marker. Cancers (Basel) 13(12):3102
Inhorn RC, Aster JC, Roach SA et al (1995) A syndrome of lymphoblastic lymphoma, eosinophilia, and myeloid hyperplasia/malignancy associated with t(8;13)(p11;q11): description of a distinctive clinicopathologic entity. Blood 85(7):1881–1887
Bain BJ (2010) Myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1. Haematologica 95(5):696–698
Chan J, Khoury JD (eds) (2022) WHO Classification of Haematolymphoid Tumours 5th edn. Accessed online on 12–10–2022
Cree IA (2022) The WHO classification of haematolymphoid tumours. Leukemia 36(7):1701–1702
Reiter A, Gotlib J (2017) Myeloid neoplasms with eosinophilia. Blood 129(6):704–714
Pacini F, Cantara S (2016) Chapter 10 - Molecular diagnosis of thyroid cancer. In: Weiss RE, Refetoff S (eds) Genetic diagnosis of endocrine disorders, 2nd edn. Academic Press, p 153–162. ISBN 9780128008928
Ballerini P, Struski S, Cresson C et al (2012) RET fusion genes are associated with chronic myelomonocytic leukemia and enhance monocytic differentiation. Leukemia 26(11):2384–2389
Telford N, Alexander S, McGinn OJ, Williams M, Wood KM, Bloor A, Saha V (2016) Myeloproliferative neoplasm with eosinophilia and T-lymphoblastic lymphoma with ETV6-LYN gene fusion. Blood Cancer J 6(4):e412
Antic DA, Vukovic VM, Milosevic Feenstra JD, Kralovics R, Bogdanovic AD, DencicFekete MS, Mihaljevic BS (2016) 8p11 myeloproliferative syndrome: diagnostic challenges and pitfalls. J BUON 21(3):745–9
Kasbekar M, Nardi V, Dal Cin P et al (2020) Targeted FGFR inhibition results in a durable remission in an FGFR1-driven myeloid neoplasm with eosinophilia. Blood Adv 4:3136–3140
Cowell JK, Hu T (2021) Mechanisms of resistance to FGFR1 inhibitors in FGFR1-driven leukemias and lymphomas: implications for optimized treatment. Cancer Drug Resist 4:607–619
Yoshida C, Takeuchi M, Sadahira Y (2012) A novel t(1;8)(q25;p11.2) translocation associated with 8p11 myeloproliferative syndrome. Br J Haematol 156:271–273
Li F, Zhai YP, Tang YM, Wang LP, Wan PJ (2012) Identification of a novel partner gene, TPR, fused to FGFR1 in 8p11 myeloproliferative syndrome. Genes Chromosomes Cancer 51(9):890–897
Savage N, George TI, Gotlib J (2013) Myeloid neoplasms associated with eosinophilia and rearrangement of PDGFRA, PDGFRB, and FGFR1: a review. Int J Lab Hematol 35:491–500
Abruzzo LV, Jaffe ES, Cotelingam JD, Whang-Peng J, Del Duca JV, Medeiros LJ (1992) T-cell lymphoblastic lymphoma with eosinophilia associated with subsequent myeloid malignancy. Am J Surg Pathol 16:236–245
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was received.
Conflict of interest
The authors declare no competing interests.
Ethical approval
The institute board review IRB was obtained for this study.
Informed consent
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Aljabry, M.S. Myeloid and lymphoid neoplasm with novel complex translocation: unusual case report with T-lymphoblastic lymphoma, myeloid hyperplasia, eosinophilia, basophilia, and t(1;8;10)( (p31;q24;q11.2). J Hematopathol 16, 27–31 (2023). https://doi.org/10.1007/s12308-022-00528-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12308-022-00528-1