
Abstract
Background
A cyclin-dependent kinase (CDK) 4/6 inhibitor, palbociclib, has been used to treat patients with estrogen receptor (ER)-positive (+) and human epidermal growth factor receptor (HER) 2-negative (−) advanced breast cancer. To investigate the mechanisms underlying the antitumor activity of palbociclib, we conducted a preclinical study on the anti-cell growth and anti-cancer stem cell (CSC) activity of palbociclib in breast cancer cells.
Methods
The effects of palbociclib on Rb phosphorylation, cell growth, cell cycle progression, apoptosis, cell senescence and the proportion of CSCs were investigated in five human breast cancer cell lines of different subtypes. To investigate the mechanisms of the anti-CSC activity of palbociclib, small-interfering RNAs for CDK4 and/or CDK6 were used. Palbociclib dose-dependently reduced Rb phosphorylation and cell growth in association with G1-S cell cycle blockade and the induction of cell senescence, but without increased apoptosis, in all breast cancer cell lines.
Results
The anti-cell growth activity of palbociclib widely differed among the cell lines. Palbociclib also dose-dependently reduced the CSC proportion measured by three different assays in four of five cell lines. The inhibition of CDK4 expression, but not CDK6 expression, reduced the increased proportion of putative CSCs induced by estradiol in ER (+)/HER2 (−) cell lines.
Conclusions
These results suggest that palbociclib exhibits significant anti-cell growth and anti-CSC activity in not only ER (+) breast cancer cell lines but also ER (−) cell lines. CDK4 inhibition induced by palbociclib may be responsible for its anti-CSC activity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The cyclin D-cyclin-dependent kinase (CDK) 4/6-E2F signaling pathway plays an essential role in the G1-S cell cycle transition in both normal cells and tumor cells. Dysregulation of this pathway is common in cancer cells, including breast cancer cells. In particular, deregulated transcription and/or amplification of the CCND1 gene, and constitutive activation of the cyclin D-CDK4/6 complex involving genetic and/or epigenetic inactivation of the p16 are frequently observed in breast cancer. Furthermore, cyclin D1 is one of the target genes induced by the estrogen receptor (ER) pathway, which is activated in most breast cancers. Based on these findings, a new treatment strategy inhibiting both cyclin D-CDK4/6-E2F and ER pathways has been introduced at clinics to treat ER-positive (+) and human epidermal growth factor receptor (HER) 2-negative (−) breast cancer [1, 2].
Three specific CDK4/6 inhibitors, palbociclib, ribociclib and abemaciclib, have been developed and introduced at clinics, and palbociclib is the leading compound. A number of clinical trials revealed that the combined treatment of palbociclib with endocrine therapy, such as the aromatase inhibitor letrozole and steroidal antiestrogen fulvestrant, prolonged the progression-free survival (PFS) in patients with ER (+)/HER2 (−) advanced breast cancer [1]. Furthermore, these combination therapies may prolong the overall survival in some breast cancer patients [3]. There are several ongoing clinical trials using palbociclib with different inhibitors of signal transduction, such as anti-HER2 agents and PI3K inhibitors, or immune-check point inhibitors such as an anti-PD-1 antibody [1]. These studies have suggested palbociclib to be a useful antitumor agent for the treatment of not only ER (+)/HER2 (−) breast cancer but also breast cancer of other subtypes.
According to preclinical studies, CDK4/6 inhibitors have at least four distinct action mechanisms responsible for their antitumor activity. The first mechanism is maintaining cells in a quiescent state induced by G1-S blockade. The second is cell senescence after cell cycle arrest, which makes cells unable to enter the cell cycle. The third mechanism is changing cellular metabolism and inducing apoptosis. The fourth is immune-modulatory effects by enhancing the antigenicity of the tumor cells and suppressing the negative regulatory cells [1]. In addition, it has been suggested that CDK4/6 inhibitors reduce the proportion of cancer stem cells (CSCs) via inhibition of CSC self-renewal [4]. The reduction of CSCs in cancer tissues may prolong the antitumor response to CDK4/6 inhibitors.
Understanding the action mechanisms responsible for the antitumor activity of CDK4/6 inhibitors is important to develop new combined treatment strategies for the management of breast cancer patients. Therefore, we conducted a systematic study to investigate the action mechanisms of palbociclib in breast cancer cells of different subtypes. In particular, the anti-CSC effects and molecular mechanisms of palbociclib remain to be elucidated. Therefore, we evaluated its anti-CSC activity using three different CSC assays. Additionally, we conducted knock-down of CDK4 and/or CDK6 expression using respective small interfering (si) RNA to clarify which CDK is responsible for the anti-CSC effects of palbociclib.
Materials and methods
Reagents
Palbociclib was obtained from AdooQ BIOSCIENCE (Irvine, CA, USA). 17β-estradiol (E2) was obtained from Sigma-Aldrich (St. Louis, MO, USA).
Breast cancer cell lines and cell culture
KPL-1 and KPL-4 breast cancer cell lines were established at our laboratory. Their biological characteristics were described previously [5, 6]. MCF-7 and MDA-MB-231 cell lines were provided by the late Dr. Robert B. Dickson (Lombardi Cancer Research Center, Washington DC, USA). The BT-474 cell line was purchased from the American Type Culture Collection (Manassas, VA, USA). All cell lines were maintained in Dulbecco’s modified Eagle’s medium (D-MEM, Sigma Co.) supplemented with 10% fetal bovine serum (FBS). We and others previously demonstrated that MCF-7 and KPL-1 cells express a high level of ER-α but no detectable HER2, BT-474 cells express a high level of both ER-α and HER2, KPL-4 cells express a high level of HER2 but no detectable ER-α, and MDA-MB-231 cells express neither ER-α nor HER2.
Western blot analysis
Cells were lysed for protein extraction using Pierce RIPA Buffer with protease inhibitor and phosphatase inhibitor (Thermo Fisher Scientific, Waltham, MA, USA). The total protein concentration was measured using the Pierce BCA Protein Assay kit (Thermo Fisher Scientific). Isolated proteins were separated by 5–20% SDS-PAGE and transferred to an Amersham Hybond PVDF (GE Healthcare UK, Buckinghamshire, UK). Membranes were blocked with blocking buffer at room temperature for 1 h and then subjected to immunoblotting using primary antibodies at 4 °C overnight, followed by incubation with secondary antibodies at room temperature for 1 h. Labeled protein was visualized using the ECL Prime Western Blotting Detection Reagent (GE Healthcare Japan, Tokyo, Japan) with the expression of β-actin as the internal standard. The expression levels were measured using Quantity One 1-D software ver.4.5 (BIORAD, Tokyo, Japan) [7].
Rabbit antibodies against Rb (mAb #9313) and phosphorylated Rb (mAb #8516) were purchased from Cell Signaling Technologies (Danvers, MA, USA). Mouse polyclonal antibody against β-actin was from Sigma-Aldrich. Secondary antibodies, goat anti-rabbit lgG-HRP and goat anti-mouse lgG-HRP, were purchased from Santa Cruz Biotechnology (Dallas, Texas, USA).
Anti-cell growth activity
To examine cell growth, breast cancer cells were seeded on 24-well plates (SB Medical, Tokyo, Japan) and grown in D-MEM supplemented with 10% FBS at 37 ℃ in a 5% CO2 atmosphere for 1 day. After washing with phosphate-buffered saline (PBS, Nissui Co., Tokyo, Japan), the cells were cultured in estrogen-deprived medium consisting of phenol red-free RPMI1640 (Life Technologies, Carlsbad, CA, USA) supplemented with 5% dextran-coated charcoal-treated FBS (GE Healthcare HyClone, Tokyo, Japan) plus or minus 1 nM E2 and the indicated concentrations of palbociclib for 3 days. Thereafter, the cells were harvested and counted with a Coulter counter (Coulter Electronics, Harpenden, UK) [7].
Cell cycle and apoptosis assays
To investigate cell cycle progression, harvested cells were stained with propidium iodide using the CycleTest Plus DNA Reagent kit (Becton–Dickinson, San Jose, CA, USA). Apoptotic cells were stained using an Annexin-V-FLUOS staining kit (Roche Diagnostics GmbH, Penzberg, Germany) according to the manufacturer’s recommendations. Flow cytometry was performed on a FACSCalibur flow cytometer (Becton–Dickinson), and the DNA histogram was analyzed using CELLQuest version 6.0 (Becton–Dickinson) [7].
Cell senescence assay
Senescence was measured by the SA-β gal staining kit (Millipore, Billerica, MA, USA) according to the manufacturer’s protocol. In brief, cells were plated at a low density of 2000–4000 cells in each well of 12-well plates, and treated with 1 nM E2 and/or the indicated concentrations of palbociclib for 72 h. Cells were then washed with PBS, fixed and stained with SA-β gal solution for 4 h or overnight. Senescent cells were quantified by counting 100 cells in three different fields for each replicate [8].
CSC analysis by the CD44/CD24 assay
To analyze cell surface markers, harvested cells were treated with two fluorescence-labeled antibodies: a PE-conjugated anti-CD24 antibody (clone G44-26, Becton–Dickinson) and FITC-conjugated anti-CD44 antibody (clone L5, Becton–Dickinson). Flow cytometry was performed using a FACSCalibur flow cytometer (Becton–Dickinson), and analyzed using CELLQuest Software version 6.0 (Becton–Dickinson). Cells that expressed CD44 high and CD24 low were considered CSCs [7].
CSC analysis by the Aldefluor assay
The ALDEFLUOR kit (StemCell Technologies, Durham, NC, USA) was used to isolate the cell population exhibiting strong aldehyde dehydrogenase (ALDH) activity. Harvested cells were suspended in Aldefluor assay buffer containing ALDH substrate (BODIPYTM-aminoacetaldehyde) in duplicate and incubated at 37 ℃ for 30 min. As a negative control, cells were treated with 50 mM diethylaminobenzaldehyde, a specific ALDH inhibitor [7].
CSC analysis by the mammosphere assay
Breast cancer cells were seeded on 35-mm dishes (SB Medical) and grown in D-MEM supplemented with 10% FBS at 37 ℃ in a 5% CO2 atmosphere for 2 days. After washing with PBS, cells were treated with the estrogen-deprived medium supplemented with 1 nM E2 and the indicated concentrations of palbociclib for 3 days. These cells were then dispersed, and single-cell suspensions (5 × 103 cells/well) were incubated in MammoCult basal medium (StemCell Technologies Co.) supplemented with 10% MammoCult proliferation supplements (StemCell Technologies Co.) in non-adhesive 6-well plates (CORNING Co., NY, USA) for 7 days. Mammospheres larger than 60 μm in size were counted using an Olympus phase-contrast microscope [7].
RNA isolation and quantitative reverse-transcription polymerase chain reaction (RT-PCR)
The total RNA from the cells was extracted using an RNeasy MiniKit (QIAGEN GmbH, Hilden, Germany) according to the manufacturer’s instructions, and cDNA synthesis was performed using a ReverTra Ace qPCR RT kit (TOYOBO, Tokyo, Japan). Quantitative real-time PCR analysis of CDK4 and CDK6 mRNA was performed on cDNA using TaqMan gene expression assays according to the manufacturer’s instructions (Applied Biosystems, Life Technologies, Waltham, MA, USA) and a 7500 Real-Time PCR System (Applied Biosystems). Each amplification reaction was performed in duplicate, and the average of the two threshold cycles was used to calculate the amount of transcripts in the sample. The mRNA quantification was expressed, in arbitrary units, as the ratio of the sample quantity to the calibrator or to the mean values of the control samples. All values were normalized to an endogenous control, ACTB [7].
Transfection of siRNA for CDK4 and CDK6
Cells were seeded in 6-well plates and incubated at 37 °C for 1 day. Then, the cells were treated with the siRNA solution with or without 1 nM E2 for 3 days. The siRNA solution consisted of the estrogen-deprived medium with CDK4 and/or CDK6 siRNA at a final concentration of 25 pmol/well and 7.5 μl of lipofectamine/well. All siRNA transfection reagents and siRNAs were purchased from Thermo Fischer Scientifics, and we followed the experimental protocol provided by the manufacturer.
Statistical analysis
All values are expressed as mean ± SE. Analysis of variance with StatView computer software (ATMS Co., Tokyo, Japan) was used to compare differences between two groups. A two-sided P value less than 0.05 was considered significant.
Results
Inhibitory effects of palbociclib on Rb phosphorylation
The expression levels of phosphorylated Rb were increased by E2 but dose-dependently reduced by palbociclib in ER (+)/HER2 (−) MCF-7 and KPL-1 and ER (+)/HER2 (+) BT-474 cell lines (Online Resource 1). Similarly, they were dose-dependently reduced by palbociclib in ER (−)/HER2 (+) KPL-4 and ER (−)/HER2 (−) MDA-SMB-231 cell lines (Online Resource 1). In contrast, the expression level of Rb was not significantly altered by palbociclib or E2 in all cell lines tested (Online Resource 1).
Anti-cell growth effects of palbociclib
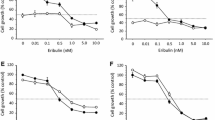
In estrogen-dependent MCF-7 cells, palbociclib dose-dependently inhibited the cell growth stimulated by E2 but not in the estrogen-deprived medium (Fig. 1a). In estrogen-sensitive but estrogen-independent KPL-1 cells, palbociclib dose-dependently inhibited cell growth in either E2-added or estrogen-deprived medium (Fig. 1b). In ER (+)/HER2 (+) BT-474 cells, palbociclib modestly and dose-dependently reduced cell growth in either E2-added or estrogen-deprived medium (Fig. 1c). In ER (−)/HER2 (+) KPL-4 and ER (−)/HER2 (−) MDA-MB-231 cells, palbociclib dose-dependently inhibited cell growth (Fig. 1d, e).

Anti-cell growth effects of palbociclib in MCF-7 (a), KPL-1 (b), BT-474 (c), KPL-4 (d) and MDA-MB-231 (e) cell lines. All cell lines were cultured in the estrogen-deprived medium plus (filled circle) or minus (unfilled circle) 1 nM E2 supplemented with 0–20 μM palbociclib for 3 days. The values are expressed as mean ± SE of the % control
The mean ± SE of the 50% growth inhibitory concentration of palbociclib (μM) was 0.21 ± 0.06 for MCF-7 cells, 0.10 ± 0.05 for KPL-1 cells, 11.28 ± 2.75 for BT-474 cells, 0.50 ± 0.18 for KPL-4 cells and 0.20 ± 0.05 for MDA-MB-231 cells, respectively. These concentrations significantly differed among the cell lines.
Effects of palbociclib on cell cycle progression and apoptosis
In ER (+) MCF-7, KPL-1 and BT-474 cells, E2 promoted the G1-S transition but palbociclib dose-dependently inhibited the G1-S transition stimulated by E2 (Online Resource 2). In ER (−) KPL-4 and MDA-MB-231 cells, palbociclib dose-dependently inhibited the G1-S transition (Online Resource 2). In contrast, E2 significantly reduced the fraction of apoptotic cells in ER (+) /HER2 (−) cell lines, but palbociclib did not significantly affect this fraction in any cell line (Online Resource 3).
Effects of palbociclib on cell senescence
In ER (+) MCF-7, KPL-1 and BT-474 cells, E2 significantly reduced the number of β-Gal-positive cells, whereas palbociclib dose-dependently increased their number (Fig. 2a–c). In ER (−) KPL-4 and MDA-MB-231 cells, palbociclib dose-dependently increased the number of β-Gal-positive cells (Fig. 2d, e). Representative microphotographs for MCF-7 cells are shown in Fig. 2f.

Effects of palbociclib on cell senescence in MCF-7 (a), KPL-1 (b), BT-474 (c), KPL-4 (d) and MDA-MB-231 (e) cell lines. Values are mean ± SE. White bars, control; the lightest grey bars, E2 alone; the second lightest grey bars, E2 plus 50 or 100 nM palbociclib; and the third lightest grey bars, E2 plus 100 or 500 nM palbociclib. *P < 0.05; **P < 0.01 in comparison with cells treated with or without E2. Representative microphotographs for MCF-7 cells are shown in f
Effects of palbociclib on the proportion of putative CSCs
According to the CD44/CD24 assay, E2 markedly increased the proportion of CD44-high/CD24-low cells in all ER (+) cell lines, but palbociclib dose-dependently reduced the proportion in MCF-7 and KPL-1 cells but not in BT-474 cells (Fig. 3a–c). In ER (−) KPL-4 and MDA-MB-231 cells, palbociclib dose-dependently reduced the proportion (Fig. 3d, e).

Effects of palbociclib on the proportion of CD44-high/CD24-low cells in MCF-7 (a), KPL-1 (b), BT-474 (c), KPL-4 (d) and MDA-MB-231 (e) cell lines. The values are mean ± SE. White bars, control; the lightest grey bars, E2 alone; the second lightest grey bars, E2 plus 50 or 100 nM palbociclib; and the third lightest grey bars, E2 plus 100 or 500 nM palbociclib. *P < 0.05; **P < 0.01 in comparison with cells treated with E2 alone in a–c, and with control cells in d and e
According to the Aldefluor assay, E2 increased the proportion of ALDH1-positive cells, whereas palbociclib dose-dependently reduced this proportion in ER (+)/HER2 (−) MCF-7 and KPL-1 cells (Fig. 4a, b). In contrast, neither E2 nor palbociclib altered the proportion in ER (+)/HER2 (+) BT-474 cells (Fig. 4c). In ER (−) KPL-4 and MDA-MB-231 cells, palbociclib dose-dependently reduced this proportion (Fig. 4d, e).

Effects of palbociclib on the proportion of ALDH1-positive cells in MCF-7 (a), KPL-1 (b), BT-474 (c), KPL-4 (d) and MDA-MB-231 (e) cell lines. The values are mean ± SE. White bars, control; the lightest grey bars, E2 alone; the second lightest grey bars, E2 plus 50 or 100 nM palbociclib; and the third lightest grey bars, E2 plus 100 or 500 nM palbociclib. *P < 0.05; **P < 0.01 in comparison with cells treated with E2 alone in a–c, and with control cells in d and e
According to the mammosphere assay, E2 increased the number of mammospheres, but palbociclib dose-dependently reduced the number in all ER (+)/HER2 (−) cell lines (Fig. 5a, b). In contrast, palbociclib did not affect the number in the ER (+)/HER2 (+) BT-474 cells (Fig. 5c). In ER (−) KPL-4 and MDA-MB-231 cells, palbociclib dose-dependently reduced the number (Fig. 5d, e).

Effects of palbociclib on the number of mammospheres/1000 cells seeded in MCF-7 (a), KPL-1 (b), BT-474 (c), KPL-4 (d) and MDA-MB-231 (e) cell lines. The values are mean ± SE. White bars, control; the lightest grey bars, E2 alone; the second lightest grey bars, E2 plus 50 or 100 nM palbociclib; and the third lightest grey bars, E2 plus 100 or 500 nM palbociclib. *P < 0.05; **P < 0.01 in comparison with cells treated with E2 alone in a–c, and with control cells in d and e
Effects of CDK4 and/or CDK6 knock-down on the proportion of putative CSCs
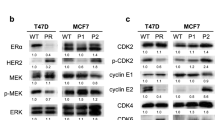
To further explore the mechanisms underlying the anti-CSC effects of palbociclib, the changes in the proportion of CD44-high/CD24-low CSCs were measured after the knock-down of CDK4 and/or CDK6 using the respective siRNAs. In ER (+)/HER2 (−) MCF-7 and KPL-1 cell lines, each siRNA specifically inhibited the respective mRNA expression level in either E2-treated or non-treated cells (Fig. 6a–d). Of note, E2-treatment reduced CDK6 expression in ER (+)/HER2 (−) cells (Fig. 6b, d).

Effects of siRNA for CDK4 and/or siRNA for CDK6 on mRNA expression of CDK4 and CDK6 in MCF-7 (a, b) and KPL-1 (c, d) cells. *P < 0.05; **P < 0.01 in comparison with control cells
CDK4 inhibition alone and combined CDK4 and CDK6 inhibition, but not CDK6 inhibition alone, significantly reduced the increased proportion of CSCs induced by E2 in ER (+)/HER2 (−) MCF-7 and KPL-1 cells (Fig. 7a, b). In contrast, no significant change was observed in these cells without E2 supplementation (Fig. 7a, b).

Effects of siRNA for CDK4 and/or siRNA for CDK6 on the proportion of CD44-high/CD24-low putative CSCs in MCF-7 and KPL-1 cells (a, b). *P < 0.05; **P < 0.01 in comparison with cells treated with E2 alone in a–c
Additionally, similar siRNA experiments were performed in the other cell lines. CDK4 inhibition alone and combined CDK4 and CDK6 inhibition paradoxically increased putative CSCs in BT-474 cells (Online Resource 4). CDK4 or CDK6 inhibition alone and the combined inhibition decreased putative CSCs in KPL-4 cells (Online Resource 5). The combined inhibition but not either alone decreased putative CSCs in MDA-MB-231 cells (Online Resource 6).
Discussion
ER (+)/HER2 (−) breast cancer is the most common type in both primary and advanced cases. Patients with breast cancer of this subtype had long been treated by endocrine therapy alone in either an adjuvant or metastatic setting. However, primary and acquired resistance to endocrine therapy often developed, and disturbed control of breast cancer of this subtype. Recently, an mTOR inhibitor, everolimus, was introduced to improve the objective response and PFS compared with endocrine therapy alone in postmenopausal patients with breast cancer of this subtype [9]. Additionally, CDK4/6 inhibitors have been introduced to improve the objective response and PFS compared with endocrine therapy alone in post- and premenopausal patients with metastatic breast cancer of this subtype [1, 2].
Preclinical and clinical findings have suggested that everolimus functions by overcoming the endocrine resistance induced by up-regulation of the mTOR pathway under long-term exposure to endocrine therapy in ER (+)/HER2 (−) metastatic breast cancer [9, 10]. In contrast, CDK4/6 inhibitors enhance the antitumor activity of endocrine therapy through the concurrent inhibition of the cyclin D-CDK4/6-E2F and ER pathways in breast cancer of this subtype [1]. It should be noted that the antitumor mechanisms between mTOR and CDK4/6 inhibitors are different.
Preclinical findings have demonstrated that CDK4/6 inhibitors block phosphorylation of Rb, inhibit the G1-S cell cycle transition and make cells quiescent. This cell cycle arrest is considered to be the most important antitumor action mechanism for CDK4/6 inhibitors [1]. The study revealed that the CDK4/6 inhibitor palbociclib can inhibit Rb phosphorylation (Online Resource 1) and induce G1-S blockade (Online Resource 2) in cell lines of different subtypes. Of note, apoptosis was not induced by palbociclib (Online Resource 3), as previously reported [11]. Another CDK4/6 inhibitor, abemaciclib, was reported to induce apoptosis in breast cancer cells [12]. The differences in molecular mechanisms responsible for the induction of apoptosis between these two CDK4/6 inhibitors remain to be elucidated.
Recent preclinical studies also found that some tumor cells undergo quiescence and others undergo senescence when treated with CDK4/6 inhibitors. It is well known that senescent cells will not return to the cell cycle. Induction of senescence by CDK4/6 inhibitors may be an important antitumor mechanism [1]. In the present study, palbociclib dose-dependently increased the proportion of β-galactosidase-positive senescent cells in all the examined breast cancer cell types (Fig. 2). In particular, the decrease in senescent cells induced by E2 was reversed by palbociclib in ER (+) breast cancer cells (Fig. 2). These findings suggest that the concomitant inhibition of the ER pathway and the cyclin D-CDK4/6-E2F pathway synergistically induce cell senescence in ER (+) breast cancer.
There are few studies on the anti-CSC effects of palbociclib in breast cancer cells. Dai et al. reported that blocking CDK4 expression using siRNA for CDK4 or kinase activity using palbociclib prevented stem cell self-renewal and the epithelial–mesenchymal transition in ER (−)/progesterone receptor (−)/HER2 (−) triple negative breast cancer cells, which were overexpressing CDK4. They also suggested that the effects of CDK4 on breast CSC self-renewal are, at least in part, mediated through the down-regulation of bone morphogenetic protein-4, a transforming growth factor-β family member known to act as an important growth factor for CSC tumorigenic capacity and a differentiation factor in CSCs [4]. Bonuccelli et al. also reported that palbociclib specifically blocked the propagation of breast, lung and ovarian CSCs, and they concluded that palbociclib may be used to target telomerase-high proliferative CSCs in multiple cancer types [13].
In the present study, we confirmed that palbociclib dose-dependently reduced the proportion of putative CSCs measured by three different CSC assays in four of the five breast cancer cell lines tested (Figs. 3, 4, 5). Of note, because estrogen-less sensitive KPL-1 cells seemed to be more sensitive to palbociclib than estrogen-dependent MCF-7 cells in terms of the growth inhibitory effect, we used lower concentrations of palbociclib for the CSC assays for KPL-1 cells. KPL-1 cells seemed to be more sensitive than MCF-7 cells in terms of the CSC inhibitory effect. On the other hand, in the ER (+)/HER2 (+) BT-474 cells, which were relatively resistant to palbociclib in terms of anti-cell growth activity (Fig. 1), it did not exhibit similar anti-CSC activity (Figs. 3, 4, 5). A poor anti-cell growth effect of palbociclib on the BT-474 cells and a cross-talk between HER2 and ER signaling might influence on the CSC regulation by palbociclib in the BT-474 cells. It has been known that up-regulation of HER2 signaling increases the expression levels of genes related to stem cell control and renders a higher self-renewal potential [14]. Additionally, it is well known that HER2 overexpression or hyper-activation is one of common causes of endocrine resistance [15, 16]. These findings support that a cross-talk between HER2 and ER signaling might influence on the CSC regulation. However, detailed action mechanisms remain to be elucidated.
CSCs have been suggested to play important roles in tumor recurrence, metastasis and drug resistance, and these subpopulations of cancer cells have emerged as potential cellular targets for clinical therapeutic strategies [17]. Therefore, the anti-CSC effects of palbociclib may enhance its antitumor activity and prolong the PFS of patients with ER (+)/HER2 (−) breast cancer.
To further explore the anti-CSC mechanisms of palbociclib, we knocked down CDK4 and/or CDK6 using the respective siRNAs. Dai et al. previously reported that CDK4 inhibition, but not CDK6 inhibition, significantly reduces the proportion of putative CSCs in CDK4-overexpressing triple negative breast cancer cells [4]. In our study, CDK4 inhibition, but not CDK6 inhibition, resulted in a reduced proportion of putative CSCs induced by E2 in ER (+)/HER2 (−) breast cancer cells (Fig. 7). Therefore, CDK4 inhibition, but not CDK6 inhibition, induced by palbociclib may be responsible for its anti-CSC effects in ER (+)/HER2 (−) breast cancer cells. Furthermore, the differences in inhibitory effects between CDK4 and CDK6 activity induced by different CDK4/6 inhibitors may influence their anti-CSC effects on breast cancer cells.
Results of the similar siRNA experiments in the other cell lines were somewhat complicated. In ER (+)/HER2 (+) BT-474 cells which did not respond to palbociclib in terms of CSC regulation, CDK4 inhibition alone and combined CDK4 and CDK6 inhibition increased putative CSCs (Online Resource 4). In ER (−)/HER2 (+) KPL-4 cells, combined CDK4 and CDK6 inhibition decreased putative CSCs but not CDK4 or CDK6 inhibition alone (Online Resource 5). In ER (−)/HER2 (−) MDA-MB-231 cells, either CDK4 or CDK6 inhibition alone and the combined inhibition decreased putative CSCs (Online Resource 6). Because a limited number of breast cancer cell lines tested in this study, it is difficult to understand action mechanisms responsible for these complicated results. Action mechanisms of CSC regulation may differ among breast cancer cell lines of different subtypes.
Unexpectedly, E2 treatment significantly reduced CDK6 expression in ER (+)/HER2 (−) MCF-7 and KPL-1 cells. According to the paper by Lucas et al., CDK6 restrains rather than stimulates breast epithelial cell proliferation and that its loss or down-regulation could play a role in breast tumor development [18]. An inhibitory effect of estradiol on CDK6 expression might promote cell proliferation in ER (+) breast cancer cells. These interesting phenomena should be further explored.
Interestingly, a knock-down of CDK4 mRNA expression resulted in a higher mRNA expression levels of CDK6 but a knock-down of CDK6 did not affect the mRNA expression levels of CDK4 in most cell lines (Fig. 6 and Online Resources 4–6). CDK4 and CDK6 are not only essential for initiation of the cell cycle but also regulatory factors responsible for the cell survival [1]. It has been indicated that CDK4 is dispensable for the cell proliferation but CDK6 plays a compensatory role in it [19]. These findings may, at least in part, explain these phenomena but action mechanisms responsible for these phenomena remain to be elucidated.
In conclusion, our systematic study demonstrated that palbociclib exhibits significant antitumor activity in not only ER (+) breast cancer cell lines but also ER (−) cell lines, which is mediated through its inhibitory effects on Rb phosphorylation, G1-S cell cycle progression, cell senescence and the proportion of CSCs. Additionally, CDK4 inhibition induced by palbociclib may be responsible for its anti-CSC activity.
References
Klein ME, Kovatcheva M, Davis LE, et al. CDK4/6 inhibitors: the mechanism of action may not be as simple as once thought. Cancer Cell. 2018;34:9–20.
VanArsdale T, Boshoff C, Arndt KT, et al. Molecular pathways: targeting the cyclin D-CDK4/6 axis for cancer treatment. Clin Cancer Res. 2015;21:2905–10.
Turner NC, Slamon DJ, Ro J, et al. Overall survival with palbociclib and fulvestrant in advanced breast cancer. N Engl J Med. 2018;379:1926–36.
Dai M, Zhang C, Ali A, et al. CDK4 regulates cancer stemness and is a novel therapeutic target for triple-negative breast cancer. Sci Rep. 2016;6:35383.
Kurebayashi J, Kurosumi M, Sonoo H. A new human breast cancer cell line, KPL-1 secretes tumour-associated antigens and grows rapidly in female athymic nude mice. Br J Cancer. 1995;71:845–53.
Kurebayashi J, Otsuki T, Tang CK, et al. Isolation and characterization of a new human breast cancer cell line, KPL-4, expressing the Erb B family receptors and interleukin-6. Br J Cancer. 1999;79:707–17.
Kurebayashi J, Koike Y, Ohta Y, et al. Anti-cancer stem cell activity of a hedgehog inhibitor GANT61 in estrogen receptor-positive breast cancer cells. Cancer Sci. 2017;108:918–30.
Vijayaraghavan S, Karakas C, Doostan I, et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat Commun. 2017;8:15916.
Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366:520–9.
Martin LA, Pancholi S, Farmer I, et al. Effectiveness and molecular interactions of the clinically active mTORC1 inhibitor everolimus in combination with tamoxifen or letrozole in vitro and in vivo. Breast Cancer Res. 2012;14:R132.
Finn RS, Dering J, Conklin D, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11:R77.
Torres-Guzmán R, Calsina B, Hermoso A, et al. Preclinical characterization of abemaciclib in hormone receptor positive breast cancer. Oncotarget. 2017;8:69493–507.
Bonuccelli G, Peiris-Pages M, Ozsvari B, et al. Targeting cancer stem cell propagation with palbociclib, a CDK4/6 inhibitor: Telomerase drives tumor cell heterogeneity. Oncotarget. 2017;8:9868–84.
Shah D, Osipo C. Cancer stem cells and HER2 positive breast cancer: The story so far. Genes Dis. 2016;3:114–23.
Kurebayashi J. Resistance to endocrine therapy in breast cancer. Cancer Chemother Pharmacol. 2005;56(Suppl 1):39–46.
Nayar U, Cohen O, Kapstad C, et al. Acquired HER2 mutations in ER+ metastatic breast cancer confer resistance to estrogen receptor-directed therapies. Nat Genet. 2019;51:207–16.
Liu S, Wicha MS. Targeting breast cancer stem cells. J Clin Oncol. 2010;28:4006–122.
Lucas JJ, Domenico J, Gelfand EW. Cyclin-dependent kinase 6 inhibits proliferation of human mammary epithelial cells. Mol Cancer Res. 2004;2:105–14.
Malumbres M, Sotillo R, Santamaría D, et al. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell. 2004;118:493–504.
Acknowledgements
We thank Mrs. Kaoru Tsuboi and Megumi Kuriyama for their technical assistance. All authors read and approved the final manuscript, and agree to be accountable for the integrity of the work.
Funding
This study was partially supported by Research Project Grants from Kawasaki Medical School and MEXT/JSPS KAKENHI Grant number JP17K10566.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
J. Kurebayashi received advisory/consultation fees and research funding from Takeda Pharmaceutical Co. J. Kurebayashi also received research funding from Eisai Co. The other authors declare that they have no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
12282_2019_1035_MOESM1_ESM.pdf
The protein expression levels of Rb and phosphorylated Rb altered by E2 and/or palbociclib in five breast cancer cell lines (a, MCF-7 cells; b, KPL-1 cells; c, BT-474 cells, d, KPL-4 cells and e, MDA-MB-231 cells). Values were analyzed after normalization to the controls and expressed as the mean ± SE. The expression level of each molecule in control cells was defined as 1. White bars, control; lightest grey bars, E2 alone; the second lightest grey bars, E2 plus 50 or 100 nM palbociclib; and the third lightest grey bars, E2 plus 100 or 500 nM palbociclib. * P < 0.05; **P < 0.01 significantly different from the E2-treated cells in ER (+) cells and from control cells in ER (-) cells. (PDF 279 kb)
12282_2019_1035_MOESM2_ESM.pdf
Effects of palbociclib on cell cycle progression in MCF-7 (a), KPL-1 (b), BT-474 (Cc KPL-4 (d) and MDA-MB-231 (e) cell lines. The values are the mean ± SE. White bars, control; lightest grey bars, E2 alone; the second lightest grey bars, E2 plus 50 or 100 nM palbociclib; and the third lightest grey bars, E2 plus 100 or 500 nM palbociclib. * P < 0.05; **P < 0.01 in comparison with cells treated with E2 alone. (PDF 252 kb)
12282_2019_1035_MOESM3_ESM.pdf
Effects of palbociclib on apoptosis in MCF-7 (a), KPL-1 (b), BT-474 (c), KPL-4 (d) and MDA-MB-231 (e) cell lines. Values are the mean ± SE. White bars, control; the lightest grey bars, E2 alone; the second lightest grey bars, E2 plus 50 or 100 nM palbociclib; and the third lightest grey bars, E2 plus 100 or 500 nM palbociclib. No significant difference was observed between palbociclib-treated and control cells. (PDF 233 kb)
12282_2019_1035_MOESM4_ESM.pdf
Effects of siRNA for CDK4 and/or siRNA for CDK6 on mRNA expression of CDK4 and CDK6 (a, b) and the proportion of CD44-high/CD24-low putative CSCs (c) in BT-474 cells. * P < 0.05; **P < 0.01 in comparison with control cells. (PDF 234 kb)
12282_2019_1035_MOESM5_ESM.pdf
Effects of siRNA for CDK4 and/or siRNA for CDK6 on mRNA expression of CDK4 and CDK6 (a, b) and the proportion of CD44-high/CD24-low putative CSCs (c) in KPL-4 cells. * P < 0.05; **P < 0.01 in comparison with control cells. (PDF 227 kb)
12282_2019_1035_MOESM6_ESM.pdf
Effects of siRNA for CDK4 and/or siRNA for CDK6 on mRNA expression of CDK4 and CDK6 (a, b) and the proportion of CD44-high/CD24-low putative CSCs (c) in MDA-MB-231 cells. * P < 0.05; **P < 0.01 in comparison with control cells. (PDF 228 kb)
About this article

Cite this article
Kishino, E., Ogata, R., Saitoh, W. et al. Anti-cell growth and anti-cancer stem cell activity of the CDK4/6 inhibitor palbociclib in breast cancer cells. Breast Cancer 27, 415–425 (2020). https://doi.org/10.1007/s12282-019-01035-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12282-019-01035-5