
Abstract
The aim of present research work was to develop self microemulsifying drug delivery system (SMEDDS) to improve the oral bioavailability of lercanidipine hydrochloride and to evaluate the effect of surfactant on the microemulsion existing area in the pseudo ternary diagram. Based on the solubility studies, capmul MCM C8 as oil, brij 35 and cremophor EL as surfactants, and propylene glycol as a co-surfactant were selected. Pseudo ternary phase diagrams were developed with two surfactants individually and the concentration of each surfactant on oil solubilization, existence of the monophasic area in a phase diagram was evaluated. A wider microemulsion existing area with greater amount of oil solubilization (37.0 %) and lesser globule size (15.02 nm) was observed with cremophor EL compared to brij 35. Formulation composed of lercanidipine hydrochloride (3.23 %), capmul MCM C8 (16.13 %), cremophor EL (53.76 %), and propylene glycol (26.88 %) was optimized based on the self-emulsification time, globule size analysis, and in vitro dissolution studies. Optimized formulation was evaluated further for UV spectra, cloud point, viscosity, robustness to dilution, transmission electron microscopy, and ex vivo permeation studies. SMEDDS was found to be promising in improving solubility and permeability of lercanidipine hydrochloride that are proven by in vitro dissolution and permeation studies.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Most of the upcoming drug molecules synthesized by the contribution of combinatorial chemistry are insoluble and demand special delivery strategies for the expected therapeutic benefit. As oral delivery is most preferred, now-a-days research is mainly focused to bring many bioavailable formulations into the market. Among various drug delivery strategies, lipid-based delivery systems, in which a drug is usually delivered by dissolving or dispersing in lipids, are being paid more attention these days. Lipid contributes to the improvement of bioavailability of poorly soluble drugs by diverse mechanisms such as improved solubility [1], improved permeability [2], stimulation of lymphatic transport [3], etc. Self micro emulsifying drug delivery systems (SMEDDS), one of the lipid-based delivery systems, is composed of lipid, surfactant and co-surfactant. SMEDDS are pre concentrates of microemulsions, as these contain all the components of microemulsions except water and produce microemulsion upon dilution with water instantaneously [4] with droplet size less than 100 nm [5]. The advantages associated with SMEDDS like ease of preparation, minimal number of unit operations, stability, and low cost offered a prime research tool for formulation scientists [4, 6].
Lercanidipine hydrochloride (LER) is an antihypertensive agent categorized under calcium channel blockers. It belongs to class IV of the Biopharmaceutical Classification (BCS) System. The bioavailability of lercanidipine hydrochloride was reported to be 10 % [7]. SMEDDS is a promising approach to improve the bioavailability of lercanidipine hydrochloride owing to the reasons stated in the following paragraphs.
Improved Solubility
Lercanidipine hydrochloride is insoluble in water with aqueous solubility of 5 μg/ml [8]. Poor water soluble drugs with dissolution rate limited absorption can be absorbed efficiently by SMEDDS with consequent stable plasma time profile [4]. Constant plasma drug levels by SMEDDS is due to presentation of poorly soluble drug in dissolved form which can bypass the dissolution step [9].
Improved Permeability
The absorption of lercanidipine hydrochloride is also limited by its poor permeability [8]. The fluidization of intestinal cell membrane by lipids and surfactants causes opening of tight junctions that contribute to the increased permeability of the drug [2, 10].
Affect on First-Pass Metabolism
Lercanidipine hydrochloride undergoes extensive metabolism in the liver as it is a substrate for CYP450IIIA4 isoenzyme [8]. Surfactants like polysorbate 80, cremophor EL, cremophor RH 40, d-α-tocopherol polyethyleneglycol (1000) succinate (TPGS) are reported to have inhibitory action on CYP 450 enzymes [11]. As the developed formulation comprises CYP inhibitory surfactant, this may contribute to the improved bioavailability of the drug.
Variable Bioavailability
Absorption of lercanidipine increases by three to four times with co-administration of food, particularly food containing high fat content. This food-dependent-improved bioavailability is not desirable and may lead to inter subject variability [8]. SMEDDS is a promising approach not only to improve oral bioavailability but also to overcome the variability in bioavailability caused by co-administration of food. As the delivery system is lipid based and hardly affected by the presence of bile salts, the effect of the co-administration of food is not significant [12, 13].
Hence, the present study aimed to develop SMEDDS of lercanidipine hydrochloride using capmul MCM C8 as oil phase, brij, and cremophor EL as surfactants and propylene glycol as a co-surfactant. With each surfactant, the effect of surfactant and co-surfactant concentration on emulsification area in the pseudo ternary phase diagram was investigated. Suitable surfactant was identified by considering the amount of oil solubilized. Self-emulsification efficiency, droplet size, percentage transmittance and enhanced in vitro dissolution profile with the lowest possible amount of surfactant were considered during optimization of formulation. Optimized formulation was evaluated further for robustness to dilution, UV spectra, cloud point, viscosity, and ex vivo permeation.
Experimental
Materials
Lercanidipine hydrochloride was obtained as a gift sample from Cipla Limited, Mumbai, Maharashtra, India. Capmul MCM C8, Miglyol 810 was a gift sample from Strides Arcolab, Bangalore, India. Brij 35 was purchased from Sigma Chemicals, St Louis, MO, USA. Tween 80 was purchased from National Chemicals, Gujarat, India. Tween 20 was purchased from SISCO Research Laboratories, Mumbai, India. Cremophor EL was obtained as gift sample from Signet Chemical Corporation Pvt. Ltd., Mumbai, India. Vegetable oils like olive oil, peanut oil, corn oil, cotton seed oil, sun flower oil, soyabean oil were purchased from Rajesh Chemicals, Mumbai, India. Capryol PGMC, Labrasol, Labrafac PG were gift samples from Gattefosse, Mumbai, India. Miglyol 517 N, Crodamol GTCC were gift samples from Dr. Reddy’s laboratories, Hyderabad, India. Lauroglycol 90, Labrafil M2125CS were gift samples from Ranbaxy Laboratories, India. Propylene glycol was purchased from Suvidinath Laboratories, Baroda, India. Polyethylene glycol 400 and Polyethylene glycol 600 were purchased from Merck Specialities Pvt. Ltd., Mumbai, India.
Methodology
Saturation Solubility Studies
Solubility studies are preliminary tests in the development of SMEDDS and are performed to select suitable excipients that show maximum solubility for the drug [14]. Drug was added in an excess amount to each excipient and mixed initially by vortexing and allowed to be shaken for 48 h using a water bath shaker at 25 °C. After 48 h, samples were subjected to centrifugation at 10,000 rpm for 5 min. The drug present in the supernatant was completely extracted into mobile phase by vortexing and analyzed for drug content with suitable dilution using HPLC [15].
HPLC Analysis of Lercanidipine Hydrochloride
A RP-HPLC method was developed and validated to determine the concentration of drug using LC 2010cHT system equipped with dual wavelength UV spectrometric detector. The stationary phase was Hibar®C18 column (250 × 4.6 mm, 5 μ) maintained at 25 °C. Mobile phase composed of acetonitrile and pH 3.5 phosphate buffer (70:30 v/v) was delivered isocratically with a flow rate 1.0 ml/min. The samples were analyzed with UV detector at wavelength of 242.0 nm. Chromatographic data was processed using LC solution 1.24 SP1 software. Typical chromatogram was depicted in Fig. 1 and validation data was provided in Table 1.

Typical chromatogram of lercanidipine hydrochloride
Construction of Pseudo Ternary Phase Diagram
Ternary diagram denotes the phase behavior of a system composed of three components. When the system to be evaluated contains three components, one of which is a combination of two different components, it is called as the pseudo ternary phase diagram [16]. These pseudo ternary phase diagrams are needed to select suitable proportion of excipients (Smix = surfactant:co-surfactant) to achieve maximum self-emulsification efficiency.
Pseudo ternary phase diagrams were developed with two surfactants i.e. brij 35 and cremophor EL individually. Two systems were evaluated, i.e., System A: capmul MCM C8, brij 35, propylene glycol; System B: capmul MCM C8, cremophor EL, propylene glycol. Water titration method was employed for construction of pseudo ternary phase diagrams. In this method, different mixtures of Smix (surfactant:co-surfactant) were prepared like 1:1, 2:1, 3:1, 4:1, 1:2 in weight ratio.
Smix ratios were selected in increasing concentration of surfactant with respect to co-surfactant (1:1, 2:1, 3:1, 4:1) and increasing concentration co-surfactant with respect to surfactant (1:1, 1:2) in order to evaluate the effect of both surfactant and co-surfactant on changes in phase behavior. For each phase diagram, Smix was mixed with oil in different weight ratios like 9:1, 8:2, 7:3, 6:4, 1:1, 4:6, 1:2, 3:7, 1:3, 1:4, 1:5, 1:6, 1:7, 1:8, and 1:9. These ratios were selected in order to cover the entire region of the phase diagram for a detailed evaluation of the system. For each combination of oil, surfactant, and co-surfactant, water was added in 5 % increments. For every incremental addition of water, the solution was kept aside for 10 min and checked for clarity, dispersibility, and flowability [14]. Then pseudo ternary phase diagram was plotted with the help of CHEMIX School v3.51 software. Compositions which formed clear solution without phase separation were denoted by symbols (dots) in the phase diagram [17]. The area formed when these points are joined signifies the microemulsion existing area [18].
Preparation of SMEDDS
SMEDDS was prepared by dissolving the drug in a mixture of oil, Smix (surfactant and co-surfactant) with initial vortex mixing followed by mixing using rotospin to ensure homogeneous mixing which was then tested for the signs of turbidity. A clear solution of SMEDDS was stored in a glass vial until use. Different formulations were prepared with brij 35 and cremophor EL separately [19]. Composition of various formulations with both systems was presented in Table 2.
Characterization of SMEDDS
Time for Emulsification and Drug Precipitation Assessment
Measurement of time required for emulsification indicates the self-emulsification capacity of the system. Precipitation assessment is helpful to predict the maintenance of solubilized form of the drug in vivo [20]. Time required for self-emulsification was assessed by adding the formulation drop wise to the beaker containing 250 ml of distilled water. The time required for the formation of clear solution was noted and was tested for any sign of drug precipitation for 12 h [21].
Percentage Transmittance
The test is indicative of the clarity of microemulsion. Percentage transmittance of diluted SMEDDS was determined by using water as blank. Water was scanned in the range of 400–800 nm and percent transmittance at 560 nm was recorded [14].
Droplet Size and Zeta Potential Analysis
SMEDDS was added to 250 ml of distilled water and the diluted formulation was subjected to droplet size, polydispersity index, and zeta potential analysis using Zetasizer (Nano ZS, Malvern Instruments, UK) at 25 °C [21]. Dynamic light scattering technique was employed for droplet size analysis. The polydispersity index (PDI) of formulation gives information regarding the pattern of size distribution and uniformity of droplet size. M3-PALS technique that utilizes both laser Doppler velocimetry and phase analysis light scattering (PALS) was used for the measurement of zeta potential. Disposable polystyrene cuvette and disposable capillary cell were used for globule size and zeta potential measurements respectively [22].
Dissolution
Drug release from the formulations was studied using USP type I dissolution apparatus. Capsules were placed into baskets of the apparatus; 0.01 N HCl was selected as dissolution media [23] and the system was maintained at 37 ± 0.5 °C. Baskets were rotated at 50 rpm [24]. Formulation was filled into the size 0 hard gelatin capsules prior to dissolution. Samples (5.0 ml) were withdrawn at regular time intervals and replaced with fresh media to maintain the sink conditions. Samples were filtered (0.22 μm) and analyzed for drug content using UV spectrophotometer at 242 nm [25]. Samples from the placebo were used as blank to avoid interferences from the components of the formulation in recording the UV absorbance.
Robustness to Dilution
Formulation was subjected to the different extent of dilutions, i.e. 50 fold, 100 fold, 1000 fold with water to evaluate the effect of volume of a medium on the dispersion, drug precipitation and phase separation. The formulation was also tested for clarity and drug precipitation in 0.1 N hydrochloric acid, 6.8 pH phosphate buffer, and 7.4 pH phosphate buffers to evaluate the effect of pH of the dispersion media. The resultant solutions were observed visually for any changes initially and after 12 h [26].
Transmission Electron Microscope
Droplets produced upon dilution of SMEDDS were examined under the transmission electron microscope (TEM; Hitachi H 7650, Canada). Sample stained with uranyl acetate was placed on formvar-coated copper grids and allowed to dry. The dried specimen was examined under microscopy at an accelerated voltage of 80 kV [26].
Thermodynamic Stability Studies
Formulation was subjected to thermodynamic stability studies, i.e., centrifugation and freeze thaw tests to evaluate the phase separation and drug precipitation after dilution. Centrifugation was performed at 5000 rpm for 5 min. Formulations were subjected to three freeze thaw cycles that included freezing at −4 °C for 24 h and thawing at +40 °C for 24 h [21].
Ultra Violet Spectra of Microemulsion
Entrapment of drug with locus can be assessed by recording UV spectra [27]. Spectra were recorded for individual excipients containing the dissolved drug, i.e., capmul MCM C8, propylene glycol using pure excipients as blank. A spectrum of diluted SMEDDS was recorded by scanning the placebo for baseline. The spectra were recorded from 200–400 nm range using UV–Visible spectrophotometer (UV-1601 Shimadzu spectrophotometer, Japan).
Cloud Point
The effect of temperature on the performance of SMEDDS can be evaluated by the determination of cloud point. The diluted formulation was kept in a water bath (Julabo TW20, USA) and temperature was increased gradually. The formulation was allowed to keep for 10 min at each incremental temperature (30, 35, 40, 45, 50, 60, 70, and 80 °C). Percentage transmittance was determined at each increment in temperature and compared with initial value. The temperature at which the percentage transmittance drops significantly was noted [26].
Viscosity
Viscosity of the SMEDDS was determined by LVDV-III Ultra programmable rheometer (Cone/plate rheometer, Brookfield Engineering Laboratories, Middleboro, MA, USA) using spindle 40 at 25 ± 1 °C. In this, the spindle was immersed in the fluid whose viscosity is to be tested and is driven through a spring. The spring’s deflection gives an indication about the resistance of the fluid to flow against the spindle. The viscosity of the sample was measured at maximum torque, i.e., 100 % [28].
Ex Vivo (Non-Everted Intestinal Sac) Permeation Studies
The non-everted intestinal sac method was employed to evaluate the permeability of the drug from the diluted formulation and from the drug solution through the gastrointestinal tract. The animals were fasted overnight and had only access to water to clear the gastric contents. Male Wistar rats were sacrificed by intra peritoneal administration of excess anaesthetic agent. Intestinal segment between the upper end of the duodenum and ileum was isolated and cut into small parts having 7 cm length. Each part is washed with normal saline solution and filled with diluted formulation and pure drug solution (5 mg/0.5 ml) individually. Both ends were tied, and 5 cm of effective length was maintained for drug permeation. Then, it was placed in a beaker containing the media (Phosphate buffer saline pH 7.4) with aeration (10–15 bubbles/min) maintained at 37.0 ± 1 °C on a magnetic stirrer with a speed of 50 rpm. At regular intervals, 2.0 ml of sample was withdrawn and sink conditions were maintained by adding the same amount of fresh media. Cumulative amount of drug permeated per unit area of sac and flux were determined assuming intestine in cylindrical shape. The results expressed were averages of triplicate experiments [29].
Statistical Analysis
All the measurements in the study were carried out three times and the data was expressed as mean ± standard deviation (SD). A two-tailed unpaired student’s t test was performed to test statistical significance. Statistical probability (p), p < 0.01 was considered as statistically significant (99 % CI).
Stability Studies
The optimized formulation was subjected to stability studies by storing the samples at accelerated stability storage conditions of 40 ± 2 °C/75 ± 5 % RH for 3 months as per ICH guidelines. SMEDDS was filled in a glass vial sealed with rubber cap and crimped for storage in the stability chamber (Thermolab, Mumbai, India). Samples were analyzed for self-emulsification and globule size [27, 30].
Results and Discussion
Solubility Studies
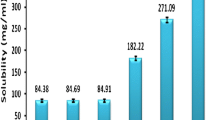
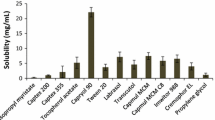
Solubility studies play an important role in the design of SMEDDS as drug loading, drug precipitation, and total volume of the formulation depend on the proper selection of excipients. Solubility of the drug in various excipients was shown in Fig. 2. Selection of excipients in which the drug exhibits superior solubility would avoid drug precipitation in vitro and in vivo, improve drug loading, and help to present the drug in a solubilized form in the gastrointestinal tract for efficient absorption. The capacity of oil to hold the drug and the capacity of surfactant to emulsify maximum amount of oil can be improved by using a proper combination of excipients [14].

Solubility of lercanidipine hydrochloride in various oils (a), surfactants (b) and co-surfactants (c); Data represented as mean ± SD (n = 3)
The solubility of lercanidipine hydrochloride was studied in various oils and maximum solubility was observed with capmul MCM C8 which is a medium chain mono glyceride (glyceryl monocaprylate). Oil phase is the important component of the formulation as maintenance of drug in solubilized form depends on the type and amount of oil. Solubility of lercanidipine hydrochloride was found to be less in various fixed oils tested, which might be due to lower solubilization capacity of long chain glycerides compared to medium chain glycerides. Capmul MCM C8 belongs to small molecular volume oils in which case emulsification was investigated to be easy and also found to have good solubilization capacity [16, 31].
Brij 35 and cremophor EL were chosen as surfactants as these excipients are found to have superior solubility for the drug. The selected surfactants belong to the category of non-ionic surfactants, which are proven to be safe and effective [32]. The hydrophilic and lipophilic balance (HLB) of two surfactants was high by which they form O/W microemulsion upon dilution as hydrophilicity of surfactant is the prime parameter to be considered for the formation of O/W emulsions. The drug was found to exhibit greater solubility in propylene glycol among various co-surfactants tested, which is non volatile and compatible with gelatin capsules compared to alcoholic co-surfactants. Among the tested co-surfactants, propylene glycol is relatively non polar and high solubilization of lercanidipine hydrochloride might be due to its non polar nature. Solubilization of oil can be improved by using propylene glycol as the improved solubility in the presence of polyols such as propylene glycol and glycerol was well investigated [33].
Construction of Pseudo Ternary Phase Diagram
Construction of a ternary phase diagram requires careful observation after each addition of water. The change in phase behavior of the system according to the change in composition can be evaluated by pseudo ternary phase diagrams. Pseudo ternary phase diagrams are helpful to select suitable concentrations of excipients that form mono phasic microemulsions [24]. In both systems, capmul MCM C8 was taken as oil and propylene glycol as co-surfactant.
During water titration, the mixtures were kept aside for 10 min after each addition of water to evaluate the appearance of metastable forms [34]. The emulsification area was found greater with cremophor EL compared to brij 35. The area represents the emulsification ability of the system and the higher the area, the higher the emulsification ability. Pseudo ternary phase diagrams were depicted in Fig. 3. With brij 35 (system A), the microemulsion existing area was increased with an increased amount of surfactant relative to co-surfactant. This was confirmed by the phase diagrams drawn with surfactant to co-surfactant ratios of 1:1, 2:1, 3:1. However, when the surfactant was increased from 3:1 to 4:1, the microemulsion area was decreased. From the results, it is evident that the maximum amount of oil solubilized was about 23.0 % and the highest microemulsion existing area was found with Smix ratio of 3:1. With cremophor EL (system B), microemulsion existing area was increased from Smix ratio of 1:1 to 1:2 and when the surfactant was increased beyond this, the area was decreased. The maximum amount of oil solubilized was about 37.0 % with cremophor EL. The highest microemulsion existing area was found with Smix ratio of 2:1.

a Pseudo ternary phase diagrams of system A (formulations using brij 35) with varying Smix ratios (1:1, 2:1, 3:1, 4:1, and 1:2). Shaded region represnets microemulsion existence region. b Pseudo ternary phase diagrams of system B (formulations using cremophor EL) with varying Smix ratios (1:1, 2:1, 3:1, and 1:2). Shaded region represents microemulsion existence region
The increase in microemulsion existing area with an increased Smix indicates the efficiency of surfactant in reducing interfacial tension between oil and water. In both the systems, increase in the concentration of surfactant above certain range decreased microemulsion existing area. This is attributed to an increase in the viscosity of the system hindering self-emulsification [35]. This might be the reason for decreased monophasic area from Smix ratio of 3:1 to 4:1 in case of system A and from 2:1 to 3:1 in case of the system B.
As the amount of co-surfactant increased, the self-emulsification area was found decreased in both the systems, and dispersion yielded cloudy solutions with subsequent phase separation. Because of the polar nature of the co-surfactants, they get partitioned into the aqueous phase upon dispersion and may lead to decrease in self-emulsification efficiency [36]. In the case of phase diagrams prepared with an increased amount of co-surfactant, the clear zone was not found to be extended towards the apex of the water, which clearly signifies the loss in emulsification efficiency with infinite dilution as formulations in which the clear zone extended towards the water apex can only be diluted to the maximum extent [34].
From the phase diagrams, it can be concluded that optimal ratio of surfactant and co-surfactant is necessary for the achievement of superior self-emulsification. The property of the surfactant that makes the interfacial film condense and the property of the co-surfactant that expands the interfacial film have effects on the self-microemulsifying property of the dosage form, and both are concentration dependent. Concentrations of surfactant and co-surfactant should be optimized to achieve the lowest possible droplet size and to impart robustness to the formulation upon dilution.
Characterization of SMEDDS
Time for Emulsification and Drug Precipitation Assessment
The time required for self-emulsification of the prepared formulations for both systems was presented in Table 3. As the free energy of formation of microemulsion is low and negative, the formation of microemulsion from SMEDDS is instantaneous. The presence of co-surfactant in suitable proportion imparts flexibility to the interface by which spontaneity in formation of microemulsion is made possible. Emulsification time was shorter for F3 of the system A and F13 and F14 of the system B. No drug precipitation was observed for 12 h [19]. The absence of drug precipitation indicates the optimum proportion of oil and surfactant in the formulation.
Percentage Transmittance
Percentage transmittance of the prepared formulations with both brij and cremophor EL was presented in Table 3. The transmittance values near 100 % indicate the transparency of the formulations which in turn indicate the formation of microemulsion with nano size droplets [14]. The transmittance values were near to the pure water except for F1 of system A and F8, F9, and F10 of system B.
Droplet Size and Zeta Potential Analysis
Droplet size plays a vital role in the drug release profile and thereby absorption of the drug through the gastrointestinal membrane. Droplets of nano size range impart higher surface area for partitioning of the drug [37]. In system A, different formulations were prepared using Smix 3:1 as this ratio has shown higher emulsification region. Formulations were prepared by varying oil to Smix ratio, i.e. from F1–F7 and subjected to globule size and zeta potential analysis after dilution with aqueous phase. In system B, different formulations were prepared using Smix 2:1, as this ratio has shown higher emulsification region. Formulations were prepared with different oil to Smix ratio, i.e., from F8–F16 and subjected to globule size and zeta potential analysis after dilution with aqueous phase. Droplet size and zeta potential of prepared formulations of both systems are presented in Table 3.
In both systems, the globule size was found to be decreased from oil to Smix ratios of 4:6 to 1:5 (1:3, 1:4, 1:5) and beyond this, the size was found to be constant up to 1:9. As it is desirable to use surfactant concentration as minimum as possible, oil to Smix ratio of 1:5 was selected as optimum in both systems based on globule size. Globule size and zeta potential graphs of optimized formulations of both systems are shown in Fig. 4.

Size (a) and zeta potential (b) distribution graphs of optimized formulation of System A (F3 - formulation using brij 35); size (c) and zeta potential (d) distribution graphs of optimized formulation of system B (F13—formulation using cremophor EL)
The decreased globule size with an increased amount of surfactant is due to the functional property of surfactant, i.e., reduction in interfacial tension and increased availability of surfactant at the interfacial region for droplet stabilization [38].
Less globule size was observed with system B than system A and was thought to be due to greater emulsification efficiency of cremophor EL than brij 35. The results of polydispersity index indicate the uniformity in droplet size distribution as these values lie below 0.327. As the magnitude of zeta potential indicates the stability of the colloidal system, the stable dispersed system was attributed to higher zeta values and also indicated the absence of aggregation [39].
When two systems are compared, cremophor EL (system B) was found to solubilize high amount of oil (37 %) compared to brij 35 (system A) (23 %) which has an impact on drug loading. Due to high solubilization capacity of oil, system B can incorporate a high amount of drug without precipitation and the optimized Smix include high surfactant increase of system A (Smix 3:1 = 75 %) compared to system B (Smix 2:1 = 66 %).
As the use of high amount of surfactant may cause undesirable effects, it is important to reduce the concentration of surfactant to the minimum extent possible [40]. When droplet size was compared, the lowest possible droplet size was observed with formulation (F13) of system B (15.84 nm) that is lower than that of system A (F3) (52.02 nm). High solubilization capacity and less globule size with relatively low surfactant concentration was observed with system B compared to the system A. The advantages associated with system B made it to be preferred than the system A. Hence, system B employing cremophor EL was selected and analyzed further.
Dissolution Studies
From system B, F10, F11, F12, F13, F14, and F15 were selected for in vitro dissolution studies based on droplet size and self-emulsification time. Release rate from all the formulations was high compared to plain drug. In vitro dissolution profile of lercanidipine from SMEDDS in comparison to pure drug was depicted in Fig. 5. Among tested formulations, drug release rate was increased from F10 to F13, and F14 has shown similar release profile with F13. The increase in release rate might be due to decreased droplet size by which surface area for drug partition increases significantly [27, 34]. Drug release from pure drug was only 25.72 % in 60.0 min whereas about 90.0 % release was observed from all formulations in 60 min indicates increased release rate. F13 and F14 has shown the complete release of drug with enhanced release rate indicating the efficiency of SMEDDS in improving both rate and extent of drug release. From the results, F13 (oil:Smix 1:5) was optimized as less droplet size, less self-emulsification time, and better drug release profile were observed with the said ratio.

Dissolution profile of lercanidipine hydrochloride (mean percent release ± SD, n = 3) from formulations (F10–F14) of system B (formulations using cremophor EL) and pure drug
Robustness to Dilution
Optimized formulation was subjected to different dilutions, i.e., 50-, 500-, and 1000-fold with water. Droplet size was found to be 18.73 ± 3.5 nm, 16.77 ± 2.54 nm, 20.80 ± 3.05 nm for 50, 500, and 1000 mL dilutions, respectively. The dispersion was found to be clear without drug precipitation at all dilutions and no significant change in droplet size was observed. The dispersions were found to be stable for 12 h indicating robustness of formulation to dilution [26]. It was investigated that the presence of co-surfactant in the optimum amount ensures flexibility to interface and makes formulation to be stable and robust to dilution [41]. The formulation was also found to form a stable dispersion in 0.1 N hydrochloric acid, 6.8 pH phosphate buffer, and 7.4 pH buffers without any sign of precipitation of drug for 12 h indicating the stability in performance of SMEDDS in vivo as the pH varies along the gastrointestinal tract.
Transmission Electron Microscopy
Droplets produced upon dilution of SMEDDS were visualized under high magnification (Fig. 6). TEM photograph confirmed that the globules are of nano size. The droplet size is in agreement with the results obtained from droplet size analysis using the zetasizer

Transmission electron microscopy of optimized formulation (F13)
Thermodynamic Stability Studies
The formulation was found to be stable without any phase separation. Stability of diluted formulation upon stress imparted by centrifugation and freeze thaw tests might be due to low free energy involved in the formation of microemulsion [21].
Ultra Violet Spectra of Microemulsion
The maximum wavelengths of UV absorption were 359.0 nm, 239.2 nm for drug dissolved in capmul MCM C8 and propylene glycol respectively (Fig. 7). The maximum wavelength for microemulsion was 240.0 nm and is closer to that of drug dissolved in propylene glycol. The closer λmax of the two solutions indicates the presence of the drug in the interfacial region of microemulsion [27].

Ultraviolet spectra indicating the location of drug in diluted formulation of SMEDDS (a) and drug dissolved in propylene glycol (b)
Cloud Point
Temperature has an effect on functional property of surfactant, especially surfactants containing polyoxyethylene moiety as these undergo dehydration at high temperature that causes phase separation of emulsion with subsequent cloudiness [26]. The diluted formulation was found to be clear and transparent up to 70 °C and above this temperature, it turned cloudy with a sudden drop in percentage transmittance value to 65.3 from initial 99.6. Drop in percentage transmittance was represented in Fig. 8 From the test, it is evident that the formulation was stable without drug precipitation and phase changes up to 70 °C indicating its stability at body temperature.

Drop in percentage transmittance at 70 °C indicating the absence of drug precipitation at body temperature
Viscosity
The ease of emulsification is governed by viscosity of the formulation. Viscosity is an important in vitro physical parameter to evaluate the filling feasibility and filling rate of the fluid into soft gelatin capsules during its production [28]. Optimized SMEDDS has shown a viscosity of 304.7 cP. The lesser viscosity of SMEDDS (<10,000 cps) indicates the feasibility of filling in large-scale capsule production [42].
Ex Vivo (Non-Everted Intestinal Sac) Permeation Studies
Cumulative amount of drug permeated with respect to time from the pure drug solution (control) and diluted SMEDDS was shown in Fig. 9. The amount of drug permeated per unit area of sac (permeability) were 974.89 and 430.80 μg/cm2 with diluted SMEDDS and pure drug solution, respectively in 3 h (p < 0.01). Increase in amount of drug permeation for about 2.26 times was observed with diluted SMEDDS compared to pure drug. The rate of permeation (flux) was found to be 324.96 and 143.60 μg/cm2/h from the diluted SMEDDS and control respectively. The enhanced permeation is due to less droplet size and also due to the surfactant partitioning effect through the intestinal barrier for drug permeation [4]. The result indicates that the absorption of drug can be improved significantly from SMEDDS. As lercanidipine hydrochloride belongs to class IV of the BCS classification, permeability problem associated with oral administration of the drug can be avoided with SMEDDS.

Ex vivo permeation profile (mean percent release ± SD, n = 3) of pure drug and optimized formulation (F13)
Stability Studies
Stability samples were evaluated for self-emulsification efficiency and droplet size. There was no significant change in the droplet size and self-emulsification capacity. Stability parameters are presented in Table 4. No change in physical appearance and droplet size upon dilution with initial samples indicates the stability of SMEDDS.
Conclusion
The present study demonstrated that lercanidipine hydrochloride, a poorly soluble drug, can be formulated as SMEDDS to improve the oral bioavailability by improving both the solubility and permeability. Cremophor EL was found to solubilize capmul MCM C8 to a significant extent compared to brij 35 at relatively low concentration. Formulation with cremophor EL was optimized based on the results of amount of oil solubilized, globule size analysis, self-emulsification efficiency, percentage transmittance, in vitro dissolution, and ex vivo permeation studies. As cremophor EL is known to inhibit CYP enzymes, which are responsible for the first-pass metabolism of the drug, the optimized formulation would be a promising dosage form to overcome all associated problems of the oral delivery of the drug, i.e., solubility, permeability, presystemic metabolism, and variable bioavailability with co-administration of food.
References
Humberstone AJ, Charman WN. Lipid-based vehicles for the oral delivery of poorly water soluble drugs. Adv Drug Deliv Rev. 1997;25:103–28.
Jannin V, Musakhanian J, Marchaud D. Approaches for the development of solid and semi solid lipid based formulations. Adv Drug Deliv Rev. 2008;60:734–46.
Stegemann S, Leveiller F, Franchic D, De Jong H, Lind´en H. When poor solubility becomes an issue: from early stage to proof of concept. Eur J Pharm Sci. 2007;31:249–61.
Gursoy RN, Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother. 2004;58:173–82.
Sarciaux JM, Acar L, Sado PA. Using microemulsion formulations for oral drug delivery of therapeutic peptides. Int J Pharm. 1995;120:127–36.
Constantinides PP, Scalart J, Lancaster C, Marcello J, Arks G, Ellens H, et al. Formulation and intestinal absorption enhancement evaluation of water-in-oil micro emulsions incorporating medium-chain glycerides. Pharm Res. 1994;11:1385–90.
Charde S, Mudgal M, Kumar L, Saha R. Development and evaluation of buccoadhesive controlled release tablets of lercanidipine. AAPS PharmSciTech. 2008;9:182–90.
G.D. Mahendra, C. Anil, K.P. Ram, R.A. Todd, A. Wattanaporn, R.K. Suneel, inventor; Forest Laboratories, Inc., assignee. Lercanidipine pH dependent pulsatile release compositions. United States Patent 20060165788 A1. 2006 July 27.
Narang AS, Delmarre D, Gao D. Stable drug encapsulation on micelles and micro emulsions. Int J Pharm. 2007;345:9–25.
Buyukozturk F, Benneyan J, Carrier RL. Impact of emulsion-based drug delivery systems on intestinal permeability and drug release kinetics. J Control Release. 2010;142:22–30.
Christiansen A, Backensfeld T, Denner K, Weitschies W. Effects of non-ionic surfactants on cytochrome P450-mediated metabolism in vitro. Eur J Pharm Biopharm. 2011;78:166–72.
Kawakami K, Yoshikawa T, Hayashi T, Nishihara Y, Masuda K. Microemulsion formulation for enhanced absorption of poorly soluble drugs. II. In vivo study. J Control Release. 2002;81:75–82.
Woo JS, Song YK, Hong JY, Lim SJ, Kim CK. Reduced food-effect and enhanced bioavailability of a self-microemulsifying formulation of itraconazole in healthy volunteers. Eur J Pharm Sci. 2008;33:159–65.
Singh AK, Chaurasiya A, Awasthi A, Mishra G, Asati D, Khar RK, et al. Oral bioavailability enhancement of exemestane from self-microemulsifying drug delivery system (SMEDDS). AAPS PharmSciTech. 2009;10:906–16.
Yin YM, Cui FD, Mu CF, Choi MK, Kim JS, Chung SJ, et al. Docetaxel microemulsion for enhanced oral bioavailability: preparation and in vitro and in vivo evaluation. J Control Release. 2009;140:86–94.
Lawrence MJ, Rees GD. Micro emulsion based media as novel drug delivery systems. Adv Drug Deliv Rev. 2000;45:89–121.
Zhang H, Cui Y, Zhu S, Feng F, Zheng X. Characterization and antimicrobial activity of a pharmaceutical microemulsion. Int J Pharm. 2010;395:154–60.
Ghosh PK, Majithiya RJ, Umrethia ML, Murthy RSR. Design and development of micro emulsion drug delivery system of acyclovir for improvement of oral bioavailability. AAPS PharmSciTech. 2006;7:172–77.
Balakrishnan P, Lee BJ, Oh DH, Kim JO, Lee YI, Kim DD, et al. Enhanced oral bioavailability of coenzyme Q10 by self emulsifying drug delivery systems. Int J Pharm. 2009;374:66–72.
Grove M, Mullertz A, Nielsen JL, Pedersen GP. Bioavailability of seocalcitol II: development and characterisation of self-microemulsifying drug delivery systems (SMEDDS) for oral administration containing medium and long chain triglycerides. Eur J Pharm Sci. 2006;28:233–42.
Patel AR, Vavia PR. Preparation and in vivo evaluation of SMEDDS (self-microemulsifying drug delivery system) containing fenofibrate. The AAPS J. 2007;9:344–52.
Sharma G, Wilson K, Vander Walle CF, Sattar N, Petrie JR, Ravi Kumar MNV. Micro emulsions for oral delivery of insulin: design, development, evaluation in streptozocin induced diabetic rats. Eur J Pharm Biopharm. 2010;76:159–69.
G.D. Mahendra, C. Anil, R.K. Suneel, inventors; Forest Laboratories, Inc., assignee. Lercanidipine immediate release compositions. United States Patent: US 2006/0134212 A1. 2006, June 22.
Cui J, Yu B, Zhao Y, Zhu W, Li H, Lou H, et al. Enhancement of oral absorption of curcumin by self-microemulsifying drug delivery systems. Int J Pharm. 2009;371:148–55.
Basalious EB, Shawky N, Badr-Eldin SM. SNEDDS containing bioenhancers for the improvement of dissolution and oral absorption of lacidipine: development & optimization. Int J Pharm. 2010;391:203–11.
Elnaggar YS, El-Massik MA, Abdallah OY. Self-nanoemulsifying drug delivery systems of tamoxifen citrate: design and optimization. Int J Pharm. 2009;380:133–41.
Li X, Yuan Q, Huang Y, Zhou Y, Liu Y. Development of silymarin self-microemulsifying drug delivery system with enhanced oral bioavailability. AAPS PharmSciTech. 2010;11:672–78.
Bali V, Ali M, Ali J. Nanocarrier for the enhanced bioavailability of a cardiovascular agent: in vitro, pharmacodynamic, pharmacokinetic and stability assessment. Int J Pharm. 2011;403:46–56.
Shishu K, Maheshwari M. Development and evaluation of novel microemulsion based oral formulations of 5-fluorouracil using non-everted rat intestine sac model. Drug Dev Ind Pharm. 2012;38:294–300.
Borhade V, Nair H, Hegde D. Design and evaluation of self-microemulsifying drug delivery system (SMEDDS) of tacrolimus. AAPS PharmSciTech. 2008;9:13–21.
Bandivadekar MM, Pancholi SS. Self-microemulsifying smaller molecular volume oil (Capmul MCM) using non-ionic surfactants: a delivery system for poorly water-soluble drug. Drug Dev Ind Pharm. 2011;1–10.
Hauss DJ. Oral lipid based formulations. Adv Drug Deliv Rev. 2007;59:667–76.
Karamustafa F, Celebi N. Development of an oral microemulsion formulation of alendronate: effects of oil and co-surfactant type on phase behavior. J Microencapsul. 2008;25:315–23.
Shafiq S, Shakeel F, Talegaonkar S, Ahmad FJ, Khar RK, Ali M. Development and bioavailability assessment of ramipril nanoemulsion formulation. Eur J Pharm Biopharm. 2007;66:227–43.
Pouton CW. Formulation of self-emulsifying drug delivery systems. Adv Drug Deliv Rev. 1997;25:47–58.
Müllertz A, Ogbonna A, Ren S, Rades T. New perspectives on lipid and surfactant based drug delivery systems for oral delivery of poorly soluble drugs. J Pharm Pharmacol. 2010;62:1622–36.
Gershanik T, Benita S. Self-dispersing lipid formulations for improving oral absorption of lipophilic drugs. Eur J Pharm Biopharm. 2000;50:179–88.
Barakat SN. Enhanced oral bioavailability of etodolac by self-emulsifying systems: in-vitro and in-vivo evaluation. J Pharm Pharmacol. 2010;62:173–80.
Parmar N, Singla N, Amin S, Kohli K. Study of cosurfactant effect on nanoemulsifying area and development of lercanidipine loaded (SNEDDS) self nanoemulsifying drug delivery system. Colloids Surf B. 2011;86:327–38.
Constantinides PP. Lipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects. Pharm Res. 1995;12:1561–72.
Bagwe RP, Kanicky JR, Palla BJ, Patanjali PK, Shah DO. Improved drug delivery using microemulsions: rationale, recent progress, and new horizons. Crit Rev Ther Drug Carrier Syst. 2001;18:77–140.
Gupta S, Chavhan S, Sawant KK. Self-nanoemulsifying drug delivery system for adefovir dipivoxil: design, characterization, in vitro and ex vivo evaluation. Colloids and Surfaces A: Physicochem Eng. 2011;392:145–55.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Akula, S., Gurram, A.K., Devireddy, S.R. et al. Evaluation of Surfactant Effect on Self Micro Emulsifying Drug Delivery System (SMEDDS) of Lercanidipine Hydrochloride: Formulation and Evaluation. J Pharm Innov 10, 374–387 (2015). https://doi.org/10.1007/s12247-015-9233-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12247-015-9233-6