
Abstract
Rebamipide is marketed as a peptic ulcer agent under the trade name MucostaR. The objective of this work was to investigate the existence of polymorphs and pseudopolymorphs of rebamipide. Two crystal forms of rebamipide were isolated by recrystallization and characterized by differential thermal analysis (DTA), thermogravimetric analysis (TG), powder X-ray diffractometry, infrared spectrometry, and nuclear magnetic resonance. The DTA curve of Form 1 showed one endothermic peak at 305.2 °C, and that of Form 2 showed one endothermic peak at 307.3 °C. The TG curve of Form 1 showed a single weight loss at 305.2 °C, which corresponded to melting. The TG curve of Form 2 also showed a single weight loss at 307.3 °C, which corresponded to melting. The melting point of Form 2 was higher than that of Form 1. In the dissolution studies in pH 6.8 buffer at 37 ± 0.5 °C, the two crystal forms showed no significant differences in dissolution. After 3 months of storage at 0, 52, and 95 % RH, the two crystal forms were not transformed. After milling with a Specamill for 2 h, the two crystal forms were not transformed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pharmaceutical solids can exist in different crystal forms, such as crystalline, amorphous, or glass, and also in solvated or hydrated states (Chadha et al. 2013; Perlovich et al. 2013). Polymorphism is defined as the ability of a substance to exist as two or more crystalline phases with different arrangements and/or conformations of the molecules in the crystal lattice. Polymorphs share the same chemical composition but have different crystal structures. Because of their structural differences, polymorphs can have different physicochemical properties. For example, polymorphs can have different densities, habits, melting properties, vapor pressures, solubilities, dissolution rates, and tableting and mechanical properties (Haleblian and McCrone 1969; Haleblian 1975; Chadha et al. 2013; Drebushchak et al. 2013; Perlovich et al. 2013; Shin and Sohn 2014).
The crystal forms include polymorphs, solvates, and amorphous forms, as defined in International Conference on Harmonization (ICH) Guideline Q6A. The crystal form affects properties such as drug absorption, rate of dissolution, elimination rate, and stability in galenic preparations (Otsuka 1993; Griesser et al. 2004; Zhang et al. 2004; Petit et al. 2007; Dichi et al. 2014; Seo and Sohn 2015). The successful utilization of a crystal form with significantly greater thermodynamic activity (i.e., solubility) than the stable modification can provide, in some instances, therapeutic blood levels of otherwise inactive drugs (Brittain and Grant 1999).
Companies have experienced market shortages because they have experienced unpredicted changes in the crystal forms of drugs, which ultimately resulted in problematic release and stability testing of the finished dosage form (Nicolai et al. 2007; de Oliveira et al. 2012; Gana et al. 2013; Terada et al. 2013). A thorough understanding of the way in which solid state properties influence solubility, stability, and other properties of the drug substance is critical in the development of profiling strategies and in the establishment of criteria for developability assessment (Huang and Tong 2004).
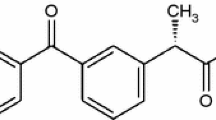
Rebamipide, (N-(4-chlorobenzoyl)-3-(2-oxo-1,2-dihydro-quinolin-4-yl)-alanine) (Fig. 1), is marketed by Otsuka Pharmaceutical Co. Ltd. (Tokyo, Japan) as a peptic ulcer agent under the trade name Mucosta®. It is used for mucosal protection, healing of gastroduodenal ulcers, and treatment of gastritis. It functions by enhancing mucosal defense, scavenging free radicals, and temporarily activating genes encoding cyclooxygenase-2 (Arakawa et al. 1995, 1998; Tarnawski et al. 2004). In a randomized controlled trial, it was also shown to alleviate the signs and symptoms of dry eye; although this formulation is not yet widely available clinically (Kinoshita et al. 2013). Rebamipide belongs to BCS Class IV and has low solubility and low permeability (Zhi et al. 1995).

Chemical structure of rebamipide
In the case of a new drug substance, it is important that crystal form data be generated prior to initiation of the pivotal clinical studies and primary stability batches. Thus, the thorough investigation of new solid states of a drug molecule is recognized as an essential part of preformulation studies (Rodriguez-Spong et al. 2004).
The aim of this study was to investigate the existence of polymorphs of rebamipide.
Materials and methods
Materials
Rebamipide was provided by Estech Pharma Co. Ltd. (Hwaseong-Si, Korea). All of the other chemicals were of reagent grade and were used without additional purification.
Preparation of crystal forms
Form 1
For drug preparation, 10 g of 2-amino-3-(1,2-dihydro-2-oxo-quinoline-4-yl)propionic acid and 5.13 g of sodium hydroxide were dissolved in 100 mL water and cooled to 10 °C. Then, 6.66 g of 4-chlorobenzoyl chloride was slowly added dropwise, and the mixture was stirred at room temperature for 3 h. To neutralize the solution, concentrated hydrochloric acid was added, and the solution was stirred for 2 h. The resultant crystal was dried at 50 °C for 10 h and at 90 °C for 12 h to yield Form 1.
Form 2
For preparation of Form 2, 10 g of 2-amino-3-(1,2-dihydro-2-oxo-quinoline-4-yl)propionic acid and 5.13 g of sodium hydroxide were dissolved in 100 mL water and cooled to 10 °C. Then, 6.66 g of 4-chlorobenzoyl chloride was slowly added dropwise, and the mixture was stirred at room temperature for 3 h. To neutralize the solution, concentrated hydrochloric acid was added, and the solution was stirred for 2 h. The resultant crystal was dissolved in a methanol solution [methanol:water (10:1)] at room temperature for 12 h, stirred, and filtered. The crystalline product was dried at 50 °C for 10 h and at 90 °C for 12 h to yield Form 2.
Thermal analysis
The thermal analysis methods used in this study included differential thermal analysis (DTA) and thermogravimetric analysis (TG). The DTA and TG data were collected using a model SEIKO EXSTAR DTA6100 module and a model SEIKO EXSTAR 6100 TGA module within the temperature range of 50–450 °C at a heating rate of 10 °C min−1 using highly purified nitrogen gas at a flow rate 30 mL min−1. DTA data were obtained in a closed aluminum pan using a sample weight of about 5 mg. TGA data were also collected using a sample weight of about 5 mg.
Powder X-ray diffraction
Powder X-ray diffraction patterns under ambient conditions were collected on a Rigaku DMAX-IIIA (Japan) diffractometer using graphite monochromatized CuKα radiation (λ = 1.54178 Å). The isothermal measurement conditions were: target, Cu; voltage, 30 kV; and current, 10 mA. The powder X-ray diffractometry (PXRD) patterns of the samples were compared with respect to peak position and relative intensity, peak shifting, and absence of peaks in certain angular regions.
Nuclear magnetic resonance
Proton nuclear magnetic resonance (NMR) spectra were obtained on a Varian Unity Inova 500NB. Solution proton NMR was performed in DMSO-d6 using a Varian 500 MHz instrument. The samples were not ground but were packed into 7-mm silicon nitride rotors fitted with Torlon caps and spun at the rate of 5 kHz. The Hartman-Hahn match and the contact times were adjusted to optimize conditions for each crystal form. Tetramethylsilane was used as a chemical shift reference.
Infrared spectroscopy
Fourier transform infrared spectra (FT-IR) of solid rebamipide were generated using a JASCO model 460 spectrometer. Two milligrams of rebamipide were dispersed into 200 mg of dry KBr. The spectrum was collected from 4000 to 500/cm.
Dissolution
The dissolution rate of rebamipide was measured using the paddle method of USP XXIV using a dissolution tester (Erweka DT-D, Germany). A fixed amount (100 mg, 250–600 µm) of rebamipide crystals was placed into 900 mL of buffer (pH 6.8) and stirred at 100 rpm at 37 ± 0.5 °C. Next, 2 mL of the dissolution samples were collected at predetermined time intervals of 5, 10, 15, 30, and 60 min and replaced with an equal volume of fresh media in order to maintain sink conditions. All of the dissolution samples were filtered using a syringe filter (0.45 μm) and were then spectrophotometrically analyzed at 326 nm.
Transformation
Storage conditions: A specific amount (20 mg) of the crystal form was placed in a weighing dish. Samples were stored in desiccators at 0 % RH (silica gel, 20 °C), 52 % RH (a saturated solution of Na2Cr2O .7 2H2O/20 °C), and 95 % RH (a saturated solution of Na2HPO4/20 °C). The transformation behavior of the crystal forms was monitored by PXRD analysis, DTA, and TG.
Milling: A specific amount (2 g) of the crystal form was milled with a Specamill for 2 h. The transformation behavior of the crystal forms was monitored by PXRD analysis, DTA, and TG.
Results
The DTA and TG curves of Forms 1 and 2 are illustrated in Fig. 2. The DTA curve of Form 1 showed one endothermic peak at 305.2 °C, and that of Form 2 showed one endothermic peak at 307.3 °C. The TG curve of Form 1 showed a single weight loss at 305.2 °C, which corresponded to melting. The TG curve of Form 2 also showed a single weight loss at 307.3 °C, which corresponded to melting. The powder X-ray diffraction patterns of Forms 1 and 2 are illustrated in Fig. 3 and showed differences. The FT-IR patterns of Forms 1 and 2 are illustrated in Fig. 4. The FT-IR pattern of Form 1 showed a single peak at 1645 cm−1, and that of Form 2 showed two peaks at 1640 cm−1 and 1670 cm−1. The NMR spectra of Forms 1 and 2 are illustrated in Fig. 5 and were virtually identical. Although there were some very subtle differences between the two forms, it was difficult to make qualitative determinations between the two crystal forms with NMR.

DTA and TG curves of Form 1 (a) and Form 2 (b)

PXRD patterns of Form 1 (a) and Form 2 (b)

Infrared spectra of Form 1 (a) and Form 2 (b)

Proton NMR spectra of Form 1 (a) and Form 2 (b)
The DTA, TG, FT-IR, NMR, and PXRD results confirmed the existence of two crystal forms of rebamipide.
The dissolution patterns of the two crystal forms of rebamipide are illustrated in Fig. 6. In the dissolution studies in buffer (pH 6.8) at 37 ± 0.5 °C, the dissolution rate of Form 1 was greater than that of Form 2, but the difference was not significant (p < 0.5).

Dissolution patterns of Form 1 and Form 2 (n = 3)
After three months storage of at 0 % RH (silica gel, 20 °C), 52 % RH (a saturated solution of Na2Cr2O .7 2H2O/20 °C) and 95 % RH (a saturated solution of Na2HPO4/20 °C), the two crystal forms showed no change in DTA, TG, or PXRD pattern (not shown).
To study the effect of the pharmaceutical process on the transformation of the crystal forms, the two crystal forms of rebamipide were milled with a Specamill for two hours. The two crystal forms showed no changes in DTA, TG, or PXRD pattern (not shown).
Discussion
There have been no previous reports on polymorphisms of rebamipide. In this study, we prepared two such polymorphs. Currently, one of the most widely used methods for polymorph screening is recrystallization in different solvents. Slow solvent evaporation is a valuable method for producing crystals (Guillory 1999). In this method, solutions, preferably saturated, of the material being crystallized are filtered to remove most nuclei and then left undisturbed for some period of time. All compounds, organic and inorganic, can crystallize in different crystal forms or polymorphs. One question that is likely to arise during a polymorphism study is: “What assurance can be provided that no other crystalline forms of this compound exist?” (Guillory 1999). One can never be certain that no additional forms will be identified in the future. The prediction probability on the generation of polymorphs is not particularly high.
Of all the methods available for the physical characterization of solid materials, it is generally agreed upon that crystallography, thermal analysis, solubility studies, vibrational spectroscopy, and nuclear magnetic resonance are the most useful for the characterization of polymorphs and solvates. However, it cannot be overemphasized that the defining criterion for the existence of polymorphic types must always be a nonequivalence of crystal structures. For compounds of pharmaceutical interest, this ordinarily implies that a nonequivalent X-ray powder diffraction pattern needs to be observed for each suspected polymorphic variation. All other methodologies must be considered to be sources of supporting and ancillary information; they cannot by themselves be considered definitive proof of the existence of a polymorphism. One technique that is becoming increasingly important for the characterization of materials is solid-state NMR spectroscopy (Brittain 1999). The methodologies for the physical characterization of polymorphs in this study included powder X-ray diffraction, thermal analysis (DTA and TG), IR spectroscopy, NMR, and solubility. It was possible to characterize the polymorphs of rebamipide with X-ray diffraction, thermal analysis, IR spectroscopy, and solubility. However, the NMR spectra of the two crystal forms were virtually identical. Powerful as the technique has been shown to be, one must remember that the ultimate arbiter of polymorphism is crystallography, not spectroscopy (Brittain 1999).
Since a variety of phase conversions are possible upon exposure to the energetic steps of bulk material (storage, milling), we studied the effect of pharmaceutical processing on the transformation of the crystal forms of rebamipide. After storage for 3 months at 0 % RH, 52 % RH, or 95 % RH and milling with a Specamill for 2 hours, the two crystal forms had not transformed; this indicates that the two crystal forms of rebamipide are very stable.
Rebamipide belongs to BCS Class IV and has low solubility and low permeability (Zhi et al. 1995). In those specific instances where the absorption rate of the active ingredient in a solid dosage form depends upon the rate of drug dissolution, the use of different polymorphs would be expected to affect the bioavailability. However, in this study, the two crystal forms of rebamipide showed no significant differences in dissolution. Therefore, in order to improve the solubility and dissolution rate, other techniques should be used. For example, conversion of the drug’s crystal lattice into an amorphous form, micronization, use of surfactants, inclusion complex, nanoparticle formation, and solid dispersion could all be evaluated (Tung et al. 2011; Pradhan et al. 2014). The dissolution rate of rebamipide is strongly dependent on pH.
References
Arakawa T, Watanabe T, Fukuda T, Yamasaki K, Kobayashi K (1995) Rebamipide, novel prostaglandin-inducer accelerates healing and reduces relapse of acetic acid-induced rat gastric ulcer. Comp Cimet Dig Dis Sci 40:2469–2472
Arakawa T, Kobayashi K, Yoshikawa T, Tarnawski A (1998) Rebamipide: overview of its mechanisms of action and efficacy in mucosal protection and ulcer healing. Dig Dis Sci 43(9 Suppl):5S–13S
Brittain HG (1999) Methods for the characterization of polymorphs and solvates. In: Brittain HG (ed) Polymorphism in pharmaceutical solids. Marcel Dekker, New York, pp 227–278
Brittain HG, Grant DJW (1999) Effects of polymorphism and solid-state solvation on solubility and dissolution rate. In: Brittain HG (ed) Polymorphism in pharmaceutical solids. Marcel Dekker, New York, pp 279–330
Chadha R, Arora P, Garg M, Bhandari S, Jain DS (2013) Thermoanalytical and spectroscopic studies on different crystal forms of nevirapine. J Therm Anal Calorim 111:2133–2142
de Oliveira GGG, Ferraz HG, Severino P, Souto E (2012) Analysis of phase transition and dehydration process of nevirapine. J Therm Anal Calorim 108:53–57
Dichi E, Legendre B, Sghaier M (2014) Physico-chemical characterization of a new polymorph of caffeine. J Therm Anal Calorim 115:1551–1561
Drebushchak VA, Drebushchak TN, Boldyreva EV (2013) New interpretation of heat effects in polymorphic transitions. J Therm Anal Calorim 113:419–424
Gana I, Ceolin R, Rietveld IB (2013) Bicalutamide polymorphs I and II. J Therm Anal Calorim 112:223–228
Griesser UJ, Weigand D, Rollinger JM, Haddow M, Gstrein E (2004) The crystal polymorphs of metazachlor. J Therm Anal Calorim 77:511–522
Guillory JK (1999) Generation of polymorphs. In: Brittain HG (ed) Polymorphism in pharmaceutical solids. Marcel Dekker, New York, pp 183–226
Haleblian J (1975) Characterization of habits and crystalline modification of solids and their pharmaceutical applications. J Pharm Sci 64:1269–1288
Haleblian J, McCrone W (1969) Pharmaceutical applications of polymorphism. J Pharm Sci 58:911–929
Huang LF, Tong WQ (2004) Impact of solid state properties on developability assessment of drug candidates. Adv Drug Deliv Rev 56:321–334
Kinoshita S, Oshiden K, Awamura S, Suzuki H, Nakamichi N (2013) A randomized, multicenter phase 3 study comparing 2 % rebamipide (OPC-12759) with 0.1 % sodium hyaluronate in the treatment of dry eye. Ophthalmology 120:1158–1165
Nicolai B, Espeau P, Ceolin R, Perrin MA, Zaske L, Giovanni J, Leveiller F (2007) Polymorph formation from solvate desolvation. J Therm Anal Calorim 90:337–339
Otsuka M (1993) Effects of environmental temperature and compression energy on polymorphic transformation during tableting. Drug Dev Ind Pharm 19:2241–2269
Perlovich GL, Blokhina SV, Manin NG, Volkova TV, Tkachev VV (2013) Polymorphism and solvatomorphism of bicalutamide. J Therm Anal Calorim 111:655–662
Petit S, Mallet F, Petit MN, Coquerel G (2007) Role of structural and macrocrystalline factors in the desolvation behavior of cortisone acetate solvates. J Therm Anal Calorim 90:39–47
Pradhan R, Tran TH, Choi JY, Choi IS, Choi HG, Yong CS, Kim JO (2014) Development of a rebamipide solid dispersion system with improved dissolution and oral bioavailability. Arch Pharm Res 38:522–533
Rodriguez-Spong B, Price CP, Jayasankar A, Matzger AJ, Rodriguez-Hornedo N (2004) General principles of pharmaceutical solid polymorphism: a supramolecular perspective. Adv Drug Deliv Rev 56:241–274
Seo HO, Sohn YT (2015) Crystal transformation of a flavonoid derivative DA-6034. J Therm Anal Calorim 120:749–757
Shin JY, Sohn YT (2014) Solid state of a new flavonoid derivative DA-6034. J Therm Anal Calorim 115:2457–2461
Tarnawski AS, Chai J, Pai R, Chiou SK (2004) Rebamipide activates genes encoding angiogenic growth factors and Cox2 and stimulates angiogenesis: a key to its ulcer healing action? Dig Dis Sci 49:202–209
Terada K, Kurobe H, Ito M, Yoshihashi Y, Yonemochi E, Fujii K, Uekusa H (2013) Polymorphic and pseudopolymorphic transformation behavior of acyclovir based on thermodynamics and crystallography. J Therm Anal Calorim 113:1261–1267
Tung NT, Park CW, Oh TO, Kim JY, Ha JM, Rhee YS, Park ES (2011) Formulation of solid dispersion of rebamipide evaluated in a rat model for improved bioavailability and efficacy. J Pharm Pharmacol 63:1539–1547
Zhang GGZ, Law D, Scmitt EA, Qiu Y (2004) Phases transformation considerations during process development and manufacture of solid oral dosage forms. Adv Drug Deliv Rev 56:371–390
Zhi J, Melia AT, Eggers H, Joly R, Patel IH (1995) Review of limited systemic absorption of orlistat, a lipase inhibitor, in healthy human volunteers. J Clin Pharmacol 35:1103–1108
Acknowledgments
This work was supported by a research grant from Duksung Women’s University (2015).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest with any person or any organization.
Rights and permissions
About this article

Cite this article
Jeon, S.H., Sohn, Y.T. The solid state of rebamipide: preparation, characterization, and dissolution. Arch. Pharm. Res. 39, 508–515 (2016). https://doi.org/10.1007/s12272-016-0723-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-016-0723-y