
Abstract
Purpose
Gastrointestinal hydrodynamics are poorly replicated in vitro and can significantly alter the release kinetics of drug products due to compressive forces in the stomach and peristaltic movement in the intestines. In this work, we describe the development and application of a predictive in vitro dissolution device that simulates gastrointestinal forces for the testing of oral drug products. The peristaltic dissolution device developed herein is designed as an addition to the common USP Apparatus 2 that applies repetitive compressive forces via a piston during dissolution testing of a product to replicate in vivo conditions.
Methods
A dissolution testing device was designed, fabricated, and evaluated against human in vivo pharmacokinetic data to better mimic the physical forces present in the gastrointestinal tract. An optimized compression protocol to predict in vivo dissolution was developed using clinical data from two modified release carvedilol drug products. The apparatus was further evaluated using data from an additional modified release drug product. Finally, additional dissolution studies were performed to evaluate the utility of the apparatus for in vitro analysis of medicated gums, gastric retentive formulations, and long-acting injectable drug depots.
Results
The device was successfully implemented and the protocol to use the device was optimized using two initial drug products and further evaluated using an additional three drug products. The optimized protocol included a 1-h lag time (applicable in the fed state), followed by a cycle of 3 s of compression with 6 s intervals between compressions. Additional applications of the peristaltic dissolution device were also demonstrated through small exploratory studies, with continued potential for further optimization of the testing protocols following further research.
Conclusion
This simple compressive device referred to as the “peristaltic dissolution device” was successfully proven to better predict in vivo performance of modified release drug products, as gastrointestinal mechanical forces have been observed to significantly impact and occasionally cause complete dose dumping of controlled release formulations. In addition, it has proven to be easily adapted for evaluation of other drug products such as medicated gums, gastric retentive formulations, and ex vivo long-acting injectable drug depots.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dissolution testing is a requirement in the pharmaceutical industry and its use is divided into two main areas: quality control and as a predictive tool. Quality control has been a frequent and continual application of dissolution testing, with standardized protocols and test conditions; however, with rising costs of clinical studies, ethical concerns associated with frequent animal testing, and the increasing emphasis on quality by design (QbD), the industry has been driven to adapt the testing to be used as a predictive tool as much as possible [1].
Unfortunately, the predictive ability of traditional dissolution testing is limited due to the lack of many of the dynamic in vivo factors that can affect oral drug product performance. A 2012 review article on biorelevant in vitro dissolution has highlighted that although our knowledge of biorelevant media composition has increased dramatically over the past 15 years, our ability to simulate biorelevant hydrodynamics and other mechanical factors has lagged behind [2]. Furthermore, modified release dosage forms add an additional layer of complexity as the interplay of various physiological and drug product factors dictate the in vivo performance [3]. The ability to better simulate biorelevant hydrodynamics and predict in vivo performance, especially for modified release dosage forms, would help minimize the number of in vivo studies required to develop a commercial product and gain regulatory approval, or alternatively, enable better optimization of the formulations that are taken into in vivo studies, enhancing the probability of success.
During the gastrointestinal (GI) movement of dosage forms, there is not always a continuous phase of free fluid available in the intestinal lumen in which the drug product is transported through the intestine by simple convection. Rather, there is significant physical contact between the walls of the GI system and drug products, and the drug product is moved through the GI tract based on peristalsis (waves of compression) rather than the dosage form being suspended in a river of fluid. The mechanical stresses applied to the drug product by this peristaltic action can contribute to various pharmacokinetic phenomena observed such as dose dumping [3]. This highlights a clear gap between current dissolution techniques—namely the widely used USP Apparatus 2, which does not simulate the interaction between drug product and the gut lumen—and the actual in vivo environment. The forces that the lumen applies to the dosage forms can have a significant impact on in vivo performance. As a result, there have been many research efforts to develop a “biorelevant” dissolution technique which can bridge the gap between traditional dissolution methods and accurate predictions of performance in vivo.
The United States Pharmacopeia has included the Apparatus 3 Biodis®, a reciprocating cylinder apparatus, which offers a small degree of biological relevance as compared to the standard basket (USP Apparatus 1) or paddle apparatus (USP Apparatus 2) in that it enables adjustment of chemical and physical conditions over the course of the experiment. As such, combining this apparatus with biorelevant dissolution media and varying testing times and hydrodynamic conditions to mimic gastrointestinal passage conditions offer some temporal in vitro modeling of in vivo performance. Apparatus 3 can also achieve more aggressive hydrodynamics than Apparatus 1 or 2 and therefore may provide a better simulation of the gastrointestinal environment, where peristaltic contractions can significantly affect a drug product’s performance. The USP Apparatus 4—the flow-through cell—also enables one to vary the dissolution media and media flow rates to offer more predictive modeling of GI dissolution. However, the amount of media used in Apparatus 4 is typically much greater than the corresponding amount of fluid found in the GI tract as seen by the dosage form, which prevents saturation effects that can happen in vivo from occurring in vitro [3].
More importantly, however, neither Apparatus 3 nor Apparatus 4 device simulates any of the mechanical forces that a drug product can experience in vivo. The peristaltic compressions that move solids through the GI tract can be highly variable and powerful, at times moving dosage forms forward at rates as fast as 50 cm/s and generating mechanical forces as high as 300 mbar during events such as gastric emptying [3, 4]. With such pressures, these events can be highly destructive of solid-dose products, which can significantly affect dissolution and absorption rates, leading to very different in vivo pharmacokinetic behavior than would be predicted by standard in vitro tests [1].
An early device designed by Simmons et al. sought to address this in vitro–in vivo disparity [5]. This setup uses a cylindrical plastic tube dissolution chamber in which the oral solid-dosage product is contained between a stopper at the base and a septum at the top, with two hypodermic needles run through the septum that are in turn coupled to an automated pipetting system. The sides of the cylindrical chamber containing the drug product have portholes which allow fluid mixing and transfer of disintegrated particles from the dissolution chamber into a larger 1000 mL beaker of dissolution medium in which the dissolution chamber is submerged. During dissolution experiments, peristaltic fluid dynamics are generated within the dissolution chamber by the pipetting device cycle, which drives fluid both into and out of the chamber simultaneously through the hypodermic needles, generating a peristaltic effect with turbulent fluid mechanics that better replicate those observed in vivo. The device has shown in vitro–in vivo correlation with tested tolbutamide, prednisone, ibuprofen, allopurinol, meprobamate, and indomethicin drug products, all with a 3-to-1 in vivo to in vitro time differential attributed to absorption and distribution time in vivo [6]. This device appears to offer substantially better prediction of in vivo drug product performance compared to USP Apparatus 1 or 2 as a result of the turbulent peristaltic hydrodynamics generated within the dissolution chamber. Comparing to the device presented in this paper, the device by Simmons et al. uses fluid jets to create additional turbulence that aids in the disintegration of tested drug products, whereas the device presented herein uses a piston arrangement to generate peristaltic turbulence as well as to physically interact with the drug product, as the GI environment does during dynamic events such as gastric emptying [4].
A few newer dissolution devices have also been prepared to better simulate the physical and chemical complexities of in vivo gastrointestinal dissolution and absorption. One such device is the “fed stomach model,” which is an extension of the USP Apparatus 2 paddle device that also incorporates an additional side chamber, called a “gastric vessel,” where the dosage form is placed (see Fig. 1) [7]. Dissolution media is continuously pumped between the two chambers, and an inflatable balloon in the gastric vessel intermittently applies mechanical stresses to the dosage form as controlled by a pressure regulation module. Meanwhile, the dosage form sits atop glass beads which are consistently agitated by the motion of two blades at the top of the gastric vessel which swing in a pendular motion. The pressure, pumping rate, and stirring speeds can all be varied by software settings. By utilizing the USP Apparatus 2 device, the fed stomach model enables biorelevant pressures to be applied to dosage forms in multiple simultaneous dissolution experiments, as allowed by the dissolution device used. However, the requirement of the separate chamber and pumping system makes this system require more bench space than the standard dissolution apparatus and leaves many variables to be optimized to achieve good biorelevant correlation. A similar device was also developed by Garbacz et al., which applies compressive forces on the dosage form by pulsatile inflation and deflation of a balloon within a mesh chamber; in this case, the chamber itself rotates into and out of the dissolution medium such that it is immersed 50% of the time and in the air for 50% of the time in order to generate additional mechanical motion and stresses [8]. And most recently, Gao et al. published on the use of a texture analyzer in combination with a USP Apparatus 2, which enables similar application of pressures to the tested dosage form during dissolution as the device used in this paper, concluding that the physical interactions between the formulation and the gastrointestinal tract do indeed significantly alter the performance of polymer matrix-based drug products, supporting the need for devices such as the one used in this paper that mimic gastrointestinal compressions [9].

Fed stomach model dissolution system. a Enlarged view of the top of FSM gastric vessel. b Overall system schematic operating in closed-loop configuration. Reprinted with permission from Elsevier [7]
A far more intricate device is the TNO TIM-1, which is a multicompartmental device designed to simulate the various steps of digestion in the stomach, duodenum, jejunum, and ileum. The various parts of the digestive tract are modeled by a flexible-walled tube passing through a series of individual glass compartments connected by peristaltic valve pumps (see Fig. 2) [1, 10]. Flow of water through the glass compartment outside the flexible tube allows for temperature control at each stage, as well as computer-controlled, alternating pressure to be placed on the tube to facilitate mixing and simulate mechanical pressure placed on food or dosage forms by peristaltic action. Furthermore, the staged compartment design enables input of hydrochloric acid, enzymes, sodium bicarbonate, and various salts in each section to control pH, ionic strength, and digestive enzymes at each stage of the process to best simulate physiological conditions. Transit time is programmable to enable the system to simulate different fed- or fasted-state digestive models or to model different species’ digestive processes.

TNO TIM-1 apparatus. A) Stomach compartment; B) pyloric sphincter; C) duodenum compartment; D) peristaltic valve; E) jejunal compartment; F) peristaltic valve; G) ileum compartment; H) ileo-cecal sphincter; I) stomach secretion; J) duodenum secretion; K) jejunum/ileum secretion; L) prefilter; M) semipermeable membrane; N) filtrate pump; P) pH electrodes; Q) level sensors; R) temperature sensor; S) pressure sensor. Reprinted with permission from Springer Nature [10]
The complexity of the TNO TIM-1 device allows it to rather closely mimic the gastrointestinal environment, which has enabled it to be used as an effective in vitro tool for accurate prediction of in vivo performance. Numerous publications have shown its use for predicting absorption of various nutritional and pharmaceutical products [11,12,13,14]. Likewise, Naylor et al. have demonstrated improved in vivo prediction of oral dosage formulations by deconvoluting data taken from the TIM-1 device and then using the deconvoluted absorption data as inputs for GastroPlus simulations, as compared to standard dissolution data using the USP Apparatus 2 dissolution model [15]. However, the TIM-1 device does have some notable drawbacks. Namely, the relative complexity of the system requires a very large laboratory footprint, being housed in a large cabinet (see Fig. 3), that necessarily reduces throughput. Whereas a standard USP Apparatus 2 device can enable up to 6 simultaneous dissolution tests, a TIM device takes up more room in the lab and can only run one test at a time. Furthermore, although the external water pressure pulses help simulate mixing and mechanical stresses experienced in vivo, the TIM-1 device cannot fully reproduce the complex fluid mechanics and significant mechanical forces that can be encountered in vivo [16]. Finally, the TIM device relies on dialysis across a 10-kDa molecular weight cutoff membrane to simulate absorption of water-soluble products. While this molecular weight cutoff is useful for simulating in vivo levels of absorption, it prevents effective correlation of lipophilic products, which form micelles too large to pass through the membrane [10]. It also will not provide accurate correlation with any products that may experience active transport across the intestinal epithelium [14, 16].

TNO TIM-1 digestion model device. Reprinted with permission from John Wiley & Sons [3]
Because the lack of mechanical forces in a standard USP Apparatus 2 dissolution device can lead to poor in vitro prediction of in vivo behavior, while more advanced devices such as the TNO TIM-1 device are highly complex, we sought to develop a simple device that can be used in combination with traditional USP dissolution apparatuses to allow for more predictive dissolution behavior that mimics peristaltic compression forces. Herein, we describe experimental results demonstrating the lack of a correlation between USP Apparatus 2 dissolution results and in vivo results for an oral extended release dosage form, describe the development and application of a peristaltic dissolution device that simulates peristaltic forces to better mimic the in vivo environment, and discuss some additional applications of the device for modeling other administration routes such as intramuscular injections or chewable dosage forms.
Materials and Methods
Carvedilol Bilayer Modified Release Tablets
Carvedilol bilayer tablets used for dissolution testing herein were manufactured in a GMP environment with validated equipment. The immediate release layer of the bilayer tablets was produced by fluid bed granulation of carvedilol free base with mannitol powder, sucrose, microcrystalline cellulose, and crospovidone using an aqueous povidone solution. The wet granules were dried to an LOD of less than 1% moisture. The dried granules were then milled, blended with crospovidone, amorphous colloidal silicon dioxide, and magnesium stearate to create a final immediate release compression mix. The modified release layer of the bilayer tablets was produced by high shear wet granulation of carvedilol phosphate with mannitol powder, hypromellose K4M, and microcrystalline cellulose using an aqueous povidone solution. The hypromellose K4M excipient is used to slow the dissolution of the carvedilol. The wet granules were dried to an LOD of less than 1% moisture. The dried granules were milled, blended with microcrystalline cellulose, amorphous colloidal silicon dioxide, and magnesium stearate to create a final modified release compression mix. The compression mixes were loaded into separate feeders of a Korsch XL-400 bilayer tablet press, followed by compression into a tablet containing 10 mg of carvedilol free base in the immediate release layer and 40 mg carvedilol free base equivalents as carvedilol phosphate in the modified release layer. The bilayer tablet was then film coated with Opadry clear coat to create the final clinical product. These carvedilol bilayer tablets were part of a series of carvedilol modified release products evaluated in a clinical study, and further details of the manufacture can be found in Burke et al. [17]. In particular, example 32 of the cited work follows the exact same processing steps outlined above. Two additional manufacturing steps after the Opadry coating are also mentioned in example 32 but are not applicable for the carvedilol drug products described in this paper. The overall compositions of the bilayer tablets are shown in Table 1.
A USP Apparatus 2 (Hanson) was used to evaluate the dissolution of the carvedilol bilayer drug products. The method used was the validated quality control dissolution method used in regulatory submissions to release the batch for clinical dosing and to determine the product shelf life. The paddle speed was constant at 75 rpm and a temperature of 37 °C. The dissolution medium was 700 mL of 0.1 N HCl for the first 2 h, then 200 mL of a concentrated phosphate buffer with sodium dodecyl sulfate was added to adjust the pH to 7.0 with a final phosphate buffer concentration of 20 mM and a 0.5% SDS concentration. Samples were evaluated by UV spectroscopy at 332 nm with background correction at 380 nm.
Peristaltic Dissolution Device
A schematic of the peristaltic dissolution device as well as photographs of it attached to a USP Apparatus 2 is shown in Fig. 4, and details of the dimensions of the apparatus can be found in patent WO 2006_052742 [18]. The prototype peristaltic dissolution device was constructed of electropolished stainless steel parts that were machined at GSK, with food-grade silicone injection molded over the parts as required. The chamber which holds the tablet has an inner diameter of 25 mm, which was selected to accommodate most standard tablet sizes. The tablets were loaded into the device by removing a mesh screen at the base of the chamber (Super Corrosion Resistant 316 SS wire cloth, 20 × 20 mesh size, 0.018 in. wire diameter, cut to size (McMasters 9319T558)), placing the tablet against the silicone-coated piston, and then replacing the mesh screen. To standardize the initial piston depth at the start of the dissolution run, the depth was adjusted for each individual tablet height and set to ensure contact by compressing 20% into the dry height of the tablet, as measured by calipers. However, during dissolution, the tablet is expected to change dimensions. The flexibility of the bottom mesh screen and silicone coating on the piston head were designed to accommodate the changes while ensuring consistent performance. The movement of the piston was controlled by a pneumatic cylinder in this initial design, which was set at a supply pressure of 1 bar, which demonstrated consistent movement. The device was designed to not interfere with the operation of a conventional USP Apparatus 2, and instead complements the existing equipment. Unless specified, the peristaltic dissolution device was added to the existing USP Apparatus 2 method without any modification other than adjusting the paddle speed to 75 rpm to prevent coning of insoluble material at the bottom of the dissolution vessel. Three tablet samples (n = 3) were tested to create the mean dissolution profiles shown in all figures unless otherwise specified.

Schematic and pictorial representation of the peristaltic dissolution apparatus affixed to a conventional USP Apparatus 2 dissolution vessel. a Numbered components: 150—USP Apparatus 2 glass dissolution vessel; 200—USP Apparatus 2 impeller; 250—dissolution media sampler; 300—force application system (comprised of parts 310–355); 310—dosage form chamber’s external housing; 320—force-imparting mechanism (in this case, a piston [350]); 330—dosage form cylindrical chamber; 340—detachable mesh screen bottom of dosage form chamber; 350—piston; 355—drilled holes to enable fluid transfer [15]. b, c Pictures of a two-vessel peristaltic dissolution setup in a USP Apparatus 2
TNO TIM-1 Method
The high-fat procedure described by Minekus [10] was used to mimic the fed-state human dosing of the carvedilol bilayer tablets with the exception of a lower meal size. Two different meal sizes were evaluated previously by Burke et al. [19], and it was found that for carvedilol, a 60 g fed meal size best predicted the performance with the TNO TIM-1.
Clinical Trials
It is important to note that all pharmacokinetic data presented in the paper were obtained from fed-state clinical trials. The clinical trials were performed by GlaxoSmithKline and followed a similar procedure to Henderson et al. [20] and Johnson et al. [21]. Healthy adult volunteers between the ages of 18 and 55 years of age, body weight > 60 kg, and within − 20% or + 35% of ideal weight based on height and body frame were enrolled with the goal to complete dosing of at least 12 volunteers. Volunteers were administered one tablet after a standard FDA meal with water given ad libitum. Following an overnight fast of at least 10 h, volunteers were provided a standard FDA meal (~ 900 cal) 30 min prior to administration of the drug product. The drug product was administered with 240 mL (8 fl oz) of water. No food was allowed for 4 h post-dose. Water was allowed as desired except for 1 h before and after drug administration.
Plasma samples were withdrawn at regular intervals over 72 h periods for determination of drug content, thereby enabling profiles to be constructed. For the carvedilol study, one conventional, immediate release dosage form (commercial Coreg tablet) containing 25 mg of drug was additionally dosed at an interval of 12 h (giving a total dose of 50 mg) to provide comparative data. Blood samples were analyzed by a validated clinical trial method. Mean plasma profiles are shown for the reference immediate release products and the investigative drug products are shown in the appropriate figures.
Deconvolution
The pharmacokinetic data was deconvoluted using a Wagner–Nelson (1 compartment) model for carvedilol as described by George et al. [22, 23]. The elimination rate data was modeled from either IV human PK data from internal reports.
Carvedilol Phosphate Monolayer Modified Release Tablets
The manufacture of carvedilol phosphate monolayer modified release tablets followed the same procedure as the modified release layer from the carvedilol bilayer tablets with the hypromellose included in the granulation with microcrystalline cellulose, colloidal silicon dioxide, and magnesium stearate added extragranularly. The composition of the carvedilol phosphate monolayer is shown in Table 2. USP Apparatus 2 was used to evaluate the carvedilol phosphate monolayer drug products. The paddle speed was 100 rpm and the temperature was 37 °C, in 900 mL of 0.1 N HCl. Samples were evaluated by UV spectroscopy at 332 nm, per a validated quality control analytical method used in regulatory submissions to release the batch for clinical dosing and to determine the product shelf life. When the peristaltic dissolution device was added to USP Apparatus 2 to evaluate the product, there were no other changes to the method to create the potential for its use as a quality control method with the advantage of predicting clinically relevant performance.
Nicotine Gum
The drug release from a nicotine gum drug product was also assessed using the peristaltic dissolution device. The nicotine product evaluated was the commercially available 4 mg Nicorette Gum Original, Nicotine Polacrilex gum, lot HC411A. The peristaltic dissolution device was placed in a 250 mL glass vessel, from a USP 3 Apparatus, to use a smaller volume vessel than the typical USP 2 Apparatus. In this case, 200 mL of 0.01 M potassium dihydrogen phosphate buffer at pH 6.8 was the media used, which was magnetically stirred at medium speed. Sample analysis followed the method of Tambwekar et al. [24].
GSK2838232 Long-Acting Injectable
The GSK2838232 long-acting injectable formulation was created using a suspension of micronized API (D50 of ~ 10 μm) at a concentration of 200 mg/mL, in a vehicle containing 0.2% polysorbate 80, 2% PEG3350, 4.5% mannitol, and 10 mM phosphate buffered saline. An intramuscular injection (right gastrocnemius) of 0.1 mL was performed in male Sprague–Dawley rats, and the injection site depot was excised after 2 months, being careful not to compromise the depot structure. Pharmacokinetic data from the in vivo portion of the experiment was presented previously, along with further rat study details and the API molecular structure [25].
A Distek 2100 dissolution system with the peristaltic dissolution device was used for further ex vivo study. A drug release experiment from excised tissue sample was carried out in 1.0 L of PBS buffer containing 20.00 mg/mL bovine serum albumin (BSA) and 0.50 mg/mL sodium azide for 7 days at 37 °C with 75 rpm stirring speed throughout the experiment. For the first 2 days, both depots were placed inside the peristaltic compression basket, but the compression was not used. At the 48 h time point, the tissue samples were punctured using an 18G needle through the center of the sample and remained in buffer for continued dissolution without peristaltic compression to test the effect of the granuloma on drug release. Finally, at approximately the 5.9 day time point, the peristaltic compression was added and operated continuously in a cycle of 3 s of compression followed by 6 s between compressions through the end of the experiment; 1.0 mL samples of the dissolution media were collected approximately daily throughout the experiment. At the end of the 7 days, the tissue samples were removed and homogenized in 45 mL PBS with 20 mg/mL BSA in order to determine the total amount of API in the sample, enabling calculation of percent release of drug over the studied time frame. Concentrations of GSK2838232 were determined using a solid-phase extraction and UHPLC/MS method (sensitivity 10 pg/mL).
To numerically compare drug release rates between the in vivo and ex vivo conditions, the rat in vivo long-acting injectable pharmacokinetic data was deconvoluted using the Wagner–Nelson method with the elimination rate constant calculated from oral dosing in rats. The last seven data points (between 336 and 1344 h) were used to estimate the constant in vivo release rate. The percent release obtained from the Wagner–Nelson deconvolution was converted to micrograms released by multiplying the percent value by the total drug injected minus the remaining quantity in the ex vivo depot. The r2 value was 0.9939 with a slope of 3.848 μg/h or 92.4 μg/day released in vivo during the last ~ 1000 h of the in vivo study, just prior to depot extraction.
Results and Discussion
Traditional Dissolution and Pharmacokinetics of Carvedilol Bilayer
Dissolution is a common tool to guide the formulation development to an “optimal” formulation, yet it is well known that in vitro dissolution is often not predictive of in vivo performance. This can be especially true in the case of matrix tablet dosage forms for slow release of the active ingredient. For a matrix tablet, it has been concluded that gastric and intestinal hydrodynamics can be strong enough to destroy the integrity of the matrix and result in dose dumping of the active. This is more commonly observed in the fed state [26].
Figure 5 shows the dissolution profile of two modified release formulations of carvedilol. These are bilayer tablets, with an immediate release layer and a sustained release layer using hypromellose K4M as the matrix-forming polymer. The key difference between the formulations is the amount of K4M in the formulations—one formulation contained 20% K4M, while the other contained 25% K4M, with increasing K4M content expected to slow the drug release rate. The overall goal was to create a once-a-day product to replace the commercial twice-a-day immediate release product.

In vitro USP Apparatus 2 dissolution of carvedilol bilayer matrix tablets containing two different levels of hypromellose K4M
As expected, the immediate release layer provides a burst of the drug followed by a slow prolonged release of the active from the sustained release hypromellose matrix layer. The rate of release from the matrix tablet layer is dependent upon the quantity of hypromellose used. For the 25% K4M tablet, the release profile appears to be approximately linear (zero-order) with respect to time over an approximately 16 h time frame. Based on the prolonged release of the active ingredient from this in vitro data, it would be expected that the in vivo pharmacokinetic profile would be quite flat with a relatively small Cmax to Cmin ratio.
A clinical study was subsequently performed to evaluate these prototype modified release formulations in humans. The resulting pharmacokinetic profile is shown in Fig. 6, where the in vivo performance revealed a significantly faster drug release than was predicted by the in vitro data. In fact, the result could be described as dose dumping, which occurred with both of the “sustained release” formulations and mirrored the behavior of an immediate release tablet with the majority of the drug having been released within 6 h. The immediate release tablets were dosed two times, at 0 h and 12 h, yielding the second response curve that is not observed in the bilayer tablets. However, over the initial 6 h, the shape of the concentration profile is very similar between the IR and SR formulations, despite the in vitro dissolution data shown in Fig. 5 suggesting sustained release behavior for ≥ 16 h. This discrepancy demonstrates that the traditional in vitro dissolution method was failing as a predictive tool for in vivo performance.

Human pharmacokinetic clinical data for carvedilol bilayer matrix tablets. a The mean data for immediate release (IR) tablets were given at t = 0 h and 12 h. Mean data for modified release tablets were given at t = 0 h only. b The individual data for the carvedilol bilayer matrix tablets containing 20% K4M demonstrating the variability and potential for high individual Cmax events
The above clinical data was deconvoluted using the Wagner–Nelson method to yield a plot of percent drug absorbed versus time. The resulting data is shown and compared to the in vitro dissolution profiles in Fig. 7, which shows that both tablets exhibited nearly complete drug release over an approximately 5 h time period in humans. However, as the in vitro dissolution predicted approximately a 16 h dissolution time, there must be a critical variable that the USP Apparatus 2 does not simulate which results in the ~ 3× faster rate of dissolution and absorption in vivo.

Comparison of in vitro dissolution profile and deconvoluted human pharmacokinetic clinical data for carvedilol bilayer matrix tablets
Peristaltic Dissolution Device Development and Testing
Initial attempts to adjust existing dissolution method parameters, such as using paddle speeds of 200 rpm, did not produce predictive dissolution results, suggesting the need for additional mechanical stressing. To create a more biorelevant testing of oral dosage forms, we developed the “peristaltic dissolution” device, borrowing the word “peristaltic” from the peristalsis movements that occur throughout the gastrointestinal tract. The ubiquity of the USP Apparatus 2 in dissolution testing within the pharmaceutical industry encouraged our efforts to make a device that could be simply added to the existing apparatus to add the mechanical stresses while leveraging the existing quality control systems for sampling, mixing, and temperature control offered natively by the familiar Apparatus 2.
Various iterations of the device were tested to optimize materials, chamber size, and compression force and frequency. The final device is shown in schematic and pictorial form in the “Materials and Methods” section, Fig. 4. The device consists of a pneumatically driven, double-acting piston controlled by a computer that applies pressure on a tablet (or other dosage form) at regular intervals. The tablet is housed within a 25 mm inner diameter steel chamber with multiple vertical openings in the sides and a wire mesh bottom, which allows for good mass transfer with the surrounding medium while also keeping the tablet in place for repeated compressions. The piston and chamber are mounted onto a typical USP Apparatus 2. The level of force applied can be controlled by varying the inlet pressure of the supply gas that drives the piston down. The length of time that the compression is maintained, the length of time between compressions, and the overall length of the compression program are maintained by the electronic controller. Electropolished stainless steel with injection-molded silicone coating was chosen as the apparatus construction material, for its versatility in a wide range of buffer types and pH values and for its ability to be easily cleaned and maintained.
The first evaluation of this device utilized the previously described carvedilol bilayer matrix tablets to determine the effect of compression frequency. In this evaluation, 3 s compressions were applied to the bilayer tablets with the time interval between compressions varying from 6 to 360 s. The rate of dissolution was measured over 16 h. The resulting dissolution profiles are shown in Fig. 8 a and b.

Effect of compression frequency on dissolution of carvedilol bilayer matrix tablets—a (left) the data for the 20% K4M hypromellose product and b (right) the data for the 25% K4M hypromellose product. The numbers in the legend correspond to “time compression (s), time between compressions (s).” For example, “3, 6” corresponds to repeated cycles of 3 s where compression is applied followed by 6 s of no compression, repeated throughout the experiment. The “control” sample was simple dissolution with no compression in a USP Apparatus 2 device with the tablet at the bottom of the vessel. The “cage” sample was the bilayer matrix tablet placed inside the peristaltic device cage (mounted within a USP Apparatus 2 device) with stirring but no compression
As expected, based on the results of the in vivo PK and scintigraphy studies, the application of compression accelerates the rate of dissolution, with more frequent compressions leading to an increase in the drug release rate. Compared to the control sample, placing the dosage form in the peristaltic dissolution device cage, which is at an elevated position in the USP Apparatus 2 dissolution vessel, did accelerate dissolution. This is due to known hydrodynamic differences within the USP Apparatus 2 vessel: loose dosage forms often collect at the base of the vessel during dissolution, below the paddle, where a zone of reduced hydrodynamic mixing occurs as compared to off-center positions above the paddle [27], leading to accelerated dissolution when the dosage form is held in the peristaltic dissolution device cage. While the increased hydrodynamic forces experienced at the elevated position contributed to the increased dissolution rate, this alone was not sufficient to accurately mimic the observed in vivo dissolution rate. As seen in Fig. 8, the samples experiencing compressions from the peristaltic dissolution device reached complete dissolution much faster than the control samples—approximately 7–10 h (in 20% or 25% HPMC tablets, respectively) for the “3, 6” compression cycle versus approximately 16 h for the control. These results better mimic those from the previously described human study, where the carvedilol bilayer tablets were completely absorbed within 5 h in humans versus the predicted 16 h time frame based on standard in vitro dissolution, indicating that the addition of peristaltic compression by this device better simulates in vivo performance compared to traditional dissolution techniques.
The remaining discrepancy between the peristaltic dissolution profiles and the fed-state clinical data is the brief, approximately 0.5 h “lag time” in the clinical data. Whereas the in vitro experiment immediately places the tablet into the sampled sink and applies compression immediately, resulting in an instantaneous increase in the dissolved API, there is a small time in vivo during which the drug must transport into systemic circulation before it will be observed in the data, which is not mimicked by in vitro setups. This lag time difference was also observed with the traditional Apparatus 2 device, so it is an inherent disadvantage to the dissolution setup where the dosage form is placed directly in the sampled media. For very fast dissolving, immediate release formulations, this lag time could introduce more significant error in the dissolution. However, for extended release dosage forms, the lag time is not nearly as substantial as the overall improvement in clinical modeling by using the peristaltic dissolution device.
A slightly improved profile was obtained by introducing a 1 h lag time between the start of the dissolution test and the start of the compression cycle (Fig. 9). There is physiological justification for this as the initial mixing waves in the fed-state stomach are relatively gentle followed by more intense waves and increasing force. A direct comparison of the dissolution data using the 1 h lag with the “3, 6” compression cycle and the deconvoluted pharmacokinetic data from the human trial is shown in Fig. 10. Although there is some discrepancy between the two peristaltic dissolution profiles and the two clinical profiles at early time points, the overall rate of dissolution is quite similar to the rate of absorption in the deconvoluted clinical data, especially as compared to the vastly different profiles observed between the deconvoluted data and traditional USP Apparatus 2 dissolution shown in Fig. 7. As such, it appears that the addition of peristaltic compressions has recreated more physiologically relevant luminal hydrodynamics which translates to more clinically relevant drug release kinetics.

Evaluation of the carvedilol bilayer matrix tablet containing 20% K4M hypromellose with a “3, 6” compression cycle corresponding to repeated cycles of 3 s where compression is applied followed by 6 s of no compression, repeated throughout the experiment. The initiation of the compression cycle was varied to start at the same time as the dissolution test (0 h lag), after a 1 h lag time or after a 2 h lag time

Comparison of peristaltic dissolution data to deconvoluted human pharmacokinetic data
When the peristaltic dissolution device was applied to future oral projects, a standard protocol was used without modification which was a 1 h lag time followed by the “3, 6” compression cycle. The peristaltic device was simply added to existing dissolution method approaches without changes to the dissolution media or method. The only exception was adjustment of the paddle speed to 75 rpm.
For comparison, the carvedilol bilayer tablets were also tested using the TNO TIM-1. The results of the study are shown along with the deconvoluted clinical data in Fig. 11. Like the peristaltic dissolution device, the TNO TIM-1 data produces absorption profiles that are much more similar to the deconvoluted clinical absorption profile than the USP Apparatus 2. However, some slight deviation is observed, namely that the lag phase is overestimated by the TNO TIM-1, showing absorption only beginning to accumulate at times well after 1 h, whereas actual absorption begins to accumulate within 30 min after dosing. It then corrects by slightly overestimating the rate of absorption over 1–3 h. This is the opposite of the peristaltic dissolution device, which did not model the lag phase, instead showing significant absorption (~ 40%) within the first 30 min but then closely predicting the rate of absorption at times > 1 h. As such, the peristaltic dissolution device produced a slightly better match to the clinical absorption profile over the entire course of the experiment than the TNO TIM-1, but overestimated the initial absorption phase. Nevertheless, both devices give a good representation of actual absorption over time, especially as compared to USP Apparatus 2 which significantly underpredicts the dissolution rate.

TNO TIM-1 evaluation of the carvedilol bilayer matrix tablets. a The total concentration from all sampling ports at each time point when the tablets were administered with a small meal of 60 g. b The deconvolution of the TNO TIM-1 results compared to the deconvolution of the clinical pharmacokinetic data
Further Evidence of Applicability and Predictive Capability
The above data were generated in a retrospective manner using carvedilol bilayer matrix tablets containing both an immediate release layer and a modified release matrix layer. Here, we demonstrate the ability of the developed device and standard method to accurately predict in vivo oral pharmacokinetics with an additional carvedilol phosphate matrix tablet which was evaluated in a clinic trial. In this case, the carvedilol phosphate salt was used instead of the carvedilol free base due to a higher solubility of the salt form over the free base. Higher solubility compounds are known to be at higher risk of dose dumping in the event of tablet erosion or rupture [28], making the carvedilol phosphate product an interesting additional test case for the peristaltic dissolution device, to see how the effects of the mechanical forces affect the in vitro–in vivo correlation with a relatively high solubility compound.
Carvedilol Phosphate Sustained Release Matrix Tablets
Dissolution testing of a carvedilol phosphate matrix tablet, which contained only a sustained release matrix with no immediate release component (composition shown in Table 2), using the standard USP Apparatus 2 device produced the dissolution profile shown in Fig. 12. Clearly, there is a substantial difference between the slower, relatively flat (zero-order) dissolution profile generated by the USP Apparatus 2 device and the deconvoluted dissolution profile obtained from in vivo human data. As with the carvedilol bilayer matrix tablets, it appears the Apparatus 2 device is significantly underpredicting the rate of dissolution in vivo, especially after 1 h post-administration, when GI mechanical forces become major contributing factors. Meanwhile, the peristaltic dissolution presented in this paper produced a dissolution profile that better predicts the in vivo fed-state performance.

Comparison of a USP Apparatus 2 dissolution profile of a carvedilol phosphate matrix tablet to peristaltic dissolution data and deconvoluted human PK data
This result produces further evidence that the compressive forces of the GI tract play a significant role in the performance of oral solid-dosage forms, which the peristaltic dissolution device simulates in vitro. Further, with no immediate release component in these carvedilol phosphate tablets, the discrepancy in results using only the Apparatus 2 device is not specific to the dual layers of the previously discussed carvedilol bilayer tablets and may instead be more widely applicable to various solid-dosage forms. Being able to predict the ultimate in vivo pharmacokinetics from an in vitro tool could help reduce the number of relatively expensive and arduous animal studies required for drug product testing and, therefore, could significantly reduce the time needed to produce a working formulation. The peristaltic dissolution device developed and described herein provides a tool that achieves this in an efficient manner without requiring significant changes to dissolution media, protocols, or equipment, as it simply attaches onto an existing USP Apparatus 2.
Additional Applications
The peristaltic dissolution device presented here has been demonstrated to offer an in vitro tool that better mimics the actual gastrointestinal environment experienced by oral modified release drug products, yielding a far more accurate prior prediction of in vivo pharmacokinetics from in vitro dissolution testing. However, the design of the device also allows for use in some additional applications where compressive, mechanical forces improve the in vivo dissolution prediction.
Medicated Gums
Medicated gum products present a challenge to in vitro performance evaluation because the primary driver for drug release is the continuous mastication of the product, which is not simulated in traditional dissolution setups. Furthermore, the United States Pharmacopeia monograph for medicated gum products does not specify a drug release test or a compendial apparatus to be used for testing product performance [29]. The European Pharmacopeia does specify a compendial apparatus for testing gum products, which places the product inside a small chamber in solution and alternately applies pressure to the product using a vertical piston (simulating the tongue) and two horizontal pistons (meant to simulate chewing). This device and a similar, noncompendial device developed by Kvist et al. [30] have been utilized in numerous successful testing cases [31,32,33], but are standalone devices that may not be initially owned by many companies or research institutions and are not more broadly applicable to testing other dosage forms. Based on the adjustable, compressive piston action offered by the peristaltic dissolution device presented herein and the containment of the dosage form within the test chamber, the peristaltic dissolution device can potentially be used as a noncompendial analog of the chewing device that research institutions can use for in vitro testing of medicated gum products while also applying to peristaltic dissolution testing of other oral solid-dosage products, and attaches onto a standard USP Apparatus 2 device common to research institutions.
A demonstration was run using a nicotine gum product in the peristaltic dissolution device. The chew rate settings were selected based on a paper showing an increase in nicotine release with increased chewing rate [34]. The resulting data is shown in Fig. 13 and shows the impact of the peristaltic “chewing” action on the release of the API. With no compression, only 3% of the nicotine was dissolved after 60 min, whereas with chewing, nearly 60% of the nicotine was dissolved. Further, a compression every 4 s produces a faster release rate than less frequent chewing with a compression every 20 s. Given the vast difference in drug release between the data with no compression and that with compression, it is apparent that simulating chewing is necessary for proper testing of these drug products. Varying the compression rate can also provide insight into how robust the product design is in terms of delivering a suitable quantity of drug within a reasonable time, given the significant person-to-person variance in chewing tendencies. While this product was merely tested as a proof of concept, the results are consistent with prior clinical data that shows an increased chewing rate yields higher drug release and plasma profiles [34], suggesting that this peristaltic dissolution device can offer a relatively simple method of simulating chewing gum product performance that is consistent with the trends observed in clinical trials.

Testing of a nicotine gum product using the peristaltic dissolution apparatus where the simulated chew rate was varied
Gastroretentive Dosage Testing
Another potential application for the peristaltic dissolution device is in testing of gastroretentive dosage forms. A number of different approaches have been taken to keeping the oral dosage product in the stomach: low-density dosage forms to float atop the gastric fluid, high-density dosage forms that will sink to the retentive portion of the stomach, bioadhesion to the mucosal lining of the stomach, co-delivery of motility-slowing drugs or excipients, or expansion to a size larger than the pyloric sphincter as occurs with the Lyndra capsule formulation [35]. Recently, the expanding dosage form that remains in the stomach by swelling or unfolding to a size larger than the pyloric sphincter has become the most prominently studied gastroretentive dosage form [36].
Retention in the gastric environment will subject the dosage form to prolonged and significant compressive forces during stomach emptying. The pyloric sphincter is 12.8 (± 7) mm in diameter, and dosage forms approximately 13 mm in diameter or larger are retained approximately 3 h (approximately 1 h longer than a 7-mm tablet would be), while larger dosage forms (3+ cm diameter, or 5+ cm length) may be completely unable to pass through the pyloric sphincter and thus are retained throughout the stomach’s “housekeeper waves” until they degrade to a sufficiently small size to pass through [36]. As the product remains in the stomach, the dosage form will experience the muscular contractions of the stomach which, as demonstrated above, can contribute significantly to the pharmacokinetics of the drug release and absorption from the solid-dosage form.
In order to test such dosage forms, a small modification can be made to the peristaltic dissolution device that introduces a larger chamber to mimic the antrum of the stomach and a 13-mm “pylorus” at the base of the peristaltic test chamber, as shown in Fig. 14. As the peristaltic compressions take place during dissolution testing, the gastroretentive device will be held in the compression chamber, where it experiences simultaneous physical compression and chemical dissolution as driving forces for API release. However, as the gastroretentive device degrades or physically erodes into pieces sufficiently small to pass through the 13 mm pylorus, the smaller fragments are pushed out into the surrounding medium where they will continue to experience dissolution in the test media, but without continuing to experience the significant compressive forces encountered in the compression chamber. As such, this setup could be used to mimic the digestive environment that would be experienced by a gastroretentive device if simulated gastric fluid is used as the test media and potentially predict the time of gastric emptying by the housekeeper wave in vivo. Although the nonretained particles would pass back into gastric test fluid, not intestinal, with this setup, the removal of the eroded fragments from the intense compressions experienced in the stomach chamber nevertheless yields a more biorelevant model of the GI digestive tract for gastroretentive dosage forms than a standard dissolution apparatus. Slight optimization of the compression protocol may be needed to better model these unique dosage forms, but the simple exchange of piston and chamber preserves the customizable nature of the peristaltic dissolution device, enabling straightforward optimization of the compression timing and intensity.

a CAD drawing of an available modification to the peristaltic dissolution device for testing gastroretentive solid-dosage forms. b Fabricated compression chamber and piston for testing gastroretentive devices (right) compared to the standard compression chamber and piston head (left)
Intramuscular Injection Modeling
The ability to add intermittent compressions may also enable the modeling of additional routes of administration with some slight modification to the dissolution protocol. As an example, the drug release of GSK2838232 from an intramuscular injection depot was studied ex vivo following intramuscular injection of a suspension of the product and tissue/injection site excision. The purpose of the study was twofold: to determine the effect of granuloma formation on the drug release and to determine the effect of muscular contractions on drug release as simulated by intermittent compression of the excised muscle tissue using the peristaltic dissolution device. The data obtained from the dissolution study is shown in Fig. 15, and calculated release rates from the different phases of the experiment are compared to the prior in vivo experiment in Table 3.

Ex vivo dissolution modeling of intramuscular injection drug depot using peristaltic dissolution device and long-acting injectable suspension of GSK2838232. Initial time period—no compressions, no puncture of granuloma. Second time period—granuloma punctured with 18G needle, no compressions. Final time period—granuloma punctured, peristaltic compressions added
Compromising the integrity of the granuloma surrounding the site of injection did not appear to have a significant effect on drug release rate of GSK2838232B, whereas adding peristaltic compressions to mimic muscle contractions yielded a multifold increase in drug release rate. As a result, we conclude that the fully formed biological foreign body response (i.e., the granuloma formed around the site of injection) did not significantly affect drug release rates for this molecule, but the site of injection (intramuscular vs. subcutaneous) may cause very significant differences in the rate of drug exposure. While this result may seem surprising given that other studies have previously shown that the foreign body response has significant impact on the active release from intramuscular depots, it may yet fit with the prior knowledge when considering the granuloma effect is only being studied well after it has fully formed around the depot and most importantly fully vascularized [37,38,39]. In our case, the depot was excised after 2 months when the granuloma was in a state more open to mass transfer rather than in the very initial stages of granuloma formation, where the local area is not fully vascularized and the granuloma tissue has previously shown to present a greater barrier to dissolution and absorption. Meanwhile, muscular contractions, mimicked in this experiment by the peristaltic compressions, appear to substantially increase drug release rate through the surrounding tissue. As such, differences in degree of muscle use may be a factor affecting clinical variability between patients receiving intramuscular injections, and the simulation of muscle contractions by the use of the peristaltic dissolution device could enable more effective in vitro modeling of predicted in vivo drug release rate.
The data in Table 3 reveals that the current ex vivo depot dissolution study underpredicted the drug release rate compared to the in vivo data; nevertheless, adding the compression element significantly increased the mean drug release to a value much closer to the deconvolution result. The remaining discrepancy could be due to the lack of biologic mechanisms leading to drug removal from the depot such as macrophages or active blood flow, due to the limited amount of drug remaining in the depot slowing release, or due to the need for further optimization of the compression sequence.
Further study would be needed to determine optimal test parameters for simulating actual muscle contraction and intramuscular injection pharmacokinetics. Additionally, the use of excised muscle tissue is not ideal; development of a fully in vitro analog of muscle tissue could help create a more broadly useful in vitro tool. However, this simple study shows that the periodic deformation of the tissue around an injection site due to muscle contractions or similar events can be a significant contributing factor to drug absorption rate and patient-to-patient pharmacokinetic variability. This data suggests that the use of the peristaltic dissolution device could help better inform formulation optimization to minimize the impact of muscle contractions on release rate from the depot and better model expected performance in vivo. The simple addition of mechanical forces to evaluate a variety of dosage forms can be very useful for better probing the expected clinical profile of a molecule using in vitro tools, even if the intended administration route is not the oral pathway.
Conclusion
The peristaltic dissolution device presented here creates a more biorelevant dissolution apparatus for testing the performance of oral solid-dose products than the standard USP in vitro dissolution apparatuses, which have historically failed to correlate with human clinical pharmacokinetic data. Data presented herein detailing the development and testing of a carvedilol modified release bilayer tablet, coupled with a scintigraphy study performed in beagles, demonstrated that one of the contributing factors in this failure to correlate with clinical data is the inability of USP Apparatus 2 to apply mechanical forces that will be experienced in the gastrointestinal environment due to peristaltic contractions and gastric digestion and emptying forces. The device presented in this paper simulates these forces by adding a compression chamber for the oral solid-dosage form to the standard paddle apparatus, in which a programmable piston repeatedly applies a compressive force to the dosage form while still allowing for standard dissolution into the test media, simultaneously simulating both of the mechanisms facilitating in vivo drug dissolution and absorption.
Testing the device with the carvedilol bilayer tablets, a standard testing protocol using a 3 s compressive pulse with 6 s between compressions was selected as a suitable protocol for the device to generate in vitro drug dissolution profiles that match deconvoluted clinical absorption data. This protocol also yielded in vitro drug dissolution profiles for an additional oral modified release drug products that closely matched the deconvoluted clinical absorption data without requiring a modification to the dissolution media used, demonstrating that the device and protocol are effective in simulating the in vivo gastrointestinal environment using in vitro methods for a range of oral solid-dose drug products.
We anticipate that this device will be very useful for the future testing of oral solid-dosage forms. More accurate prediction of in vivo pharmacokinetics using only in vitro tools can help speed product development cycle time and thus greatly reduce development costs by reducing the number of iterative animal studies and formulation redesign steps needed before proceeding into human trials. Furthermore, this device attaches onto standard USP Apparatus 2 dissolution setups that are ubiquitous throughout formulation development research institutions, meaning no large additional equipment to purchase or store. As such, this peristaltic dissolution device offers a simple and effective, yet resource-efficient alternative to other biorelevant dissolution testing systems that have been described previously, such as the TNO TIM device, which are very complex, require bulky standalone equipment, and lack parallel throughput capability. Furthermore, the device’s customizable design may enable it to be further applied in the in vitro testing of additional drug products, such as medicated gums, gastroretentive dosage forms, or intramuscular injections, with only minor modifications.
Data Availability
Clinical data used in this manuscript has been previously published and citations to the published data are included herein.
Abbreviations
- API:
-
Active pharmaceutical ingredient
- BSA:
-
Bovine serum albumin
- CTAB:
-
Cetyltrimethylammonium bromide
- GI:
-
Gastrointestinal
- GSK:
-
GlaxoSmithKline
- IVIVC:
-
In vitro–in vivo correlation
- LOD:
-
Limit of detection
- PBS:
-
Phosphate buffered saline
- PEG:
-
Polyethylene glycol
- PK:
-
Pharmacokinetics
- QbD:
-
Quality by design
- TNO TIM-1:
-
TNO [organization name] intestinal model 1
- UHPLC/MS:
-
Ultra-high performance liquid chromatography–mass spectrometry
- USP:
-
United States Pharmacopeia
References
Kostewicz ES, Abrahamsson B, Brewster M, Brouwers J, Butler J, Carlert S, et al. In vitro models for the prediction of in vivo performance of oral dosage forms. Eur J Pharm Sci. 2014;57:342–66.
Reppas C, Vertzoni M. Biorelevant in-vitro performance testing of orally administered dosage forms. J Pharm Pharmacol. 2012;64:919–30.
Garbacz G, Klein S. Dissolution testing of oral modified-release dosage forms. J Pharm Pharmacol. 2012;64:944–68.
Cassilly D, Kantor S, Knight LC, Maurer AH, Fisher RS, Semler J, et al. Gastric emptying of a non-digestible solid: assessment with simultaneous SmartPill pH and pressure capsule, antroduodenal manometry, gastric emptying scintigraphy. Neurogastroenterol Motil. 2008;20:311–9.
Simmons DL, Legore AA, Picotte P, Lee KS, Joshi NN. A dissolution rate apparatus for the prediction of initial drug absorption patterns in beagles: tolbutamide tablets. J Pharmacokinet Biopharm. 1975;3:39–49.
Simmons DL. Peristaltic dissolution assembly as a bioequivalence surrogate: a review. 2016.https://doi.org/10.13140/RG.2.1.2436.6961
Koziolek M, Görke K, Neumann M, Garbacz G, Weitschies W. Development of a bio-relevant dissolution test device simulating mechanical aspects present in the fed stomach. Eur J Pharm Sci. 2014;57:250–6.
Garbacz G, Wedemeyer R-S, Nagel S, Giessmann T, Mönnikes H, Wilson CG, et al. Irregular absorption profiles observed from diclofenac extended release tablets can be predicted using a dissolution test apparatus that mimics in vivo physical stresses. Eur J Pharm Biopharm. 2008;70:421–8.
Gao Z, Ngo C, Ye W, Rodriguez JD, Keire D, Sun D, et al. Effects of dissolution medium pH and simulated gastrointestinal contraction on drug release from nifedipine extended-release tablets*. J Pharm Sci. 2019;108:1189–94.
Minekus M. The TNO Gastro-Intestinal Model (TIM). In: Verhoeckx K, Cotter P, López-Expósito I, Kleiveland C, Lea T, Mackie A, et al., editors. The Impact of Food Bioactives on Health: in vitro and ex vivo models. Cham: Springer International Publishing. 2015;37–46.https://doi.org/10.1007/978-3-319-16104-4_5
Verwei M, Freidig AP, Havenaar R, Groten JP. Predicted serum folate concentrations based on in vitro studies and kinetic modeling are consistent with measured folate concentrations in humans. J Nutr. 2006;136:3074–8.
Mateo Anson N, Havenaar R, Bast A, Haenen GRMM. Antioxidant and anti-inflammatory capacity of bioaccessible compounds from wheat fractions after gastrointestinal digestion. J Cereal Sci. 2010;51:110–4.
Larsson M, Minekus M, Havenaar R. Estimation of the bioavailability of iron and phosphorus in cereals using a dynamic in vitro gastrointestinal model. J Sci Food Agric. 1997;74:99–106.
Krul C, Luiten-Schuite A, Baan R, Verhagen H, Mohn G, Feron V, et al. Application of a dynamic in vitro gastrointestinal tract model to study the availability of food mutagens, using heterocyclic aromatic amines as model compounds. Food Chem Toxicol. 2000;38:783–92.
Naylor TA, Connolly PC, Martini LG, Elder DP, Minekus M, Havenaar R, et al. Use of a gastro-intestinal model and Gastroplus™ for the prediction of in vivo performance. J Appl Ther Res. 2006;6:15.
Kong F, Singh RP. Disintegration of solid foods in human stomach. J Food Sci. 2008;73:R67–80.
Burke M, Coffin M, Lamey K, Martini L, Oh C, Peterson H, et al. Carvedilol free base, salts, anhydrous forms or solvates thereof, corresponding pharmaceutical compositions, controlled release formulations, and treatment or delivery methods. 2006. Available from: https://patents.google.com/patent/US20060182804A1/en?oq=us20060182804a1.
Burke MD, Maheshwari CR, Zimmerman BO. Pharmaceutical analysis apparatus and method [Internet]. 2006. Available from: https://patents.google.com/patent/WO2006052742A2/en?oq=WO2006052742.
Burke M, Beato S, Barish P, Doucet D, Casazza A, Coffin M. Advanced in-vitro dissolution apparatus: novel peristaltic dissolution and TNO TIM-1. 2004.
Henderson LS, Tenero DM, Campanile AM, Baidoo CA, Danoff TM. Ethanol does not alter the pharmacokinetic profile of the controlled-release formulation of carvedilol. J Clin Pharmacol. 2007;47:1358–65.
Johnson M, Jewell RC, Peppercorn A, Gould E, Xu J, Lou Y, et al. The safety, tolerability, and pharmacokinetic profile of GSK2838232, a novel 2nd generation HIV maturation inhibitor, as assessed in healthy subjects. Pharmacol Res Perspect. 2018;6:e00408.
George JK, Singh SK, Verma P. In vivo in silico pharmacokinetic simulation studies of carvedilol-loaded nanocapsules using GastroPlus™. Ther Deliv. 2016;7:305–18.
Langenbucher F. Handling of computational in vitro/in vivo correlation problems by Microsoft Excel: III. Convolution and deconvolution. Eur J Pharm Biopharm. 2003;56:429–37.
Tambwekar KR, Kakariya RB, Garg S. A validated high performance liquid chromatographic method for analysis of nicotine in pure form and from formulations. J Pharm Biomed Anal. 2003;32:441–50.
Burke M. Transforming the patient experience through long acting injectable/implantable formulations: new opportunities and technologies [Internet]. Washington, DC; 2018. Available from: https://www.researchgate.net/publication/331044264_Keynote_Speaker_Transforming_the_Patient_Experience_through_Long_Acting_InjectableImplantable_Formulations_New_Opportunities_and_Technologies.
Abrahamsson B, Roos K, Sjögren J. Investigation of prandial effects on hydrophilic matrix tablets. Drug Dev Ind Pharm. 1999;25:765–71.
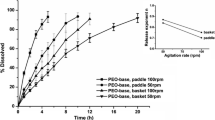
Todaro V, Persoons T, Grove G, Healy AM, D’Arcy DM. Characterization and simulation of hydrodynamics in the paddle, basket and flow-through dissolution testing apparatuses - a review. Dissolution Technol. 2017;24:24–36.
Maderuelo C, Zarzuelo A, Lanao JM. Critical factors in the release of drugs from sustained release hydrophilic matrices. J Control Release. 2011;154:2–19.
Gajendran J, Kraemer J, Knudsen SR. Product performance test for medicated chewing gums. Dissolution Technol. 2010;17:15–8.
Kvist C, Andersson SB, Fors S, Wennergren B, Berglund J. Apparatus for studying in vitro drug release from medicated chewing gums. Int J Pharm. 1999;189:57–65.
Morjaria Y, Irwin WJ, Barnett PX, Chan RS, Conway BR. In Vitro Release of Nicotine From Chewing Gum Formulations. Dissolution Technol. 2004;11:12–5
Yang X, Wang G, Zhang X. Release kinetics of catechins from chewing gum. J Pharm Sci. 2004;93:293–9.
Faraj JA, Dorati R, Schoubben A, Worthen D, Selmin F, Capan Y, et al. Development of a peptide-containing chewing gum as a sustained release antiplaque antimicrobial delivery system. AAPS PharmSciTech. 2007;8:E177–85.
Nemeth-Coslett R, Benowitz NL, Robinson N, Henningfield JE. Nicotine gum: chew rate, subjective effects and plasma nicotine. Pharmacol Biochem Behav. 1988;29:747–51.
Bellinger AM, Jafari M, Grant TM, Zhang S, Slater HC, Wenger EA, et al. Oral, ultra–long-lasting drug delivery: application toward malaria elimination goals. Sci Transl Med. 2016;8:365ra157.
Klausner EA, Lavy E, Friedman M, Hoffman A. Expandable gastroretentive dosage forms. J Control Release. 2003;90:143–62.
Anderson FD, Archer DF, Harman SM, Leonard RJ, Wilborn WH. Tissue response to bioerodible, subcutaneous drug implants: a possible determinant of drug absorption kinetics. Pharm Res. 1993;10:369–80.
Darville N, van Heerden M, Vynckier A, De Meulder M, Sterkens P, Annaert P, et al. Intramuscular administration of paliperidone palmitate extended-release injectable microsuspension induces a subclinical inflammatory reaction modulating the pharmacokinetics in rats. J Pharm Sci. 2014;103:2072–87.
Darville N, van Heerden M, Mariën D, De Meulder M, Rossenu S, Vermeulen A, et al. The effect of macrophage and angiogenesis inhibition on the drug release and absorption from an intramuscular sustained-release paliperidone palmitate suspension. J Control Release. 2016;230:95–108.
Acknowledgments
The authors would like to acknowledge the Jeff Seely and Distek team for continuing development work on the Peristaltic Dissolution Device, as well as David Curran, Stefania Beato, Nena Mistry, Paul Connolly, Richard Lloyd, Lihua Zhang, Philip Barish, Brian Zimmerman, Mark Coffin, Alan Parr, and David DeMagistris for their technical expertise and work in support of this project.
Funding
Research was funded and performed via GlaxoSmithKline. No external funding was received for this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Burke, M.D., Koetting, M.C. Development of a Clinically Relevant Dissolution Approach to Simulate Physiological Forces with a USP 2 Apparatus: “Peristaltic Dissolution”. J Pharm Innov 16, 699–714 (2021). https://doi.org/10.1007/s12247-020-09485-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12247-020-09485-7